

Key Points
LoF variants in BMF and hematologic malignancy predisposition genes occur in >4 per 1000 individuals in the general population.
Mutational constraint analysis can predict penetrance, severity, and molecular pathogenesis of rare genetic diseases.

Abstract
Inherited bone marrow failure (IBMF) syndromes are rare blood disorders characterized by hematopoietic cell dysfunction and predisposition to hematologic malignancies. Despite advances in the understanding of molecular pathogenesis of these heterogeneous diseases, genetic variant interpretation, genotype–phenotype correlation, and outcome prognostication remain difficult. As new IBMF and other myelodysplastic syndrome (MDS) predisposition genes continue to be discovered (frequently in small kindred studies), there is an increasing need for a systematic framework to evaluate penetrance and prevalence of mutations in genes associated with IBMF phenotypes. To address this need, we analyzed population-based genomic data from >125 000 individuals in the Genome Aggregation Database for loss-of-function (LoF) variants in 100 genes associated with IBMF. LoF variants in genes associated with IBMF/MDS were present in 0.426% of individuals. Heterozygous LoF variants in genes in which haploinsufficiency is associated with IBMF/MDS were identified in 0.422% of the population; homozygous LoF variants associated with autosomal recessive IBMF/MDS diseases were identified in only .004% of the cohort. Using age distribution of LoF variants and 2 measures of mutational constraint, LOEUF (“loss-of-function observed/expected upper bound fraction”) and pLI (“probability of being loss-of-function intolerance”), we evaluated the pathogenicity, tolerance, and age-related penetrance of LoF mutations in specific genes associated with IBMF syndromes. This analysis led to insights into rare IBMF diseases, including syndromes associated with DHX34, MDM4, RAD51, SRP54, and WIPF1. Our results provide an important population-based framework for the interpretation of LoF variant pathogenicity in rare and emerging IBMF syndromes.
Introduction
Inherited bone marrow failure (IBMF) and genetic syndromes predisposing to myelodysplastic syndrome (MDS) and myeloid malignancies represent a disparate group of rare diseases, linked by common downstream pathophysiology of hematopoietic stem and progenitor cell dysfunction.1-3 In these disorders, failed regulation of hematopoiesis, either at the stem cell level or in lineage-specific progenitor cells, increases the likelihood for compensatory development of somatic genetic alterations associated with malignant potential.4,5 These syndromes include inherited causes of trilineage bone marrow aplasia such as Fanconi anemia (FA) and telomere biology disorders, as well as diseases associated with single lineage failure such as Diamond-Blackfan anemia (DBA), congenital neutropenias, and inherited thrombocytopenias.6-12 They also include recently described disorders such as GATA2 haploinsufficiency and SAMD9/SAMD9L syndromes, which have variable impacts on hematopoietic function but carry a high risk of MDS transformation.13-15
IBMF and hematologic malignancy predisposition syndromes have widely variable phenotypes and penetrance, even within families, making prognostic counseling of patients and families difficult. For example, not every patient with a heterozygous loss-of-function (LoF) RTEL1 variant will progress to BMF or develop other manifestations of short telomere syndromes, including pulmonary fibrosis; however, the likelihood of these outcomes remains poorly defined.16,17 Deleterious effects of heterozygous loss of IBMF genes that mediate clinical phenotypes with biallelic inactivation (eg, genes associated with FA) also remain incompletely defined.1,18 Clarifying genotype–phenotype correlation and disease penetrance is vital to helping patients and families make informed decisions about therapeutic options such as hematopoietic stem cell transplantation or family planning. As molecular diagnostic capabilities have increased genetic testing, there is an urgent need to improve our understanding of the significance of putative pathogenic variants in IBMF genes.1,19,20
In the current study, we estimate the frequency of predicted LoF (pLoF) variants in IBMF-associated genes in the general population using a large population database, encompassing exomes of 125 748 individuals. We characterize these genes according to variant occurrence compared with an expected frequency based on gene size, mutability, and methylation status, defining evolutionary constraint against LoF alleles. We separately examine genes associated with IBMF syndromes for which heterozygous LoF is tolerated, allowing for true carrier status and the possibility of incomplete penetrance.
Methods
Genome database
The Genome Aggregation Database (gnomAD; version 2.1.1) has assembled exome sequence data from 125 748 unrelated individuals with a median age of 55 years but spanning the spectrum of adulthood.21 The gnomAD excludes individuals with severe pediatric-onset diseases, including IBMF syndromes and malignancies. Demographic characteristics of the gnomAD data set and the cohorts from which it is derived are summarized in supplemental Tables 1 and 2. For validation, we used 2 independent genome data sets of 71 702 sequenced genomes included in gnomAD version 3.0 and 15 708 genomes included in gnomAD version 2.1.1 (supplemental Tables 3 and 4).
IBMF gene selection
One hundred genes with known variants associated with IBMF/MDS predisposition were interrogated for pLoF variants. This gene panel contains genes included in the Clinical Laboratory Improvement Amendments–approved IBMF next-generation sequencing panel at the Children’s Hospital of Philadelphia (CHOP), supplemented with IBMF/MDS-associated genes reported after the creation of the CHOP IBMF panel.22 Genes that mediate disease through gain of function or similar dominant-negative mechanisms (eg, ELANE) were excluded.
LoF variants
We used the Loss-Of-Function Transcript Effect Estimator (LOFTEE), a stringent filtering process, to identify high-confidence pLoF variants in IBMF and predisposition syndrome-associated genes.21 As previously described, the variant effect predictor identifies high-confidence pLoF variants that cause premature stop, frameshift, or alter 2 essential splice site nucleotides. Putative variants were filtered through LOFTEE, and variants predicted to escape nonsense-mediated decay were removed. As quality control, variants from a subgroup of genes were manually curated, showing that the computational filtering algorithms stringently selected true LoF variants. Observed unique pLoF variants arising from single-nucleotide variants (SNVs) were reported for each gene and compared with expected number of pLoF variants by using previously described algorithms that incorporated variables such as gene size, mutability, and methylation status. The aggregate frequency of all pLoF variants in each gene was determined as a summation of SNVs and insertions/deletions predicted to cause LoF. We estimated mutational burden for each gene and, in aggregate, for each disease subgroup (eg, telomere biology disorders, inherited red blood cell disorders). Genes with a reported clinical phenotype in both haploinsufficiency and biallelic inactivation states had frequency for haploinsufficiency calculated. A diagram of the analytical workflow is shown in Figure 1.
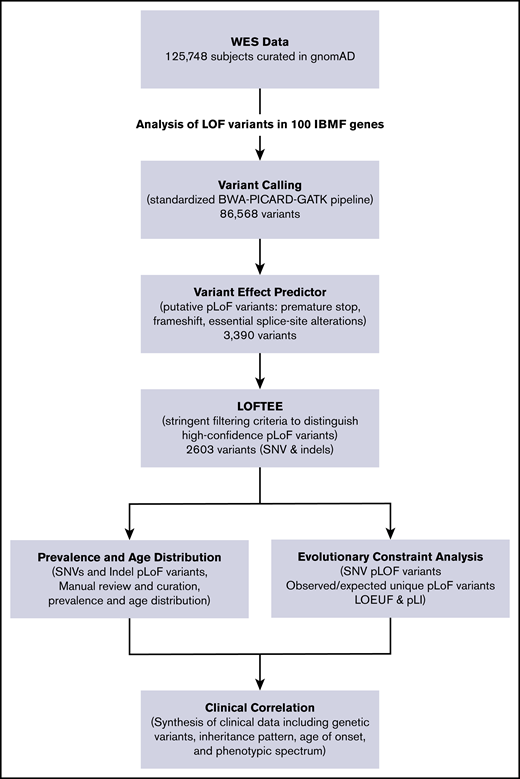
IBMF variant analysis pipeline. The flowchart of the analytical workflow for analysis of pLoF variants in 100 genes linked to IBMF and hematologic malignancy predisposition. RPS17 is located in a segmental duplication and thus is not amenable to sequence-based analysis in the gnomAD data set. indels, insertions/deletions; WES, whole-exome sequencing.
IBMF variant analysis pipeline. The flowchart of the analytical workflow for analysis of pLoF variants in 100 genes linked to IBMF and hematologic malignancy predisposition. RPS17 is located in a segmental duplication and thus is not amenable to sequence-based analysis in the gnomAD data set. indels, insertions/deletions; WES, whole-exome sequencing.
Gene tolerance/intolerance (evolutionary constraint) calculations
The proportion of haplotypes with pLoF variants was computed as previously described to determine aggregate pLoF frequency for each gene.21 These data were analyzed by using 2 metrics of mutational constraint that detect depletion of variation in recent human evolution. The first, “the loss-of-function observed/expected upper bound fraction” (LOEUF), represents a conservative estimate of the ratio of observed to expected pLoF variants. LOEUF for IBMF genes was calculated as described previously, by determining the observed/expected ratio of mutations in each gene and calculating the confidence interval around that ratio. The upper bound of the confidence interval was used as a conservative estimate of the observed/expected ratio. For ease of interpretation, the observed/expected upper bound estimates for each gene in the human genome were binned into deciles of ∼1920 genes each. The LOEUF deciles range from 0 (most depleted/evolutionarily constrained) to 9 (not depleted/constrained). Each gene was also analyzed by using the “probability of loss-of-function intolerance” score (pLI). pLI has previously been established to estimate the probability that LoF in one gene allele causes a haploinsufficient phenotype and estimates the likelihood that a gene falls into the class of LoF-haploinsufficient genes. pLI was previously shown to separate genes of adequate length into those intolerant (pLI ≥ 0.9) or tolerant (pLI ≤ 0.1) to LoF.23
Statistical analysis
The Student t test was used to compare pLoF depletion/constraint in IBMF-associated genes vs the remaining genes in the genome. Fisher’s exact test was used for sex distribution analysis. LOEUF scores were previously statistically validated.21 The gnomAD data set of 125 748 exomes provided sufficient power to enable LOEUF score calculation for all genes in which the expected pLoF variant frequency was >9.2 variants in the entire exome cohort. For genes that had expected pLoF <9.2, we included observed and expected pLoF and variant frequencies but not LOEUF decile or pLI.
Results
Prevalence of IBMF-associated LoF variants in the general population
Using the stringent algorithm LOFTEE to analyze 125 748 exomes for high-confidence pLoF variants in IBMF genes, 2603 unique pLoF variants were identified in 100 IBMF genes (Figure 1). In total, SNV and insertion/deletion pLoF variants were identified in 0.426% of the population (Figure 2; Table 1). Heterozygous pLoF variants associated with IBMF/MDS predisposition in the haploinsufficient state were identified in 0.422% of this population, whereas homozygous or compound heterozygous pLoF variants associated with autosomal recessive disease were identified in only 0.004% of the cohort. The most common putative disease–causing pLoF variants were in telomere biology disorder–associated genes, with 155 unique pLoF variants comprising 0.198% of the population, closely followed by 74 pLoF variants in primary MDS predisposition genes present in 0.178%. In contrast, frequencies of disease-causing IBMF-associated pLoF variants in DNA mismatch repair, inherited neutropenias, and inherited red blood cell disorders were very rare, at 0.001%, 0.013%, and 0.036% of the population, respectively. pLoF variants were evenly distributed between the 2 sexes. Similar high frequencies were observed in an independent validation cohort of 71 702 sequenced genomes included in gnomAD version 3.0 (supplemental Tables 3 and 4).

Mutational constraint on LoF variants in IBMF genes. The blue bar plots indicate the percentage of observed over expected unique pLoF SNVs in IBMF genes grouped as telomere biology disorders (A), inherited red blood cell disorders (B), hematologic malignancy predisposition syndromes (C), inherited neutropenias (D), and chromosomal breakage disorders (E). Genes with <9.2 expected variants are not shown. The pLI score for each gene is plotted as an area plot (peach area under the black line).
Mutational constraint on LoF variants in IBMF genes. The blue bar plots indicate the percentage of observed over expected unique pLoF SNVs in IBMF genes grouped as telomere biology disorders (A), inherited red blood cell disorders (B), hematologic malignancy predisposition syndromes (C), inherited neutropenias (D), and chromosomal breakage disorders (E). Genes with <9.2 expected variants are not shown. The pLI score for each gene is plotted as an area plot (peach area under the black line).
Frequency and constraint on LoF variants in IBMF and myeloid malignancy predisposition genes
| Gene | OMIM | Chromosome location | Disease | Inheritance | Observed unique pLoF (SNVs) | Expected unique pLoF (SNVs) | LOEUF decile | pLI | % Heterozygote (SNVs and indels) | % Biallelic (SNVs and indels) |
|---|---|---|---|---|---|---|---|---|---|---|
| MDM4 | 602704 | 1q32.1 | DKC, BMF syndrome 6 | AD | 0 | 26.7 | 0 | 1.000 | .0000 | |
| TERT | 187270 | 5p15.33 | DKC AD2, AR4 | AD, AR | 7 | 44.8 | 1 | 0.990 | .0183 | |
| NAF1 | 617868 | 4q32.2 | Telomere disorder | AD | 2 | 20.5 | 1 | 0.978 | .0056 | |
| ZCCHC8 | 616381 | 12q24.31 | Telomere-related BMF | AD | 5 | 32.5 | 1 | 0.966 | .0048 | |
| POT1 | 606478 | 7q31.33 | Telomere disorder | AD | 6 | 32.7 | 1 | 0.853 | .0310 | |
| RTEL1 | 608833 | 20q13.33 | DKC AD4, AR5 | AD, AR | 29 | 73.7 | 2 | 0.000 | .0668 | |
| PARN | 604212 | 16p13.12 | Telomere-related BMF (AD); DKC AR6 | AD, AR | 14 | 42.2 | 2 | 0.000 | .0177 | |
| TINF2 | 604319 | 14q12 | DKC AD3 | AD | 9 | 23.4 | 3 | 0.001 | .0080 | |
| ACD | 609377 | 16q22.1 | DKC AD6, AR7 | AD, AR | 21 | 25.5 | 6 | 0.000 | .0445 | |
| DKC1 | 300126 | Xq28 | DKC | X-linked | 1 | 24.9 | 0 | 0.999 | .0009 | |
| WRAP53 | 612661 | 17p13.1 | DKC AR3 | AR | 12 | 28.2 | 3 | 0.000 | 2.79 × 10−6 | |
| CTC1 | 613129 | 17p13.1 | Telomere disorder | AR | 29 | 63.0 | 3 | 0.000 | 1.96 × 10−4 | |
| STN1 | 613128 | 10q24.33 | Telomere disorder | AR | 17 | 20.8 | 6 | 0.000 | 3.6 × 10−6 | |
| NOP10 | 606471 | 15q14 | DKC AR1 | AR | 0 | 4.9 | NE | NE | 2.5 × 10−8 | |
| NHP2 | 606470 | 5q35.3 | DKC AR2 | AR | 3 | 7.2 | NE | NE | 3.6 × 10−6 | |
| Telomere biology disorders: | .1976 | 2.06 × 10−4 | ||||||||
| SRP54 | 604857 | 14q13.2 | SCN AD8 | AD | 2 | 28.7 | 0 | 0.999 | .0040 | |
| GFI1 | 600871 | 1p22.1 | SCN AD2 | AD | 4 | 16.3 | 2 | 0.255 | .0056 | |
| WAS | 300392 | Xp11.23 | SCN, WAS | X-linked | 0 | 20.0 | 0 | 0.999 | .0000 | |
| TAZ | 300394 | Xq28 | Barth syndrome | X-linked | 2 | 13.0 | 2 | 0.726 | .0008 | |
| WIPF1 | 602357 | 2q31.1 | WAS type 2 | AR | 2 | 16.3 | 1 | 0.903 | 2.53 × 10−8 | |
| LYST | 606897 | 1q42.3 | Chediak-Higashi | AR | 44 | 184.4 | 1 | 0.015 | 1.28 × 10−5 | |
| VPS45 | 610035 | 1q21.2 | SCN AR5 | AR | 15 | 35.1 | 3 | 0.000 | 2.79 × 10−6 | |
| VPS13B | 607817 | 8q22.2 | Cohen syndrome | AR | 105 | 189.9 | 3 | 0.000 | 2.48 × 10−4 | |
| DNAJC21 | 617048 | 5p13.2 | BMF syndrome 3 | AR | 17 | 35.4 | 3 | 0.000 | 3.80 × 10−5 | |
| CSF3R | 138971 | 1p34.3 | SCN AR7 | AR | 23 | 42.5 | 4 | 0.000 | 2.06 × 10−5 | |
| USB1 | 613276 | 16q21 | Poikiloderma with neutropenia | AR | 7 | 13.2 | 5 | 0.001 | 1.72 × 10−6 | |
| G6PC3 | 611045 | 17q21.31 | SCN AR4 | AR | 11 | 17.7 | 5 | 0.000 | 6.68 × 10−6 | |
| SBDS | 607444 | 7q11.21 | Shwachman-Diamond | AR | 8 | 11.8 | 6 | 0.000 | 1.71 × 10−3 | |
| HAX1 | 605998 | 1q21.3 | SCN AR3 | AR | 12 | 14.1 | 7 | 0.000 | 1.84 × 10−5 | |
| RAB27A | 603868 | 15q21.3 | Griscelli syndrome | AR | 10 | 10.0 | 8 | 0.000 | 6.08 × 10−6 | |
| LAMTOR2 | 610389 | 1q22 | Immunodeficiency (defect in MAPBP) | AR | 2 | 6.8 | NE | NE | 2.53 × 10−8 | |
| Inherited neutropenias: | .0104 | 2.07 × 10−3 | ||||||||
| KIF23 | 605064 | 15q23 | Red blood cell disorder | AD, AR | 4 | 62.8 | 0 | 1.000 | .0072 | |
| RPL5 | 603634 | 1p22.1 | DBA | AD | 0 | 17.9 | 0 | 0.998 | .0000 | |
| SLC2A1 | 138140 | 1p34.2 | GLUT1 deficiency syndrome | AD | 1 | 19.7 | 0 | 0.994 | .0016 | |
| RPL19 | 180466 | 17q12 | DBA | AD | 0 | 12.2 | 0 | 0.982 | .0000 | |
| RPL15 | 604174 | 3p24.2 | DBA | AD | 0 | 11.0 | 1 | 0.971 | .0000 | |
| RPL18 | 618310 | 19q13.33 | DBA | AD | 0 | 10.5 | 1 | 0.970 | .0000 | |
| RPS7 | 603658 | 2p25.3 | DBA | AD | 0 | 9.7 | 1 | 0.954 | .0000 | |
| RPS10 | 603632 | 6p21.31 | DBA | AD | 0 | 11.0 | 1 | 0.971 | .0016 | |
| RPL11 | 604175 | 1p36.11 | DBA | AD | 0 | 10.1 | 1 | 0.961 | .0000 | |
| KLF1 | 600599 | 19p13.13 | Dyserythropoietic anemia | AD | 7 | 11.7 | 6 | 0.000 | .0167 | |
| RPS19 | 603474 | 19q13.2 | DBA | AD | 0 | 8.1 | NE | NE | .0000 | |
| RPL26 | 603704 | 17p13.1 | DBA | AD | 0 | 8.0 | NE | NE | .0008 | |
| RPL35A | 180468 | 3q29 | DBA | AD | 0 | 7.3 | NE | NE | .0000 | |
| RPL27 | 607526 | 17q21.31 | DBA | AD | 0 | 6.2 | NE | NE | .0008 | |
| RPS26 | 603701 | 12q13.2 | DBA | AD | 0 | 6.2 | NE | NE | .0000 | |
| RPS27 | 603702 | 1q21.3 | DBA | AD | 0 | 4.7 | NE | NE | .0000 | |
| RPL31 | 617415 | 2q11.2 | DBA | AD | 0 | 6.2 | NE | NE | .0033 | |
| RPL35 | 618315 | 9q33.3 | DBA | AD | 1 | 6.6 | NE | NE | .0008 | |
| RPS15A | 603674 | 16p12.3 | DBA | AD | 0 | 5.4 | NE | NE | .0000 | |
| RPS24 | 602412 | 10q22.3 | DBA | AD | 1 | 8 | NE | NE | .0018 | |
| RPS29 | 603633 | 14q21.3 | DBA | AD | 1 | 3.9 | NE | NE | .0010 | |
| RPS28 | 603685 | 19p13.2 | DBA | AD | 0 | 3.8 | NE | NE | .0000 | |
| RPS17 | 180472 | 15q25.2 | DBA | AD | n/a* | n/a* | n/a* | n/a* | n/a* | |
| ALAS2 | 301300 | Xp11.21 | Sideroblastic anemia | X-Linked | 0 | 16.2 | 0 | 0.996 | .0000 | |
| TSR2 | 300945 | Xp11.22 | DBA | X-Linked | 0 | 5.3 | NE | NE | .0000 | |
| CDAN1 | 607465 | 15q15.2 | Dyserythropoietic anemia | AR | 30 | 60.2 | 3 | 0.000 | 1.58 × 10−5 | |
| SEC23B | 610512 | 20p11.23 | Dyserythropoietic anemia | AR | 37 | 49.8 | 5 | 0.000 | 4.79 × 10−5 | |
| CECR1 | 607575 | 22q11.1 | Vasculitis, autoinflammation, immunodeficiency, and hematologic defects syndrome | AR | 14 | 20.5 | 5 | 0.000 | 1.01 × 10−5 | |
| SLC25A38 | 610819 | 3p22.1 | Sideroblastic anemia | AR | 10 | 15.3 | 6 | 0.000 | 9.38 × 10−6 | |
| GLRX5 | 609588 | 14q32.13 | Sideroblastic anemia | AR | 0 | 4.5 | NE | NE | .0000 | |
| Inherited red blood cell disorders: | .0356 | 8.32 × 10−5 | ||||||||
| FANCB | 300515 | Xp22.2 | FA CG B | X-linked | 1 | 20.9 | 0 | 0.996 | .0000 | |
| RAD51 | 179617 | 15q15.1 | FA CG R | AD, AR† | 6 | 18.4 | 3 | 0.027 | 5.69 × 10−8 | |
| NHEJ1 | 611290 | 2q35 | SCID, sensitivity to ionizing radiation | AR | 7 | 17.8 | 3 | 0.004 | 2.67 × 10−7 | |
| FANCM | 609644 | 14q21.2 | FA | AR | 40 | 87.9 | 3 | 0.000 | 2.5 × 10−4 | |
| BRCA2 | 600185 | 13q13.1 | FA CG D1 | AR | 61 | 118.9 | 3 | 0.000 | 1.4 × 10−5 | |
| ATM | 607585 | 11q22.3 | Ataxia-telangiectasia | AR | 103 | 171.0 | 3 | 0.000 | 2.09 × 10−4 | |
| ERCC6L2 | 615667 | 9q22.32 | BMF syndrome 2 | AR | 17 | 37.7 | 3 | 0.000 | 5.36 × 10−5 | |
| FANCD2 | 613984 | 3p25.3 | FA CG D2 | AR | 60 | 83.9 | 4 | 0.000 | 5.01 × 10−5 | |
| FANCE | 613976 | 6p21.31 | FA CG E | AR | 13 | 23.2 | 4 | 0.000 | 6.08 × 10−6 | |
| SLX4 | 613278 | 16p13.3 | FA CG P | AR | 45 | 66.2 | 4 | 0.000 | 3.9 × 10−5 | |
| LIG4 | 601837 | 13q33.3 | LIG4 syndrome | AR | 13 | 25.8 | 4 | 0.000 | 4.47 × 10−5 | |
| ESCO2 | 609353 | 8p21.2 | Roberts syndrome | AR | 15 | 27.7 | 4 | 0.000 | 5.12 × 10−7 | |
| ERCC4 | 133520 | 16p13.12 | FA CG Q | AR | 25 | 39.0 | 4 | 0.000 | 1.4 × 10−5 | |
| FANCC | 613899 | 9q22.32 | FA CG C | AR | 24 | 32.3 | 5 | 0.000 | 1.68 × 10−5 | |
| FANCI | 611360 | 15q26.1 | FA CG I | AR | 63 | 75.9 | 5 | 0.000 | 9.89 × 10−5 | |
| NBN | 602667 | 8q21.3 | Nijmegen breakage syndrome | AR | 30 | 40.3 | 5 | 0.000 | 2.47 × 10−5 | |
| DDX11 | 601150 | 12p11.21 | Warsaw breakage syndrome | AR | 40 | 54.2 | 5 | 0.000 | 1.61 × 10−4 | |
| PALB2 | 610355 | 16p12.2 | FA CG N | AR | 35 | 46.1 | 5 | 0.000 | 4.61 × 10−6 | |
| BRCA1 | 113705 | 17q21.31 | FA CG S | AR | 55 | 75.2 | 5 | 0.000 | 4.63 × 10−5 | |
| FANCG | 602956 | 9p13.3 | FA CG G | AR | 25 | 32.0 | 6 | 0.000 | 1.91 × 10−5 | |
| FANCA | 607139 | 16q24.3 | FA CG A | AR | 96 | 83.3 | 7 | 0.000 | 1.15 × 10−4 | |
| RAD51C | 602774 | 17q22 | FA CG O | AR | 20 | 19.5 | 8 | 0.000 | 2.13 × 10−5 | |
| FANCL | 608111 | 2p16.1 | FA CG L | AR | 31 | 25.9 | 8 | 0.000 | 1.4 × 10−5 | |
| FANCF | 613897 | 11p14.3 | FA CG F | AR | 0 | 1.6 | NE | NE | .0000 | |
| DNA mismatch repair: | .0000 | 1.20 × 10−3 | ||||||||
| MECOM | 165215 | 3q26.2 | Radioulnar synostosis with amegakaryocytic thrombocytopenia | AD | 5 | 46 | 0 | 1.000 | .0056 | |
| PAX5 | 167414 | 9p13.2 | ALL predisposition | AD | 0 | 18.1 | 0 | 0.998 | .0080 | |
| ETV6 | 600618 | 12p13.2 | Thrombocytopenia 5 | AD | 3 | 24.4 | 1 | 0.973 | .0056 | |
| GATA2 | 137295 | 3q21.3 | MDS/AML predisposition | AD | 1 | 16.3 | 1 | 0.979 | .0010 | |
| FLI1 | 193067 | 11q24.3 | Bleeding with cancer predisposition | AD | 2 | 23 | 1 | 0.989 | .0032 | |
| SRP72 | 602122 | 4q12 | BMF syndrome 1 | AD | 13 | 41.8 | 2 | 0.002 | .0270 | |
| RUNX1 | 151385 | 21q22.12 | FPD-AML | AD | 0 | 21 | 2 | 0.654 | .0000 | |
| CBL | 165360 | 11q23.3 | Noonan-like disorder with or without AML | AD | 14 | 43.5 | 2 | 0.001 | .0310 | |
| DDX41 | 608170 | 5q35.3 | Familial myeloproliferative and/or lymphoproliferative disorders | AD | 18 | 36.3 | 3 | 0.000 | .0708 | |
| GFI1B | 604383 | 9q34.13 | Bleeding with cancer predisposition | AD | 10 | 17 | 4 | 0.001 | .0262 | |
| SAMD9L | 611170 | 7q21.2 | AD | 32 | 54.9 | 4 | n/a‡ | n/a‡ | ||
| SAMD9 | 610456 | 7q21.2 | AD | 51 | 51.8 | 6 | n/a‡ | n/a‡ | ||
| HOXA11 | 142958 | 7p15.2 | Radioulnar synostosis with amegakaryocytic thrombocytopenia | AD | 0 | 9 | NE | NE | .0000 | |
| CEBPA | 116897 | 19q13.11 | AML predisposition | AD | 0 | 2.4 | NE | NE | .0000 | |
| GATA1 | 305371 | Xp11.23 | Anemia, thrombocytopenia, neutropenia | X-linked | 0 | 9 | NE | NE | .0000 | |
| AK2 | 103020 | 1p35.1 | Reticular dysgenesis | AR | 8 | 11.8 | 6 | 0.000 | 1.15 × 10−6 | |
| Predisposition syndromes: | 0.1784 | 1.15 × 10−6 | ||||||||
| Gene | OMIM | Chromosome location | Disease | Inheritance | Observed unique pLoF (SNVs) | Expected unique pLoF (SNVs) | LOEUF decile | pLI | % Heterozygote (SNVs and indels) | % Biallelic (SNVs and indels) |
|---|---|---|---|---|---|---|---|---|---|---|
| MDM4 | 602704 | 1q32.1 | DKC, BMF syndrome 6 | AD | 0 | 26.7 | 0 | 1.000 | .0000 | |
| TERT | 187270 | 5p15.33 | DKC AD2, AR4 | AD, AR | 7 | 44.8 | 1 | 0.990 | .0183 | |
| NAF1 | 617868 | 4q32.2 | Telomere disorder | AD | 2 | 20.5 | 1 | 0.978 | .0056 | |
| ZCCHC8 | 616381 | 12q24.31 | Telomere-related BMF | AD | 5 | 32.5 | 1 | 0.966 | .0048 | |
| POT1 | 606478 | 7q31.33 | Telomere disorder | AD | 6 | 32.7 | 1 | 0.853 | .0310 | |
| RTEL1 | 608833 | 20q13.33 | DKC AD4, AR5 | AD, AR | 29 | 73.7 | 2 | 0.000 | .0668 | |
| PARN | 604212 | 16p13.12 | Telomere-related BMF (AD); DKC AR6 | AD, AR | 14 | 42.2 | 2 | 0.000 | .0177 | |
| TINF2 | 604319 | 14q12 | DKC AD3 | AD | 9 | 23.4 | 3 | 0.001 | .0080 | |
| ACD | 609377 | 16q22.1 | DKC AD6, AR7 | AD, AR | 21 | 25.5 | 6 | 0.000 | .0445 | |
| DKC1 | 300126 | Xq28 | DKC | X-linked | 1 | 24.9 | 0 | 0.999 | .0009 | |
| WRAP53 | 612661 | 17p13.1 | DKC AR3 | AR | 12 | 28.2 | 3 | 0.000 | 2.79 × 10−6 | |
| CTC1 | 613129 | 17p13.1 | Telomere disorder | AR | 29 | 63.0 | 3 | 0.000 | 1.96 × 10−4 | |
| STN1 | 613128 | 10q24.33 | Telomere disorder | AR | 17 | 20.8 | 6 | 0.000 | 3.6 × 10−6 | |
| NOP10 | 606471 | 15q14 | DKC AR1 | AR | 0 | 4.9 | NE | NE | 2.5 × 10−8 | |
| NHP2 | 606470 | 5q35.3 | DKC AR2 | AR | 3 | 7.2 | NE | NE | 3.6 × 10−6 | |
| Telomere biology disorders: | .1976 | 2.06 × 10−4 | ||||||||
| SRP54 | 604857 | 14q13.2 | SCN AD8 | AD | 2 | 28.7 | 0 | 0.999 | .0040 | |
| GFI1 | 600871 | 1p22.1 | SCN AD2 | AD | 4 | 16.3 | 2 | 0.255 | .0056 | |
| WAS | 300392 | Xp11.23 | SCN, WAS | X-linked | 0 | 20.0 | 0 | 0.999 | .0000 | |
| TAZ | 300394 | Xq28 | Barth syndrome | X-linked | 2 | 13.0 | 2 | 0.726 | .0008 | |
| WIPF1 | 602357 | 2q31.1 | WAS type 2 | AR | 2 | 16.3 | 1 | 0.903 | 2.53 × 10−8 | |
| LYST | 606897 | 1q42.3 | Chediak-Higashi | AR | 44 | 184.4 | 1 | 0.015 | 1.28 × 10−5 | |
| VPS45 | 610035 | 1q21.2 | SCN AR5 | AR | 15 | 35.1 | 3 | 0.000 | 2.79 × 10−6 | |
| VPS13B | 607817 | 8q22.2 | Cohen syndrome | AR | 105 | 189.9 | 3 | 0.000 | 2.48 × 10−4 | |
| DNAJC21 | 617048 | 5p13.2 | BMF syndrome 3 | AR | 17 | 35.4 | 3 | 0.000 | 3.80 × 10−5 | |
| CSF3R | 138971 | 1p34.3 | SCN AR7 | AR | 23 | 42.5 | 4 | 0.000 | 2.06 × 10−5 | |
| USB1 | 613276 | 16q21 | Poikiloderma with neutropenia | AR | 7 | 13.2 | 5 | 0.001 | 1.72 × 10−6 | |
| G6PC3 | 611045 | 17q21.31 | SCN AR4 | AR | 11 | 17.7 | 5 | 0.000 | 6.68 × 10−6 | |
| SBDS | 607444 | 7q11.21 | Shwachman-Diamond | AR | 8 | 11.8 | 6 | 0.000 | 1.71 × 10−3 | |
| HAX1 | 605998 | 1q21.3 | SCN AR3 | AR | 12 | 14.1 | 7 | 0.000 | 1.84 × 10−5 | |
| RAB27A | 603868 | 15q21.3 | Griscelli syndrome | AR | 10 | 10.0 | 8 | 0.000 | 6.08 × 10−6 | |
| LAMTOR2 | 610389 | 1q22 | Immunodeficiency (defect in MAPBP) | AR | 2 | 6.8 | NE | NE | 2.53 × 10−8 | |
| Inherited neutropenias: | .0104 | 2.07 × 10−3 | ||||||||
| KIF23 | 605064 | 15q23 | Red blood cell disorder | AD, AR | 4 | 62.8 | 0 | 1.000 | .0072 | |
| RPL5 | 603634 | 1p22.1 | DBA | AD | 0 | 17.9 | 0 | 0.998 | .0000 | |
| SLC2A1 | 138140 | 1p34.2 | GLUT1 deficiency syndrome | AD | 1 | 19.7 | 0 | 0.994 | .0016 | |
| RPL19 | 180466 | 17q12 | DBA | AD | 0 | 12.2 | 0 | 0.982 | .0000 | |
| RPL15 | 604174 | 3p24.2 | DBA | AD | 0 | 11.0 | 1 | 0.971 | .0000 | |
| RPL18 | 618310 | 19q13.33 | DBA | AD | 0 | 10.5 | 1 | 0.970 | .0000 | |
| RPS7 | 603658 | 2p25.3 | DBA | AD | 0 | 9.7 | 1 | 0.954 | .0000 | |
| RPS10 | 603632 | 6p21.31 | DBA | AD | 0 | 11.0 | 1 | 0.971 | .0016 | |
| RPL11 | 604175 | 1p36.11 | DBA | AD | 0 | 10.1 | 1 | 0.961 | .0000 | |
| KLF1 | 600599 | 19p13.13 | Dyserythropoietic anemia | AD | 7 | 11.7 | 6 | 0.000 | .0167 | |
| RPS19 | 603474 | 19q13.2 | DBA | AD | 0 | 8.1 | NE | NE | .0000 | |
| RPL26 | 603704 | 17p13.1 | DBA | AD | 0 | 8.0 | NE | NE | .0008 | |
| RPL35A | 180468 | 3q29 | DBA | AD | 0 | 7.3 | NE | NE | .0000 | |
| RPL27 | 607526 | 17q21.31 | DBA | AD | 0 | 6.2 | NE | NE | .0008 | |
| RPS26 | 603701 | 12q13.2 | DBA | AD | 0 | 6.2 | NE | NE | .0000 | |
| RPS27 | 603702 | 1q21.3 | DBA | AD | 0 | 4.7 | NE | NE | .0000 | |
| RPL31 | 617415 | 2q11.2 | DBA | AD | 0 | 6.2 | NE | NE | .0033 | |
| RPL35 | 618315 | 9q33.3 | DBA | AD | 1 | 6.6 | NE | NE | .0008 | |
| RPS15A | 603674 | 16p12.3 | DBA | AD | 0 | 5.4 | NE | NE | .0000 | |
| RPS24 | 602412 | 10q22.3 | DBA | AD | 1 | 8 | NE | NE | .0018 | |
| RPS29 | 603633 | 14q21.3 | DBA | AD | 1 | 3.9 | NE | NE | .0010 | |
| RPS28 | 603685 | 19p13.2 | DBA | AD | 0 | 3.8 | NE | NE | .0000 | |
| RPS17 | 180472 | 15q25.2 | DBA | AD | n/a* | n/a* | n/a* | n/a* | n/a* | |
| ALAS2 | 301300 | Xp11.21 | Sideroblastic anemia | X-Linked | 0 | 16.2 | 0 | 0.996 | .0000 | |
| TSR2 | 300945 | Xp11.22 | DBA | X-Linked | 0 | 5.3 | NE | NE | .0000 | |
| CDAN1 | 607465 | 15q15.2 | Dyserythropoietic anemia | AR | 30 | 60.2 | 3 | 0.000 | 1.58 × 10−5 | |
| SEC23B | 610512 | 20p11.23 | Dyserythropoietic anemia | AR | 37 | 49.8 | 5 | 0.000 | 4.79 × 10−5 | |
| CECR1 | 607575 | 22q11.1 | Vasculitis, autoinflammation, immunodeficiency, and hematologic defects syndrome | AR | 14 | 20.5 | 5 | 0.000 | 1.01 × 10−5 | |
| SLC25A38 | 610819 | 3p22.1 | Sideroblastic anemia | AR | 10 | 15.3 | 6 | 0.000 | 9.38 × 10−6 | |
| GLRX5 | 609588 | 14q32.13 | Sideroblastic anemia | AR | 0 | 4.5 | NE | NE | .0000 | |
| Inherited red blood cell disorders: | .0356 | 8.32 × 10−5 | ||||||||
| FANCB | 300515 | Xp22.2 | FA CG B | X-linked | 1 | 20.9 | 0 | 0.996 | .0000 | |
| RAD51 | 179617 | 15q15.1 | FA CG R | AD, AR† | 6 | 18.4 | 3 | 0.027 | 5.69 × 10−8 | |
| NHEJ1 | 611290 | 2q35 | SCID, sensitivity to ionizing radiation | AR | 7 | 17.8 | 3 | 0.004 | 2.67 × 10−7 | |
| FANCM | 609644 | 14q21.2 | FA | AR | 40 | 87.9 | 3 | 0.000 | 2.5 × 10−4 | |
| BRCA2 | 600185 | 13q13.1 | FA CG D1 | AR | 61 | 118.9 | 3 | 0.000 | 1.4 × 10−5 | |
| ATM | 607585 | 11q22.3 | Ataxia-telangiectasia | AR | 103 | 171.0 | 3 | 0.000 | 2.09 × 10−4 | |
| ERCC6L2 | 615667 | 9q22.32 | BMF syndrome 2 | AR | 17 | 37.7 | 3 | 0.000 | 5.36 × 10−5 | |
| FANCD2 | 613984 | 3p25.3 | FA CG D2 | AR | 60 | 83.9 | 4 | 0.000 | 5.01 × 10−5 | |
| FANCE | 613976 | 6p21.31 | FA CG E | AR | 13 | 23.2 | 4 | 0.000 | 6.08 × 10−6 | |
| SLX4 | 613278 | 16p13.3 | FA CG P | AR | 45 | 66.2 | 4 | 0.000 | 3.9 × 10−5 | |
| LIG4 | 601837 | 13q33.3 | LIG4 syndrome | AR | 13 | 25.8 | 4 | 0.000 | 4.47 × 10−5 | |
| ESCO2 | 609353 | 8p21.2 | Roberts syndrome | AR | 15 | 27.7 | 4 | 0.000 | 5.12 × 10−7 | |
| ERCC4 | 133520 | 16p13.12 | FA CG Q | AR | 25 | 39.0 | 4 | 0.000 | 1.4 × 10−5 | |
| FANCC | 613899 | 9q22.32 | FA CG C | AR | 24 | 32.3 | 5 | 0.000 | 1.68 × 10−5 | |
| FANCI | 611360 | 15q26.1 | FA CG I | AR | 63 | 75.9 | 5 | 0.000 | 9.89 × 10−5 | |
| NBN | 602667 | 8q21.3 | Nijmegen breakage syndrome | AR | 30 | 40.3 | 5 | 0.000 | 2.47 × 10−5 | |
| DDX11 | 601150 | 12p11.21 | Warsaw breakage syndrome | AR | 40 | 54.2 | 5 | 0.000 | 1.61 × 10−4 | |
| PALB2 | 610355 | 16p12.2 | FA CG N | AR | 35 | 46.1 | 5 | 0.000 | 4.61 × 10−6 | |
| BRCA1 | 113705 | 17q21.31 | FA CG S | AR | 55 | 75.2 | 5 | 0.000 | 4.63 × 10−5 | |
| FANCG | 602956 | 9p13.3 | FA CG G | AR | 25 | 32.0 | 6 | 0.000 | 1.91 × 10−5 | |
| FANCA | 607139 | 16q24.3 | FA CG A | AR | 96 | 83.3 | 7 | 0.000 | 1.15 × 10−4 | |
| RAD51C | 602774 | 17q22 | FA CG O | AR | 20 | 19.5 | 8 | 0.000 | 2.13 × 10−5 | |
| FANCL | 608111 | 2p16.1 | FA CG L | AR | 31 | 25.9 | 8 | 0.000 | 1.4 × 10−5 | |
| FANCF | 613897 | 11p14.3 | FA CG F | AR | 0 | 1.6 | NE | NE | .0000 | |
| DNA mismatch repair: | .0000 | 1.20 × 10−3 | ||||||||
| MECOM | 165215 | 3q26.2 | Radioulnar synostosis with amegakaryocytic thrombocytopenia | AD | 5 | 46 | 0 | 1.000 | .0056 | |
| PAX5 | 167414 | 9p13.2 | ALL predisposition | AD | 0 | 18.1 | 0 | 0.998 | .0080 | |
| ETV6 | 600618 | 12p13.2 | Thrombocytopenia 5 | AD | 3 | 24.4 | 1 | 0.973 | .0056 | |
| GATA2 | 137295 | 3q21.3 | MDS/AML predisposition | AD | 1 | 16.3 | 1 | 0.979 | .0010 | |
| FLI1 | 193067 | 11q24.3 | Bleeding with cancer predisposition | AD | 2 | 23 | 1 | 0.989 | .0032 | |
| SRP72 | 602122 | 4q12 | BMF syndrome 1 | AD | 13 | 41.8 | 2 | 0.002 | .0270 | |
| RUNX1 | 151385 | 21q22.12 | FPD-AML | AD | 0 | 21 | 2 | 0.654 | .0000 | |
| CBL | 165360 | 11q23.3 | Noonan-like disorder with or without AML | AD | 14 | 43.5 | 2 | 0.001 | .0310 | |
| DDX41 | 608170 | 5q35.3 | Familial myeloproliferative and/or lymphoproliferative disorders | AD | 18 | 36.3 | 3 | 0.000 | .0708 | |
| GFI1B | 604383 | 9q34.13 | Bleeding with cancer predisposition | AD | 10 | 17 | 4 | 0.001 | .0262 | |
| SAMD9L | 611170 | 7q21.2 | AD | 32 | 54.9 | 4 | n/a‡ | n/a‡ | ||
| SAMD9 | 610456 | 7q21.2 | AD | 51 | 51.8 | 6 | n/a‡ | n/a‡ | ||
| HOXA11 | 142958 | 7p15.2 | Radioulnar synostosis with amegakaryocytic thrombocytopenia | AD | 0 | 9 | NE | NE | .0000 | |
| CEBPA | 116897 | 19q13.11 | AML predisposition | AD | 0 | 2.4 | NE | NE | .0000 | |
| GATA1 | 305371 | Xp11.23 | Anemia, thrombocytopenia, neutropenia | X-linked | 0 | 9 | NE | NE | .0000 | |
| AK2 | 103020 | 1p35.1 | Reticular dysgenesis | AR | 8 | 11.8 | 6 | 0.000 | 1.15 × 10−6 | |
| Predisposition syndromes: | 0.1784 | 1.15 × 10−6 | ||||||||
AD, autosomal dominant; AR, autosomal recessive; DKC, dyskeratosis congenita; FPD, familial platelet disorder; indels, insertions/deletions; n/a, not applicable; NE, not evaluable; SCID, severe combined immunodeficiency; SCN, severe congenital neutropenia.
RPS17 is located in a segmental duplication and thus is not amenable to sequence-based analysis in the gnomAD data set.
RAD51 was associated with AD FA in a case of a patient with a missense mutation in RAD51, which disrupted homologous recombination by disrupting the action of the wild-type protein.54 Based on the LOEUF score that is in line with that of the other FA genes, we predict that LoF mutations in RAD51 would lead to AR inheritance of FA.
SAMD9 and SAMD9L cause a pediatric-onset IBMF through gain of function. We included LoF analysis for SAMD9 and SAMD9L variants here because LoF variants were reported in a cohort of patients with MDS15 ; because of their high prevalence in the general population, we did not include these in the aggregate frequency of IBMF/MDS disease-causing variants.
Age distribution of pLoF variants in IBMF genes reveals a surprising prevalence of occult IBMF syndromes in adults
To confirm that pLoF variants in IBMF genes were germline and not due to age-related clonal hematopoiesis, the IBMF variants were evaluated for features associated with somatic acquisition. Lower pLoF allele frequency and a greater age of individuals carrying pLoF variants compared with synonymous variants are 2 established measures previously associated with age-related clonal hematopoiesis in population studies.21 Using these measures, pLoF variants in ASXL1, DNMT3A, and TET2 genes, which are commonly mutated in aging-related clonal hematopoiesis,24-26 have age distributions consistent with somatic acquisition. When we probed gnomAD to determine if somatic acquisition was a confounding variable for IBMF pLoF variants, the distribution of these pLoF variants closely mirrored age distributions of the gnomAD cohort (Figure 3; supplemental Tables 5 and 6). No gene, including those in which malignancy-associated somatic mutations have been described (CBL, CEBPA, DDX41, ETV6, GATA2, and RUNX1), had age-associated enrichment of pLoF variants. Analyzing the age distribution of 85 462 samples for which patient age was available, pLoF variants were distributed throughout each decade, with median ages of pLoF variants ranging from 40 to 65 years. These data support the germline origin of IBMF pLoF variants in this cohort.
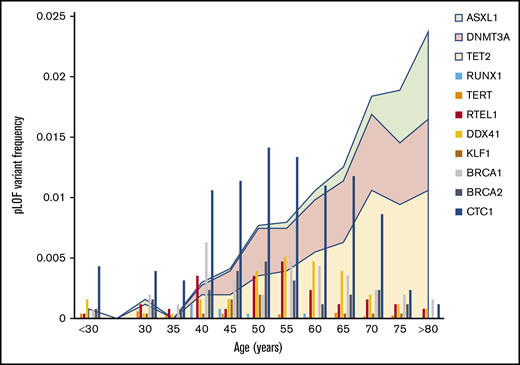
Age distribution of pLoF variants in IBMF genes. The frequency of pLoF variants as a function of age for selected IBMF genes is shown by the vertical bar graphs. These are compared with the age distribution of ASXL1, DNMT3A, and TET2, the genes commonly mutated in age-associated clonal hematopoiesis and which are depicted by the area plots.
Age distribution of pLoF variants in IBMF genes. The frequency of pLoF variants as a function of age for selected IBMF genes is shown by the vertical bar graphs. These are compared with the age distribution of ASXL1, DNMT3A, and TET2, the genes commonly mutated in age-associated clonal hematopoiesis and which are depicted by the area plots.
Although gnomAD excluded individuals with severe pediatric disease, it provides a unique opportunity to evaluate the frequency of individuals who may have a pLoF in an IBMF-associated gene but have less severe, subclinical, or no phenotypes, particularly in the adult population. We first focused on syndromes in which there were no individuals with pLoF variants in gnomAD (eg, ALAS2, RPL5, WAS) (Table 1). LoF variants in 5′-aminolevulinate synthase 2 (ALAS2) cause X-linked congenital sideroblastic anemia, an inherited syndrome of severe transfusion-dependent anemia and iron overload.11 Patients present with congenital sideroblastic anemia in infancy; in agreement with known clinical presentation, no individuals in gnomAD had ALAS2 pLoF variants. Similarly, RPL5 haploinsufficiency causes autosomal dominant DBA, manifested by severe transfusion-dependent anemia and congenital malformations. In the Canadian DBA registry, patients with RPL5 DBA presented at a median age of <1 year (range, 0-17 months) with severe anemia, and 86% had at least 1 organ malformation.9 Similar results were reported in an international cohort of patients with DBA.10 This early severe phenotype is consistent with 0 observed RPL5 pLoF variants in gnomAD. Similarly, pLoF variants in RPS19 were previously reported to be highly penetrant, and there were 0 RPS19 pLoF variants in our analysis. Both the severe hematopoietic phenotype and extra-hematopoietic manifestations of DBA such as birth defects and malignancy predisposition likely contribute to the lack of pLoF variants in DBA patients in our cohort.
Analysis of the 21 ribosomal protein genes linked to DBA10 showed a prevalence of 0.01% pLoF variants. After manual curation, 8 high-probability pLoF variants could be identified across the spectrum of DBA genes, including 3 variants in RPL26, 2 variants in RPS10, and 1 each in RPL27, RPS24, and RPS29. The age range of individuals with these DBA-associated variants was 30 to 65 years (supplemental Table 7), suggesting that there may be rare adults with DBA predisposition who either have subclinical disease or who have undergone spontaneous remission.
Inherited neutropenia syndromes frequently present in early childhood with recurrent infections, but their prevalence in adults remains undefined.12 We analyzed the gnomAD data set for pLoF variants in genes known to cause inherited neutropenias in the haploinsufficient state. Notably, because ELANE mutations associated with severe congenital neutropenia are believed to exert gain-of-function–like deleterious effects on protein folding, the prevalence of pathogenic mutations in ELANE could not be evaluated by this pLoF analysis.27,28 However, inactivating mutations in signal recognition particle 54 GTPase (SRP54) were recently identified to cause haploinsufficient severe congenital neutropenia and Shwachman-Diamond–like syndrome.29-31 Nearly all described cases of SRP54-associated neutropenia were diagnosed in infancy due to severe neutropenia and life-threatening infections.29 Interestingly, 5 individuals within our exome data set had heterozygous pLoF variants in SRP54 (supplemental Table 8). An additional pLoF variant was found in the gnomAD version 2.1.1 data set. Of the 4 individuals whose ages were available for analysis, one was aged <30 years, and the others ranged in age from 45 to 60 years. These data indicate that some SRP54 LoF may have variable penetrance or subclinical manifestations. This example provides evidence that frequencies of pLoF variants across the age spectrum can provide critical information about penetrance and phenotypes of IBMF diseases.
Spectrum of mutational constraint on IBMF genes
The expected number of variants in the absence of natural selection (expected unique pLoF) for each IBMF gene in gnomAD was previously established,21 and we compared this expected rate vs the number of observed unique pLoF variants using 2 metrics of mutational constraint: LOEUF and pLI21,23 (Table 1). LOEUF is a conservative estimate of evolutionary selection against disease-causing variants based on the upper limit of the confidence interval for the observed/expected pLoF mutation rate. Genes with lower observed/expected pLoF variant ratios are evolutionarily constrained; LOEUF scores are binned into deciles ranging from 0 being most constrained, to 9, indicating least constrained. We use LOEUF for comparative analysis of evolutionary pressure against pLoF in IBMF genes because it is a continuous function with greater resolution across the constraint spectrum. We also analyzed the IBMF genes according to pLI, which measures probability that a gene has significant evolutionary selection against its loss. In contrast to LOEUF, pLI is a dichotomous score in which pLI > 0.9 suggests that a gene is associated with severe phenotypes in the haploinsufficient state, and pLI < 0.1 suggests that a gene is not haploinsufficient.21,23
To validate our approach, we first focused on IBMF syndromes with severe presentations and well-defined clinical phenotypes. As a group, genes linked to clinical IBMF/MDS predisposition phenotype in a haploinsufficient state were significantly more constrained than the remainder of genes in the human genome (P = 1.3 × 10−11) or genes that required biallelic inactivation to cause IBMF disease. The median LOEUF decile for genes linked to haploinsufficient phenotypes was 1 (range, 0-6) vs a median LOEUF decile of 4 (range, 1-8) for genes associated with autosomal recessive IBMF syndromes, which as a group were similar to the aggregate of all genes in the genome.
Genes with the most severe constraint scores (LOEUF decile 0 or 1) were associated with highly penetrant, autosomal dominant, or X-linked pediatric syndromes. These include ribosomal protein genes linked to DBA32 ; FANCB, the loss of which causes X-linked FA33 ; and SRP54.29,30 The majority of genes associated with autosomal recessive disease without known haploinsufficient phenotypes had no evidence of evolutionary constraint.
We then examined whether constraint analysis can predict severity and age of onset of pathology in haploinsufficiency syndromes with variable penetrance. For this analysis, we selected genes associated with hematologic malignancy predisposition syndromes, for which haploinsufficient or X-linked pLoF variants had LOEUF decile scores ranging from 0 to 4.
GATA2 is a canonical example in which pLoF variants are subject to significant constraint (LOEUF decile 1). Haploinsufficiency of GATA2 is associated with immunodeficiency, lymphedema, pulmonary alveolar proteinosis, and progression to MDS/acute myeloid leukemia (AML) at a young age.34,35 According to estimates, 75% of patients with GATA2 haploinsufficiency progress to MDS/AML by ∼18 years of age.13,34,36,37 The phenotype severity and onset before and during reproductive age likely underlie significant selection against GATA2 LoF, a phenomenon we also observed in several other myeloid malignancy predisposition genes (eg, RUNX1, MECOM, ETV6). In contrast, germline heterozygous LoF variants in DDX41 have been implicated in the development of MDS/AML in older adults with a mean age of 62 years.38 The onset of pathology after reproductive age likely explains the relatively relaxed evolutionary pressure against DDX41 pLoF (LOEUF decile 3).
Similarly, although gain-of-function mutations in SAMD9 and SAMD9L genes cause early-onset familial IBMF/MDS predisposition, a recent report identified germline SAMD9 and SAMD9L LoF variants in 3% of patients with MDS.1,15 Low constraint (LOEUF decile 6 for SAMD9; LOEUF decile 4 for SAMD9L) argues against LoF as a pathogenic mechanism of SAMD9/SAMD9L–associated pediatric-onset severe IBMF syndrome. Similar to DDX41, the impact of these germline variants may be limited to aging-related MDS.38
Another potential application of this constraint analysis is the interrogation of heterozygous pLoF variants in genes associated with autosomal recessive diseases to evaluate for signs of pathogenicity with haploinsufficiency alone that could lead to evolutionary disadvantage. As an example, although biallelic mutations in Fanconi complex genes are known to cause autosomal recessive forms of FA, isolated heterozygous LoF variants in several FA genes (eg, BRCA1, BRCA2, PALB2) predispose to breast and ovarian cancer.39-41 Recently, several reports suggested that heterozygous variants in FA genes may also serve as predisposition factors for MDS/AML across a spectrum of age groups.42-45 However, FA complex genes associated with autosomal recessive FA were poorly constrained in our analyses (LOEUF deciles ranging from 3-8 and a low pLI), indicating that heterozygous LoF of FA genes does not cause clinically significant pathology at or before reproductive age.
We next applied constraint analysis to WIPF1, a gene in which mutations are known to cause disease in a biallelic fashion but the impact of heterozygous mutations are poorly characterized. Biallelic inactivating mutations in WIPF1, a gene that encodes Wiskott-Aldrich syndrome (WAS) protein-interacting protein (WIP), cause early-onset combined immunodeficiency with thrombocytopenia.46,47 Six patients from 4 different families with WIPF1 deficiency have been described. All published patients had onset of symptoms within year 1 of life. Family histories of published cases are largely unknown. We found unexpectedly strong evolutionary constraint on LoF of WIPF1 (LOEUF decile, 1; pLI, 0.903). This constraint against pLoF variants is more severe than explained by a clinical phenotype that occurs solely from biallelic inactivation, and it suggests a potential effect from heterozygous LoF.48,49 WIP stabilizes WAS protein by binding to its N terminus.50 Interestingly, although clinical phenotypes of heterozygous WIPF1 LoF variant carriers have not been reported, the parents of a patient with homozygous LoF mutations in WIPF1 WAS type 2, each heterozygous for the WIPF1 mutation, had significant reductions in WAS protein level.46 Low WAS protein was also observed in mice heterozygous for WIP null allele. The unusual clinical phenotype due to partner protein destabilization (Figure 4) is reminiscent of the von Willebrand Normandy variant, which mimics mild hemophilia A due to failure of von Willebrand factor binding and stabilizing factor VIII.51 These data suggest that family members of patients with autosomal recessive WIPF1-related disorders who carry heterozygous LoF mutations in WIPF1 should be studied in dedicated investigations of clinically significant phenotypes that could lead to this constraint.
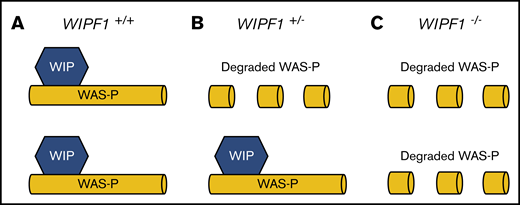
Model of molecular pathogenesis of WIPF1 haploinsufficiency. A schematic diagram for the proposed model of molecular pathogenesis of WIPF1 haploinsufficiency. WIP is the protein product of WIPF1 and is required to bind and stabilize WAS protein (WAS-P) at the N terminus. (A) In individuals with 2 wild-type copies of WIPF1, there are normal levels of WAS messenger RNA (mRNA) and WAS-P. (B) In heterozygotes for WIPF1 LoF, there is less WAS-P despite normal WAS mRNA levels. (C) Similarly, for biallelic WIPF1 LoF, there is barely detectable WAS-P in the setting of normal WAS mRNA. Individuals with biallelic WIPF1 mutations have WAS type 2.
Model of molecular pathogenesis of WIPF1 haploinsufficiency. A schematic diagram for the proposed model of molecular pathogenesis of WIPF1 haploinsufficiency. WIP is the protein product of WIPF1 and is required to bind and stabilize WAS protein (WAS-P) at the N terminus. (A) In individuals with 2 wild-type copies of WIPF1, there are normal levels of WAS messenger RNA (mRNA) and WAS-P. (B) In heterozygotes for WIPF1 LoF, there is less WAS-P despite normal WAS mRNA levels. (C) Similarly, for biallelic WIPF1 LoF, there is barely detectable WAS-P in the setting of normal WAS mRNA. Individuals with biallelic WIPF1 mutations have WAS type 2.
Constraint analyses provide framework to investigate pathogenic mechanisms of rare autosomal dominant disorders
Constraint analyses can provide insights into the molecular pathogenesis of IBMF syndromes. RAD51 FA is the only subtype of FA associated with autosomal dominant inheritance.20,52,53 Three patients with FA due to destabilizing missense RAD51 mutations with a dominant-negative phenotype, leading to the loss of RAD51 protein function, have been reported.52-54 The relatively mild evolutionary constraint (LOEUF decile, 3; pLI, 0.027) for pLoF variants in RAD51 suggests that heterozygous LoF of RAD51 would not be expected to cause severe pathology.55 We predict that, similar to other autosomal recessive forms of FA, LoF mutations in RAD51 would require biallelic inactivation to cause disease (autosomal recessive inheritance), and only gain of function/dominant-negative RAD51 mutations would cause disease in the heterozygous state.
We then applied our analysis to 2 newly discovered genes associated with autosomal dominant BMF and malignancy predisposition. Heterozygous missense mutations in DHX34, RNA helicase regulating nonsense-mediated decay (NMD), were recently reported in 4 families with familial single or multi-lineage cytopenias that progressed to aplastic anemia, MDS, and AML in the first decades of life.56 The symptom onset was a median of 10 years (range, 2-23 years). The identified DHX34 variants were shown to compromise NMD activity by abrogating the helicase’s ability to promote phosphorylation of NMD factor UPF1. Interestingly, DHX34 shows lack of constraint for pLoF (LOEUF, 4; pLI, 0.000), suggesting that the pathogenesis of DHX34-associated syndrome is likely due to altered gene function (eg, gain of function) but not due to haploinsufficiency.
MDM4-associated telomere biology disorder is another recently described syndrome caused by mutant MDM4, a negative regulator of p53. A missense hypomorphic variant in MDM4 was associated with autosomal dominant inheritance of features consistent with telomere biology disorders affecting family members aged 17 to 52 years.57 Interestingly, constraint analysis of MDM4 indicates an extreme intolerance of LoF, with LOEUF of 0 and pLI of 1.000, indicating that LoF MDM4 mutations are most likely causative of this severe phenotype.
Discussion
In this study, we analyzed 125 748 individuals in gnomAD for pLoF variants in 100 IBMF and predisposition genes. Our analysis shows that 0.426% of individuals in the general population carry variants predicted to cause IBMF disease (heterozygous pLoF in autosomal dominant and biallelic pLoF in autosomal recessive diseases). These pLoF variants occur in the general population after exclusion of patients with severe pediatric diseases and either cause disease later in life or cause subclinical or no clinical disease. Using age distribution and evolutionary constraint analyses of naturally occurring pLoF variants, we established a framework to enhance understanding of penetrance and molecular pathogenesis of rare and emerging IBMF syndromes. We made several novel insights into rare IBMF diseases, including syndromes associated with DHX34, MDM4, RAD51, SRP54, and WIPF1. Our results also provide a clinically useful framework for interpreting penetrance and pathogenicity of pLoF variants in individual IBMF-associated genes, particularly for syndromes in which only a handful of cases have been described.
Studies of variants linked to Mendelian disorders in large population cohorts suggest that frequency of Mendelian diseases in the general population may be much higher and penetrance lower than previously thought.58-61 The heterogeneity of IBMF syndromes, together with their rarity, make clinical recognition challenging. The prevalence of IBMF syndromes, particularly in older individuals, remains largely unknown. Even less is known about emerging syndromes that have been identified in small numbers of families, with limited knowledge and available materials to study molecular pathogenesis, phenotypic variation, and prevalence. Our study begins to address these gaps by defining the prevalence and evolutionary selection against LoF variants in IBMF-associated genes in a large population-based cohort. We specifically focused on LoF variants because they are frequently deleterious and because robust algorithms allow the identification of true LoF variants with high confidence, outpacing current ability to predict the functional significance for missense and noncoding genomic variation that lead to altered protein function.21,23,62
Given the exclusion of individuals with severe pediatric diseases from the gnomAD cohort, the estimated prevalence of pLoF variants in IBMF/MDS predisposition genes in our study likely represents the lower bound for the population frequency of clinically significant genetic alterations in these genes. Despite this limitation, our results indicate that germline pLoF variants associated with IBMF are much more common than previous studies have suggested, in which estimated frequencies of pathogenic mutations included 1 per million for dyskeratosis congenita,63 1 in 100 000 to 200 000 births for DBA,10 and 1 in 130 000 to 250 000 for FA.64 Our data suggest that for many individuals harboring these pLoF variants, phenotypic features may be subclinical or significantly underdiagnosed in adults. By considering each gene and all the pLoF variants associated with it as a unit, we are able to provide a denominator of people in the general population who harbor pLoF variants in the absence of severe pediatric disease. These findings provide additional data points for counseling patients and families when a variant is discovered during clinical sequencing.
Accurate data on IBMF prevalence and phenotypic spectrum are particularly important for providing anticipatory guidance to patients and families, both with respect to disorders with high severity and near-complete penetrance (eg, ALAS2, DBA, WAS) and, importantly, also for those in whom disease penetrance is found to be low in population-based studies. Although ascertainment and referral patterns to syndrome-specific registries likely bias published experience toward severe pathology, the existence of high-confidence pLoF variants in genes such as SRP54 and GATA2 in older individuals within the general population suggests broader spectrums of clinical phenotypes, as well as the need for additional studies to accurately define the complete landscape of IBMF/MDS predisposition.
The current study has limitations. Although gnomAD is the largest available population-based genome aggregation data set, the rarity and strong evolutionary pressure against IBMF disorders will require confirmation of these findings in larger, more ethnically diverse cohorts to better evaluate constraint and prevalence. Because of gnomAD inclusion criteria, our estimates of IBMF prevalence do not capture patients with severe pediatric phenotypes and lack clinical information for sequenced individuals. However, this population-based data set of presumed healthy control individuals does provide unique opportunities to capture incomplete penetrance and milder phenotypes of diseases, and it adds a new dimension to understanding the spectrum of IBMF in adult patients. Constraint analysis is a powerful tool for understanding evolutionary selection against heterozygous LoF variants; however, it cannot resolve evolutionary selection against homozygous variants and is less affected by selection after reproductive age.
To ensure reliable identification of LoF variants, we focused our analysis on SNVs and small insertions/deletions, excluding larger structural variants that are not reliably captured by short sequencing reads. Thus, haploinsufficiency resulting from large deletions, as occurs in genes such as GATA2, was not captured by this analysis. In addition to large deletions that are generally not captured by whole-exome sequencing, exon sequencing does not capture other structural variants that may not be within coding exons. Our analysis therefore does not incorporate these variants, and further studies using whole-genome sequencing would be required to incorporate large copy number changes and noncoding variants. In addition, the analysis used peripheral blood DNA, which may include somatically acquired variants; however, most IBMF variants confer a growth disadvantage and are not associated with clonal hematopoiesis. Also, we confirmed no age-associated variant enrichment.
In conclusion, our data combine population-based genomic analyses and clinical biology to provide a comprehensive framework for analyzing pLoF variants in IBMF-associated genes. Our results offer a conservative estimate of pLoF variant frequencies in IBMF genes in the general population and highlight the utility of evolutionary constraint for understanding molecular mechanisms, clinical severity, and penetrance of IBMF syndromes. These data provide a frame of reference for IBMF researchers and clinicians, and they carry important implications for interpreting variant pathogenicity and for counseling patients and families on expressivity and penetrance of IBMF syndromes across the age continuum.
The Genome Aggregation Database served as the dataset for this analysis. We used version 2.1.1 for the original analysis and version 3.0 as a comparator in the supplemental data set.
Acknowledgments
The authors thank members of the Penn/CHOP Comprehensive Bone Marrow Failure Center and the CHOP Department of Genetic Diagnostics for helpful discussions.
This work was supported by the National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) grant T32 HL715041, National Center for Advancing Translational Sciences grant KL2TR001879, an Aplastic Anemia and MDS International Foundation grant (J.H.O.), and NIH, NHLBI grant K08 HL132101 (D.V.B.). M.P. is funded by the NIH, NHLBI (R01 HL139448 and R01 HL132557) and a RUNX1 Research Program grant in association with the Alex Lemonade Foundation. T.S.O. is funded by a United States Department of Defense BMF Research Program Idea Development Award and by a Hyundai Hope on Wheels Scholar Hope Award.
Authorship
Contribution: J.H.O. and K.J.K. conceived the study; K.J.K. provided gnomAD expertise and performed the analyses for mutational constraint and statistical analyses of the gnomAD data set; J.H.O. and D.V.B. analyzed the gnomAD data set and mutational constraint within the context of BMF disorders and wrote and revised the manuscript; M.P., M.A.K., and M.P.L. provided expertise in inherited platelet disorders; T.S.O. provided expertise in IBMF and malignancy predisposition syndromes; N.W., M.P.L., and T.S.O. assisted with analysis and revised the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Joseph H. Oved, Children’s Hospital of Philadelphia, 3615 Civic Center Blvd, ARC, Room 302E, Philadelphia, PA 19104; e-mail: ovedj@email.chop.edu.

