

Key Points
MDR1 inhibitors sensitize murine leukemia stem cells to Smac-mimetics and prolong survival in AML models.
MDR1 as a predictor of Smac-mimetic treatment in AML.

Abstract
The specific targeting of inhibitor of apoptosis (IAP) proteins by Smac-mimetic (SM) drugs, such as birinapant, has been tested in clinical trials of acute myeloid leukemia (AML) and certain solid cancers. Despite their promising safety profile, SMs have had variable and limited success. Using a library of more than 5700 bioactive compounds, we screened for approaches that could sensitize AML cells to birinapant and identified multidrug resistance protein 1 inhibitors (MDR1i) as a class of clinically approved drugs that can enhance the efficacy of SM therapy. Genetic or pharmacological inhibition of MDR1 increased intracellular levels of birinapant and sensitized AML cells from leukemia murine models, human leukemia cell lines, and primary AML samples to killing by birinapant. The combination of clinical MDR1 and IAP inhibitors was well tolerated in vivo and more effective against leukemic cells, compared with normal hematopoietic progenitors. Importantly, birinapant combined with third-generation MDR1i effectively killed murine leukemic stem cells (LSCs) and prolonged survival of AML-burdened mice, suggesting a therapeutic opportunity for AML. This study identified a drug combination strategy that, by efficiently killing LSCs, may have the potential to improve outcomes in patients with AML.
Introduction
Inhibitor of apoptosis (IAP) proteins regulate cell survival in response to several stimuli. In TNF receptor (TNFR) superfamily signaling, they are necessary to activate the canonical NF-κB pathway and MAPKs. They also act as repressors of the noncanonical NF-κB pathway and apoptotic cell death.1-4 Natural IAP antagonists, such as second mitochondria–derived activator of caspases (Smac/DIABLO), can bind to IAPs to prevent their interaction with specific substrates.5,6 In certain conditions, this leads to autoubiquitylation and proteasomal degradation of IAPs.1,2
The observation that overexpression of IAPs correlates with cancer progression, poor prognosis, and treatment resistance, led to the development of small-molecule, peptidelike mimetics of Smac, termed Smac-mimetics (SMs).7 Birinapant is one of the most clinically advanced SMs and is currently in clinical trials for the treatment of certain solid and hematological cancers. Because of its limited efficacy as a single agent, birinapant is being tested in combination with chemotherapeutic drugs and immune checkpoint inhibitors (http://www.clinicaltrials.gov, registered as #NCT01188499 and #NCT02587962).8,9 Studies by us and others suggest that SMs can also synergize with several drugs, including p38-kinase inhibitors, caspase-8 inhibitors, and immunotherapy, to efficiently eliminate cancer cells.10-14 Although combinations of birinapant with other anticancer agents show promise for the treatment of several cancers, boosting their efficacy and overcoming resistance are still major challenges.
Using an unbiased high-throughput strategy (detailed description in supplemental Materials and methods), we screened a library of clinical and preclinical compounds, to identify molecules that could overcome birinapant resistance in acute myeloid leukemia (AML). From several compounds that sensitized resistant AML cells to birinapant, we selected reserpine for further study. Reserpine is an antihypertensive and antipsychotic clinical drug that also inhibits multidrug resistance protein 1 (MDR1).15-17 MDR1 or P-glycoprotein, is a member of the ATP-binding cassette (ABC) transporter family that actively exports structurally unrelated substrates out of cells, presumably to protect them from possible toxicities. MDR1 substrates include several chemotherapeutic drugs and chemical compounds, such as the fluorescent dye rhodamine-123 (Rho-123).18-21 Although MDR1 exports many xenobiotic compounds, it has not been possible to discern a common chemical feature recognized by MDR1.22 Therefore, whether a molecule is a substrate of MDR1 must be determined empirically.
MDR1 is frequently upregulated in cancer cells, and its expression correlates with treatment resistance and disease relapse.23-25 In AML, MDR1 expression has been reported in patients of all ages, with prevalence in >50% of relapsed and secondary AML.24,26 This finding led to clinical trials of MDR1 inhibitors (MDR1i) in AML. Although phase 1/2 clinical trials have proven the safety of these inhibitors in AML, limited success has been obtained because of changes in chemotherapy pharmacokinetics and increased toxicity.22,25,27,28
Our data provide strong evidence for the reevaluation of MDR1i therapy in combination with SMs, for the treatment of AML. In our study, SMs such as birinapant, synergized with third-generation MDR1i to enhance the killing of AML cells in vitro and in vivo. Importantly, murine leukemic stem cells (LSCs) derived from AML models, were highly sensitive to this combination therapy, whereas healthy hematopoietic stem/progenitor cells (HSPCs) were resistant. A shortcoming of therapies in the clinic is that, although they effectively target leukemic blasts, they fail to eradicate LSCs, leading to disease relapse.29-32 Therefore, therapies that can kill both blasts and LSCs while sparing normal HSPCs, are needed for effective treatment of AML.
In this study, we explored MDR1 as a predictor of response to birinapant treatment and determined the impact of the clinical MDR1i tariquidar and zosuquidar as novel birinapant-combination therapies that can kill AML cells. Our findings provide a rationale for testing the combination of SM/MDR1i in clinical trials for the treatment of AML.
Materials and methods
Viability assays
Primary murine leukemias, Lin−, SCA-1+, c-KIT+ (LSK) cells, and patient-derived cells were established and cultured as previously described.11 Human AML and CD34+ HSPC samples were obtained from patients after informed consent. The study was approved by the Alfred Health Ethics Committee or the Royal Adelaide Hospital Human Research Ethics Committee. Cell viability was analyzed by flow cytometry (FACSCalibur; BD Biosciences) for changes in cell volume and propidium iodide (PI) and/or annexin V (AxV) staining.
Reagents
SM drugs were obtained from TetraLogic Pharmaceuticals. Birinapant, reserpine, tariquidar, VP16, JQ1, ABT-199, and the library of compounds were purchased from Selleckchem Pharma. Rho-123, reversan, fumitremorgin-C (Fum-C), and cytarabine (Ara-c) were purchased from Sigma. Zosuquidar was from MedchemExpress. TNF was produced in house.
Western blot analysis, immunostaining, and TNF ELISA
Western blot analysis of whole-cell lysates was performed with the standard assays described in supplemental Materials and methods. Antibody details and TNF enzyme-linked immunosorbent assay protocol (ELISA; previously described)11 are detailed in the supplemental Materials and methods.
Rho-123–retention assay
Cells (1 × 106/mL) were incubated in ρ buffer (phosphate-buffered saline [PBS]+10% fetal calf serum [FCS]+3 ng/mL IL-3 for murine cells) containing 250 nM Rho-123 for 20 minutes, spun at 300g for 5 minutes at room temperature, and incubated in Rho buffer containing MDR1i for 2 hours at 37°C in 10% CO2. Rho-123 fluorescence was measured by flow cytometry (FACSCalibur; BD Biosciences).
Plasmids, cloning, transfection, and lentiviral infection
Two independent CRISPR guide RNAs (gRNAs) targeting MDR1 (human; ABCB1: 5′-TTATAGTAGGATTTACACG-3′ and 5′-AATGTTTTCAGCTATCGTGG-3′) were designed using benchling (www.benchling.com). gRNAs were cloned into LentiCRISPRv2 containing 2 expression cassettes (hSpCas9 and the chimeric gRNA), and DNA was prepared using the Qiagen Plasmid Mini Kit. Lentiviral particles were generated as previously described.33
In vivo AML treatment
All in vivo experiments were approved by the Walter and Eliza Hall Institute Animal Ethics Committee. C57BL/6 mice were injected IV with 0.3 × 105 to 5 × 105 MLL-AF9 MDR1 high (MDR1H) leukemia cells, with treatment starting 48 hours after retransplantation. Birinapant (6 mg/mL) in 12% Captisol was diluted in PBS for intraperitoneal injection. Tariquidar (10 mg/mL) in 30% propylene glycol+5% Tween 80+65% D5W was diluted in PBS for oral gavage. Upon signs of leukemia, mice were euthanized, and organs and blood were collected for cellular and histological analysis. For further details, see supplemental Materials and methods.
Quantification of intracellular birinapant
For each treatment group, cells were washed 3 times in ice-cold PBS and resuspended in 90% methanol (−20°C), divided into 4 samples, and spiked with a titration of stable isotope–labeled birinapant (heavy birinapant; birinapant-d6). Peptides were purified through C18 StageTips and analyzed by nanoflow liquid chromatography-mass spectrometry (MS) on an EASY-nLC 1200 coupled to a Q-Exactive mass spectrometer through a nanoelectrospray ion source (Thermo Fisher Scientific). Full scans (mass-to-charge ratio [m/z], 200-1000) were acquired with a resolution of 140 000 at 200 m/z. The automatic gain control target was set at 1e6, with a maximum ion time of 150 ms. Skyline software34 (version, 4.1.0.11714) was used for data analysis.
Statistical analysis
Flow cytometry data were analyzed using FlowJo 10.0.8 or 10.5 software. Unless otherwise stated in figure legends, all statistics were calculated with an unpaired 2-tailed Student t test with Welch’s correction on Prism 7/8 (GraphPad). Bliss synergy analysis was completed with the Synergy Finder program, as described.35 The drug concentration that caused 50% lethality was calculated by transforming normalized data (x = log[x]) and fitting a nonlinear log (inhibitor) vs normalized response curve.
Results
Clinical MDR1i enhance the efficacy of SM-based therapy in AML cells
To identify drugs that could enhance birinapant-mediated killing of AML cells, we conducted a high-throughput screening of >5700 bioactive compounds in 2 independent birinapant-resistant murine HoxA9/Meis1 leukemias (Figure 1A).11 The increasingly stringent tests identified 12 drugs that sensitized AML cells to birinapant (supplemental Table 1). One of the most active compounds was reserpine,15,16 which combined with birinapant and other SMs such as AT-406 and AEG-40730 to effectively kill primary HoxA9/Meis1– and MLL-AF9–driven AML cells (Figure 1B; supplemental Table 2; supplemental Figure 1A-D). However, reserpine increased only the LCL-161–induced killing of HoxA9/Meis1 cells, and the other SM, GDC-0152, did not kill cells under any condition (supplemental Figure 1E-H).

Clinical MDR1i enhance the efficacy of SM-based therapy in AML cells. (A) Approximately 5700 compounds were screened for their ability to synergize with birinapant and overcome resistance in murine HoxA9/Meis1 AML cells. Compounds that induced cell death in combination with birinapant, but not as single agents, were selected and their impact determined in a dose-response assay. (B) HoxA9/Meis1 and MLL-AF9 AML cells were treated with birinapant (Bir; 100 and 200 nM), with or without reserpine (Res; 1 µM), for 24 hours (n = 4-5). HoxA9/Meis1 (C) and MLL-AF9-NrasG12D (D) AML cells were treated with 10, 100, 500, and 1000 nM reserpine, tariquidar (Tari), or zosuquidar (Zosu), with or without Bir (200 nM), for 24 hours (n = 4). (E-F) HoxA9/Meis1 AML cells were treated with 125, 250, 500, and 1000 nM AT-406 (AT) (E) and AEG-40730 (AEG) (F), with or without Tari (1 µM), for 24 hours (n = 5). (G-H) HoxA9/Meis1 AML cells were treated with the peptide-mimetic agents ABT-199 (G) and JQ1 (H) at 100 and 1000 nM, with or without Tari or Zosu (1 μM), for 24 hours. Treatment with Bir (500 nM)+Tari and Zosu (1 µM) was used as a control (n = 4). Cell death was measured by flow cytometry analysis of PI uptake and decrease in cell volume (MLL-AF9-NrasG12D) in 3 or 4 individual tumors. Data are the mean ± SEM throughout. P values were obtained by comparison of SM alone and SM/MDR1i treatment at the indicated concentrations. *P < .05; **P < .01; ***P < .001; ****P < .0001. n.s. nonsignificant.
Clinical MDR1i enhance the efficacy of SM-based therapy in AML cells. (A) Approximately 5700 compounds were screened for their ability to synergize with birinapant and overcome resistance in murine HoxA9/Meis1 AML cells. Compounds that induced cell death in combination with birinapant, but not as single agents, were selected and their impact determined in a dose-response assay. (B) HoxA9/Meis1 and MLL-AF9 AML cells were treated with birinapant (Bir; 100 and 200 nM), with or without reserpine (Res; 1 µM), for 24 hours (n = 4-5). HoxA9/Meis1 (C) and MLL-AF9-NrasG12D (D) AML cells were treated with 10, 100, 500, and 1000 nM reserpine, tariquidar (Tari), or zosuquidar (Zosu), with or without Bir (200 nM), for 24 hours (n = 4). (E-F) HoxA9/Meis1 AML cells were treated with 125, 250, 500, and 1000 nM AT-406 (AT) (E) and AEG-40730 (AEG) (F), with or without Tari (1 µM), for 24 hours (n = 5). (G-H) HoxA9/Meis1 AML cells were treated with the peptide-mimetic agents ABT-199 (G) and JQ1 (H) at 100 and 1000 nM, with or without Tari or Zosu (1 μM), for 24 hours. Treatment with Bir (500 nM)+Tari and Zosu (1 µM) was used as a control (n = 4). Cell death was measured by flow cytometry analysis of PI uptake and decrease in cell volume (MLL-AF9-NrasG12D) in 3 or 4 individual tumors. Data are the mean ± SEM throughout. P values were obtained by comparison of SM alone and SM/MDR1i treatment at the indicated concentrations. *P < .05; **P < .01; ***P < .001; ****P < .0001. n.s. nonsignificant.
Given that reserpine can inhibit both the vesicular monoamine transporter and MDR1,16,17,36 we investigated whether the enhanced SM-mediated killing in AML is dependent on MDR1. Using the third-generation clinical MDR1i tariquidar and zosuquidar we tested the impact of specific MDR1 inhibition on birinapant-induced killing in AML cells (Figure 1C-D; supplemental Figure 2A-B). These inhibitors are molecularly distinct, but both have reached phase 2 and 3 clinical trials for a range of cancers, including AML.37-40 Tariquidar and zosuquidar sensitized several AML cell types to nanomolar concentrations of birinapant (Figure 1C-D). Moreover, this combined effect was not specific to birinapant; tariquidar also increased cell death mediated by the SMs AT-406, AEG-40730, and LCL-161, but not GDC-0152 (Figure 1E-F; supplemental Figure 2C-E).
To assess the specificity of the synergy between the SM peptidomimetics and MDR1i, we tested whether killing induced by other peptide mimetics could be enhanced by MDR1 inhibition. MDR1i did not sensitize AML cells to the BH3-mimetic BCL-2 inhibitor ABT-19941 (Figure 1G; supplemental Figure 2F) or the BET inhibitor JQ142 (Figure 1H; supplemental Figure 2G), suggesting that these drugs are not MDR1 substrates. Together these data indicate that the sensitization mediated by MDR1i is specific to SMs and is not a function of their peptidelike nature.
MDR1 and TNF are biomarkers of birinapant response in AML
To establish whether MDR1 expression is a predictor of birinapant resistance, we screened several primary murine leukemias for MDR1 expression and function. Consistent with their independent origin, the HoxA9/Meis1 and MLL-AF9-NrasG12D leukemias expressed MDR1 at various levels, whereas the MLL-ENL AMLs had no detectable MDR1 expression (Figure 2A). The expression of cellular IAP-1 (cIAP-1) and caspases was similar between the leukemias, except for XIAP, which was reduced in MLL-ENL AMLs (Figure 2A). To confirm that MDR1 was functional we used a Rho-123–retention assay.21 Consistent with the protein expression, inhibition of MDR1 increased fluorescence in MDR1H HoxA9/Meis1 AMLs, and to a lesser extent in MLL-AF9-NrasG12D leukemias, but did not affect MDR1 low (MDR1L) MLL-ENL AMLs (supplemental Figure 3A-C). In agreement with these results, tariquidar and zosuquidar synergistically increased birinapant-induced killing of HoxA9/Meis1 (Bliss synergy scores [BSS], 56.7 and 66.5, respectively) and MLL-AF9-NrasG12D AML cells (BSS, 38.6 and 49.2), but had limited effect in MLL-ENL AMLs (BSS, 11.6 and 9; Figure 2B-D; supplemental Figure 3D-L).
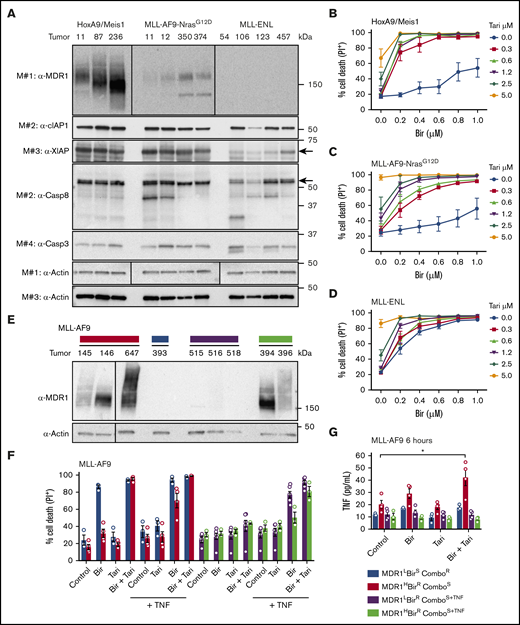
MDR1 and TNF are biomarkers of birinapant response in murine AMLs. (A) Whole-cell lysates from HoxA9/Meis1, MLL-AF9-NrasG12D, and MLL-ENL AML cells were probed with the indicated antibodies, with actin used as the loading control. M# indicates the different membranes. Tumor numbers indicate leukemias originating from individual mice. HoxA9/Meis1 (n = 5; tumors 87, 236, and 11) (B); MLL-AF9-NrasG12D (n = 4; tumors 11, 12, 350, and 374) (C); and MLL-ENL (n = 4; tumors 457, 126, 106, and 123) (D). AML cells were treated with Tari (0.3, 0.6, 1.2, 2.5, and 5 μM), with or without Bir, for 24 hours. (E) Western blot analysis of MDR1 in independent MLL-AF9 AML cells. (F) MDR1L, birinapant-sensitive (BirS), combination-resistant (ComboR) (blue bars, n = 3; tumor 393); MDR1H, birinapant-resistant (BirR), combination-sensitive (ComboS) (red bars, n = 4; tumors 145 and 647); MDR1L, BirR, ComboS+TNF (purple bars, n = 6; tumors 515, 516, and 518); and MDR1H, BirR, and ComboS+TNF (green bars, n = 3; tumors 394 and 396) AML cells were treated for 24 hours with Bir (500 nM), with or without Tari (500 nM) and with or without TNF (100 ng/mL). (G) TNF production in 4 MLL-AF9 AML subgroups was determined by ELISA. Cells were treated with Bir (500 nM), with or without Tari (500 nM), for 6 hours (1-2 individual tumors for each subgroup; 2-3 repeats). Data are the mean ± SEM throughout. Cell death was measured by flow cytometry analysis of PI uptake and decrease in cell volume. P values were obtained by comparison of control and Bir/Tari treatment. *P < .05.
MDR1 and TNF are biomarkers of birinapant response in murine AMLs. (A) Whole-cell lysates from HoxA9/Meis1, MLL-AF9-NrasG12D, and MLL-ENL AML cells were probed with the indicated antibodies, with actin used as the loading control. M# indicates the different membranes. Tumor numbers indicate leukemias originating from individual mice. HoxA9/Meis1 (n = 5; tumors 87, 236, and 11) (B); MLL-AF9-NrasG12D (n = 4; tumors 11, 12, 350, and 374) (C); and MLL-ENL (n = 4; tumors 457, 126, 106, and 123) (D). AML cells were treated with Tari (0.3, 0.6, 1.2, 2.5, and 5 μM), with or without Bir, for 24 hours. (E) Western blot analysis of MDR1 in independent MLL-AF9 AML cells. (F) MDR1L, birinapant-sensitive (BirS), combination-resistant (ComboR) (blue bars, n = 3; tumor 393); MDR1H, birinapant-resistant (BirR), combination-sensitive (ComboS) (red bars, n = 4; tumors 145 and 647); MDR1L, BirR, ComboS+TNF (purple bars, n = 6; tumors 515, 516, and 518); and MDR1H, BirR, and ComboS+TNF (green bars, n = 3; tumors 394 and 396) AML cells were treated for 24 hours with Bir (500 nM), with or without Tari (500 nM) and with or without TNF (100 ng/mL). (G) TNF production in 4 MLL-AF9 AML subgroups was determined by ELISA. Cells were treated with Bir (500 nM), with or without Tari (500 nM), for 6 hours (1-2 individual tumors for each subgroup; 2-3 repeats). Data are the mean ± SEM throughout. Cell death was measured by flow cytometry analysis of PI uptake and decrease in cell volume. P values were obtained by comparison of control and Bir/Tari treatment. *P < .05.
Our results suggested that high levels of MDR1 could be used to predict resistance to birinapant-based therapy in different AML subtypes. We therefore examined several independent MLL-AF9 leukemias that display clonal variation that mirrors the variation found in patients. MDR1 expression and activity and the sensitivity to birinapant-related therapy, was heterogeneous in these leukemias (Figure 2E-F; supplemental Figure 4A-B). MDR1L MLL-AF9 cells were sensitive to birinapant, and addition of tariquidar did not increase cell killing (Figure 2F; blue bars), whereas MDR1H MLL-AF9 cells were responsive only to birinapant in the presence of an MDR1i (Figure 2F, red bars; supplemental Figure 4C-F). However, some AMLs did not respond to birinapant (MDR1L [purple bars]) or birinapant/tariquidar (MDR1H) treatment (Figure 2F). Because SMs kill cancer cells by autocrine TNF/TNFR-1 signaling,1,2,43 we hypothesized that the inherent ability, or lack thereof, of these cells to produce TNF contributed to their response to birinapant. Indeed, whereas TNF levels were increased in MDR1H AML cells upon birinapant/tariquidar treatment (Figure 2G, red bars; supplemental Figure 4G-H), no increase in TNF was observed in MDR1H MLL-AF9 AML cells that were resistant to the combination therapy (Figure 2G, green bars; supplemental Figure 4G). Addition of exogenous TNF and an MDR1i sensitized these cells (purple and green bars) to birinapant (Figure 2F). Genetic deletion of Tnfr1 in HoxA9/Meis1 AML conferred resistance to combination treatment, thus confirming that sensitivity to SM is determined by both TNF and MDR1 expression (supplemental Figure 4I).
The correlation between MDR1 expression and birinapant sensitivity was observed in human leukemia cell lines (Figure 3A-K). Cotreatment with an MDR1i in the MDR1H leukemia cell lines KG1 and HEL potentiated killing by SM, independent of exogenous TNF (Figure 3B-C). In contrast, cells lacking MDR1 function, such as HL60, NB4, MOLM13, MV4-11, U937, OCI-AML3, and THP1 were variably sensitive to birinapant single-agent treatment and addition of tariquidar did not increase killing of the cells (Figure 3D-J; supplemental Figure 5A). OCI-AML2 cells were not sensitized to any of the treatments tested (Figure 3K), suggesting that these cells may be deficient in a component of the TNFR-1 cell death pathway.

MDR1 and TNF are biomarkers of birinapant response in human leukemic cell lines. (A) Whole-cell lysates from human leukemic cell lines were probed with the indicated antibodies, with actin used as the loading control. M# indicates the different membranes. MDR1 activity in KG1 (n = 4) (B), HEL (n = 3) (C), HL60 (n = 5) (D), NB4 (n = 4) (E), MOLM13 (n = 4) (F), MV4-11 (n = 3) (G), U937 (n = 2; error bars SD) (H), OCI-AML3 (n = 3) (I), THP1 (n = 3) (J), and OCI-AML2 (n = 2; error bars SD) (K) cells were investigated by Rho-123–retention assay, and cell viability was measured by flow cytometry analysis of PI and AxV exclusion after the cells were treated with Bir (0.016, 0.08, 0.4, 2, and 10 μM), with or without Tari (1 μM) and TNF (10 ng/mL), for 48 hours. Data are the mean ± SEM throughout, unless otherwise stated. P values were obtained by comparison of Bir alone and Bir/Tari treatment (red) or Bir/TNF and Bir/TNF/Tari treatment (purple) at the indicated concentrations and were determined by multiple t tests using the Holm-Sidak method. *P < .05; **P < .01; ***P < .001.
MDR1 and TNF are biomarkers of birinapant response in human leukemic cell lines. (A) Whole-cell lysates from human leukemic cell lines were probed with the indicated antibodies, with actin used as the loading control. M# indicates the different membranes. MDR1 activity in KG1 (n = 4) (B), HEL (n = 3) (C), HL60 (n = 5) (D), NB4 (n = 4) (E), MOLM13 (n = 4) (F), MV4-11 (n = 3) (G), U937 (n = 2; error bars SD) (H), OCI-AML3 (n = 3) (I), THP1 (n = 3) (J), and OCI-AML2 (n = 2; error bars SD) (K) cells were investigated by Rho-123–retention assay, and cell viability was measured by flow cytometry analysis of PI and AxV exclusion after the cells were treated with Bir (0.016, 0.08, 0.4, 2, and 10 μM), with or without Tari (1 μM) and TNF (10 ng/mL), for 48 hours. Data are the mean ± SEM throughout, unless otherwise stated. P values were obtained by comparison of Bir alone and Bir/Tari treatment (red) or Bir/TNF and Bir/TNF/Tari treatment (purple) at the indicated concentrations and were determined by multiple t tests using the Holm-Sidak method. *P < .05; **P < .01; ***P < .001.
MDR1 inhibition increases the intracellular concentration of birinapant in MDR1H AML
The most likely hypothesis to explain the synergistic killing between birinapant and an MDR1i is that inhibition of MDR1 enhances the intracellular bioavailability of birinapant, to increase the rate of degradation of the SM targets cIAP-1- and -2 (cIAP-1/2; Figure 4A). To test this hypothesis, we compared the degradation of cIAP-1/2, in MDR1H KG1 AML cells treated with birinapant or birinapant/tariquidar. Consistent with our hypothesis, combination treatment, but not tariquidar alone, accelerated cIAP-1/2 degradation and induced potent killing of these cells (Figure 4B-C; supplemental Figure 5B-C). The reduction in cIAP levels in AML cells at early time points after combination treatment was unlikely to be related to cell death, which was negligible during the first 6 hours of treatment (Figure 4C). Similarly, when combined with the SMs AT-406 or AEG-40730, tariquidar also induced rapid degradation of cIAP-1 and killing of AML cells (supplemental Figure 5D-I).
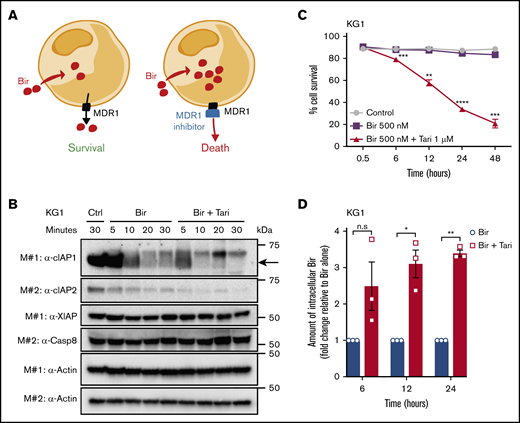
MDR1 inhibition increases the intracellular concentration of birinapant in MDR1HAML. (A) The proposed mechanism of action of SM/MDR1i combination therapy in MDR1H cells. Bir was pumped out of cells by MDR1, and inhibition of MDR1 pumps increased the intracellular levels of birinapant. (B) KG1 AML cells were treated for the indicated times with Bir (500 nM), with or without the MDR1i Tari (1 μM). Whole-cell lysates were probed with the indicated antibodies, with actin used as the loading control. M# indicates the different membranes. (C) KG1 AML cells were treated for up to 48 hours with Bir (500 nM), with or without Tari (1 μM; n = 4). Cell survival was measured by flow cytometry analysis of PI exclusion. (D) Intracellular levels of birinapant in KG1 AML cells treated with Bir (500 nM), with or without Tari (1 μM), were quantified using MS and deuterium-labeled birinapant as a standard (n = 3 independent experiments). Data are the means ± SEM throughout. P values were obtained by comparison of Bir alone and Bir/Tari treatment at the indicated time points. *P < .05; **P < .01; ***P < .001; ****P < .0001.
MDR1 inhibition increases the intracellular concentration of birinapant in MDR1HAML. (A) The proposed mechanism of action of SM/MDR1i combination therapy in MDR1H cells. Bir was pumped out of cells by MDR1, and inhibition of MDR1 pumps increased the intracellular levels of birinapant. (B) KG1 AML cells were treated for the indicated times with Bir (500 nM), with or without the MDR1i Tari (1 μM). Whole-cell lysates were probed with the indicated antibodies, with actin used as the loading control. M# indicates the different membranes. (C) KG1 AML cells were treated for up to 48 hours with Bir (500 nM), with or without Tari (1 μM; n = 4). Cell survival was measured by flow cytometry analysis of PI exclusion. (D) Intracellular levels of birinapant in KG1 AML cells treated with Bir (500 nM), with or without Tari (1 μM), were quantified using MS and deuterium-labeled birinapant as a standard (n = 3 independent experiments). Data are the means ± SEM throughout. P values were obtained by comparison of Bir alone and Bir/Tari treatment at the indicated time points. *P < .05; **P < .01; ***P < .001; ****P < .0001.
To validate that rapid degradation of cIAPs was associated with increased intracellular levels of birinapant, we performed MS on MDR1H KG1 cells treated with birinapant or birinapant/tariquidar. Using deuterium-labeled birinapant as an internal standard, we quantified the levels of intracellular birinapant and found that MDR1 inhibition resulted in a threefold increase in the concentration of birinapant after 6 hours, with SM levels remaining high up to 24 hours after treatment (Figure 4D).
Specific targeting of MDR1 sensitizes AML cells to SM-induced killing
Other ABC transporters, such as ABCC1/MRP1 and ABCG2/BCRP,44,45 can also mediate the efflux of anticancer drugs. To determine whether SMs are exclusively MDR1 substrates, we tested the impact of the MRP1 and BCRP inhibitors reversan46 and Fum-C,47 respectively, on birinapant-induced killing. Neither HoxA9/Meis1 nor MLL-AF9 leukemias were sensitized to birinapant by low doses of these inhibitors (Figure 5A; supplemental Figure 6A). However, higher concentrations of reversan increased birinapant-induced death (supplemental Figure 6A), most likely because of the off-target inhibition of MDR1 by reversan,46,48 supporting the idea that MDR1 is the primary ABC transporter responsible for regulating SM concentration in AML leukemias. To validate this hypothesis, we generated KG1 MDR1−/− cells using 2 independent CRISPR gRNAs. Protein analysis confirmed reduced MDR1 expression and activity in pools of <10 clones transduced with either MDR1 gRNA (Figure 5B-C; supplemental Figure 6B-C). Genetic loss of MDR1 led to reduced expression of cIAP-2 and sensitization of KG1 cells to birinapant single-agent treatment (Figure 5B-F). The increased killing of AML cells imparted by loss of MDR1 was specific to birinapant treatment, because KG1 control and MDR1−/− cells were similarly sensitive to the chemotherapy cytarabine (Ara-c), and the peptide-mimetics JQ1 and ABT-199 (supplemental Figure 6D-F). Furthermore, cIAP-1 degradation mediated by birinapant was faster in KG1 MDR1−/− cells, and the addition of tariquidar had no enhanced effect (Figure 5G; supplemental Figure 6G).
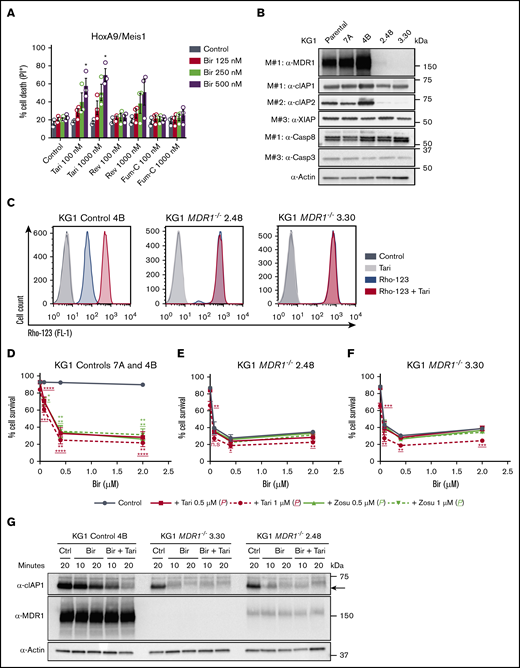
Specific targeting of MDR1 sensitizes AML cells to SM-induced killing. (A) HoxA9/Meis1 AML cells were treated with Bir (125, 250, and 500 nM), with or without Tari or the ABC transporter inhibitor Fum-C (inhibits BCRP/ABCG2) or reversan (Rev; inhibits MRP1/ABCC1 and MDR1; 100 and 1000 nM), for 24 hours (n = 3). P values were obtained by comparison of Bir alone and in combination with MDR1 or other ABC transporter inhibitors. (B) Whole-cell lysates from KG1 parental, controls 7A and 4B, and MDR1−/− 2.48 and 3.30 cell pools were probed with the indicated antibodies. (C) Rho-123–retention assay in KG1 control 4B and MDR1−/− 2.48 and 3.30 AML cells pretreated with Rho-123 (250 nM), with or without Tari (1 μM).Rho-123 fluorescence was determined by flow cytometry. KG1 controls 7A and 4B (n = 4-6) (D), MDR1−/− 2.48 (n = 2-3; error bars, SD) (E), and MDR1−/− 3.30 (n = 2-3; error bars, SD) (F) AML cells were treated with Bir (0.08, 0.4, and 2 μM), with or without Tari or Zosu (0.5 and 1 μM), for 48 hours. P values were obtained by comparison of Bir alone (blue) and in combination with Tari (red) or Zosu (green) at the indicated concentrations. Underscored P values correspond to 1 μM MDR1i. (G) KG1 control 4B and MDR1−/− 3.30 and 2.48 AML cells were treated with Bir (500 nM), with or without Tari (1 μM), for 10 and 20 minutes. Whole-cell lysates from these samples were probed with the indicated antibodies. Data are the mean ± SEM throughout, unless otherwise stated. Cell survival was measured by flow cytometry analysis of PI exclusion. Actin was used as the loading control. M# indicates individual membranes. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Specific targeting of MDR1 sensitizes AML cells to SM-induced killing. (A) HoxA9/Meis1 AML cells were treated with Bir (125, 250, and 500 nM), with or without Tari or the ABC transporter inhibitor Fum-C (inhibits BCRP/ABCG2) or reversan (Rev; inhibits MRP1/ABCC1 and MDR1; 100 and 1000 nM), for 24 hours (n = 3). P values were obtained by comparison of Bir alone and in combination with MDR1 or other ABC transporter inhibitors. (B) Whole-cell lysates from KG1 parental, controls 7A and 4B, and MDR1−/− 2.48 and 3.30 cell pools were probed with the indicated antibodies. (C) Rho-123–retention assay in KG1 control 4B and MDR1−/− 2.48 and 3.30 AML cells pretreated with Rho-123 (250 nM), with or without Tari (1 μM).Rho-123 fluorescence was determined by flow cytometry. KG1 controls 7A and 4B (n = 4-6) (D), MDR1−/− 2.48 (n = 2-3; error bars, SD) (E), and MDR1−/− 3.30 (n = 2-3; error bars, SD) (F) AML cells were treated with Bir (0.08, 0.4, and 2 μM), with or without Tari or Zosu (0.5 and 1 μM), for 48 hours. P values were obtained by comparison of Bir alone (blue) and in combination with Tari (red) or Zosu (green) at the indicated concentrations. Underscored P values correspond to 1 μM MDR1i. (G) KG1 control 4B and MDR1−/− 3.30 and 2.48 AML cells were treated with Bir (500 nM), with or without Tari (1 μM), for 10 and 20 minutes. Whole-cell lysates from these samples were probed with the indicated antibodies. Data are the mean ± SEM throughout, unless otherwise stated. Cell survival was measured by flow cytometry analysis of PI exclusion. Actin was used as the loading control. M# indicates individual membranes. *P < .05; **P < .01; ***P < .001; ****P < .0001.
The birinapant/MDR1i combination is well tolerated, efficiently kills murine LSCs, and prolongs survival in AML models
Short-term cell viability assays are indicative but not necessarily predictive of a therapeutic response, we therefore tested MDR1H MLL-AF9 leukemias in long-term assays. AML cells treated with birinapant/zosuquidar were unable to form colonies in agar culture (supplemental Figure 7A-B), suggesting that the combination therapy could be an effective way to treat MDR1–expressing AMLs. Although birinapant and MDR1i are all well tolerated as single agents, their safety in combination has not been evaluated. To test whether birinapant/tariquidar treatment is feasible in vivo, C57BL/6 mice were treated with the combined drugs 3 times a week for 4 weeks and analyzed immediately upon completion of the treatment regimen (acute toxicity) or 6 weeks after (chronic toxicity). No signs of distress were observed after the injections. Body weight, liver, and blood cell counts were mostly unaffected, although there was an increase in neutrophil counts (supplemental Figure 7C-G). Spleen weights were initially increased, but this effect subsided after 6 weeks (supplemental Figure 7H-I). Together, these results indicate a high tolerability of birinapant/MDR1i combination therapy in vivo.
A limiting factor of chemotherapies is often the toxicity to healthy HSPCs (LSKs). Notably, these cells are known to be MDR1H49,50 and thus may be particularly sensitive to MDR1i combination therapies (supplemental Figure 7J). To determine the impact of birinapant/MDR1i treatment on HSPCs, LSK cells from healthy C57BL/6 mice were treated for 48 hours with birinapant/tariquidar or the chemotherapy Ara-c. Ara-c potently killed the cells, whereas the viability of LSK cells was unaffected by birinapant or birinapant/tariquidar treatment (Figure 6A; supplemental Figure 7K), suggesting that there may be a better therapeutic window for SM/MDR1i therapy than for Ara-c.
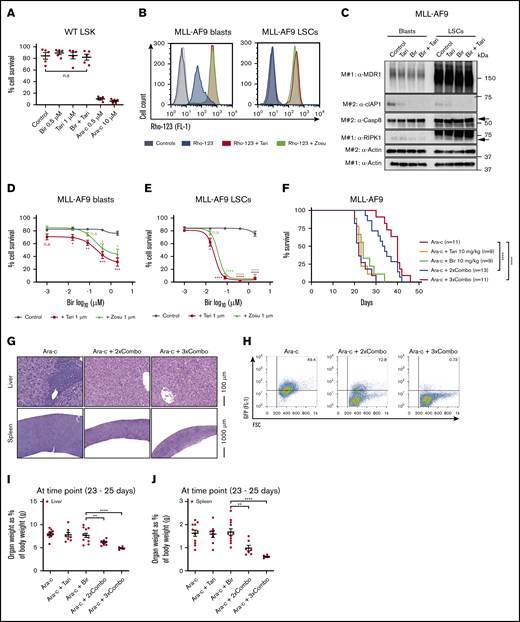
The birinapant/MDR1i combination is well tolerated, efficiently kills murine LSCs, and prolongs survival in murine AML models. (A) LSK HSPCs from wild-type (WT) C57BL/6 mice were treated with Bir (0.5 μM), with or without Tari (1 μM) or Ara-c (0.5 and 10 μM), for 48 hours (n = 3-5; 3 independent experiments). (B) MLL-AF9 blasts and LSCs were pretreated with Rho-123 (250 nM), with or without Tari (1 μM). Rho-123 fluorescence determined by flow cytometry.Histograms with different shades of gray represent control cells untreated (dark) and treated (light) with MDR1i in the absence of Rho-123. (C) MLL-AF9 blasts and LSCs were treated with Bir (500 nM), with or without Tari (1 μM), for 10 minutes. Whole-cell lysates were probed with the indicated antibodies, with actin used as the loading control. M# indicates individual membranes. MLL-AF9 AML blasts (D) and LSCs (E) were treated with Bir (0.001, 0.016, 0.08, 0.4, and 2 μM), with or without Tari or Zosu (1 μM), for 24 hours (n = 4-7). P values were obtained by comparison of Bir alone with Bir/Tari (red) or Bir/Zosu (green) treatment at the indicated time points. (F) C57BL/6 mice receiving MDR1H MLL-AF9-GFP AML cell transplants were treated with Ara-c (50 mg/kg) for 5 consecutive days, followed by either Bir single-agent treatment (10 mg/kg) or Bir/tTari (10 mg/kg) combination therapy, given 2 or 3 times a week (Ara-c+2×Combo and Ara-c+3×Combo, respectively; n = 9-13; 4 independent experiments). P values were obtained by comparison of Ara-c single-agent treatment and Bir/ Tari treatments and determined by log-rank (Mantel-Cox) test. Histology of spleens and livers (G) and percentages of GFP+ cells in the bone marrow (H) of mice. The weights of livers (I) and spleens (J) as a percentage of body weight (n = 6-11) were also determined. Cell survival was measured by flow cytometry analysis of PI exclusion. P values were obtained by comparison of Ara-c+Bir and Ara-c+2×Combo or Ara-c+3×Combo. Data are the mean ± SEM throughout. *P < .05; **P < .01; ***P < .001; ****P < .0001. FSC, forward scatter.
The birinapant/MDR1i combination is well tolerated, efficiently kills murine LSCs, and prolongs survival in murine AML models. (A) LSK HSPCs from wild-type (WT) C57BL/6 mice were treated with Bir (0.5 μM), with or without Tari (1 μM) or Ara-c (0.5 and 10 μM), for 48 hours (n = 3-5; 3 independent experiments). (B) MLL-AF9 blasts and LSCs were pretreated with Rho-123 (250 nM), with or without Tari (1 μM). Rho-123 fluorescence determined by flow cytometry.Histograms with different shades of gray represent control cells untreated (dark) and treated (light) with MDR1i in the absence of Rho-123. (C) MLL-AF9 blasts and LSCs were treated with Bir (500 nM), with or without Tari (1 μM), for 10 minutes. Whole-cell lysates were probed with the indicated antibodies, with actin used as the loading control. M# indicates individual membranes. MLL-AF9 AML blasts (D) and LSCs (E) were treated with Bir (0.001, 0.016, 0.08, 0.4, and 2 μM), with or without Tari or Zosu (1 μM), for 24 hours (n = 4-7). P values were obtained by comparison of Bir alone with Bir/Tari (red) or Bir/Zosu (green) treatment at the indicated time points. (F) C57BL/6 mice receiving MDR1H MLL-AF9-GFP AML cell transplants were treated with Ara-c (50 mg/kg) for 5 consecutive days, followed by either Bir single-agent treatment (10 mg/kg) or Bir/tTari (10 mg/kg) combination therapy, given 2 or 3 times a week (Ara-c+2×Combo and Ara-c+3×Combo, respectively; n = 9-13; 4 independent experiments). P values were obtained by comparison of Ara-c single-agent treatment and Bir/ Tari treatments and determined by log-rank (Mantel-Cox) test. Histology of spleens and livers (G) and percentages of GFP+ cells in the bone marrow (H) of mice. The weights of livers (I) and spleens (J) as a percentage of body weight (n = 6-11) were also determined. Cell survival was measured by flow cytometry analysis of PI exclusion. P values were obtained by comparison of Ara-c+Bir and Ara-c+2×Combo or Ara-c+3×Combo. Data are the mean ± SEM throughout. *P < .05; **P < .01; ***P < .001; ****P < .0001. FSC, forward scatter.
Similar to normal HSPCs, LSCs have been reported to have high levels of MDR1, and it is known that targeting these cells is crucial for the elimination of AML and prevention of relapse.24,51 The aggressive nature of the AML transplantation model makes it difficult to assess the ability of the birinapant /tariquidar therapy to target LSCs; therefore, we determined the impact of this treatment in vitro in previously established murine MLL-AF9 LSCs and matching blasts.52 Western blot and Rho-123 efflux assays confirmed high levels of functional MDR1 in these LSCs compared with leukemic blasts (Figure 6B-C). As previously observed for primary MLL-AF9 cells (Figure 2), the matched MLL-AF9 blasts were sensitized to birinapant-induced killing by the addition of an MDR1i (Figure 6D). Strikingly, MLL-AF9 LSCs were very sensitive to birinapant/MDR1i treatment but less sensitive to Ara-c chemotherapy (Figure 6E; supplemental Figure 8A-B). LSCs were more resistant to SM-mediated cIAP-1 degradation compared with blast cells, and the addition of tariquidar led to rapid cIAP-1 degradation and cell death in LSCs (supplemental Figure 8C-E). Because MDR1i have had limited efficacy in clinical trials in combination with chemotherapies that are MDR1 substrates, such as etoposide (VP16), we compared birinapant/tariquidar with VP16/tariquidar treatment. Remarkably, the birinapant/tariquidar combination was significantly more effective at killing murine LSCs than was the VP16/tariquidar combination (supplemental Figure 8F-G). These results indicate that birinapant/MDR1i combination therapy may be an effective treatment for eliminating LSCs and thereby could overcome treatment resistance and disease relapse.
Ara-c is a front-line chemotherapy for AML, and although it can dramatically reduce leukemic blast burden, disease relapse occurs in ∼50% of patients, possibly because of the presence of Ara-c–resistant LSCs.32 Because of the strong effect of birinapant/tariquidar on murine LSCs, we determined whether this therapy would prolong survival after chemotherapy. Immunocompetent mice receiving a transplant of aggressive MDR1H MLL-AF9-GFP AML cells, were treated with Ara-c (50 mg/kg) for 5 consecutive days followed by either birinapant or birinapant/tariquidar treatment, 2 or 3 times a week (supplemental Figure 9A). The birinapant/tariquidar combination (Combo) was the only treatment to give a survival benefit, doubling the median survival when compared with control or single-agent groups (median survival of Ara-c+2×Combo, 34 days; Ara-c+3×Combo, 40 days; compared with Ara-c alone, 22 days; Figure 6F). Consistently, histological analysis revealed a striking decrease in leukemic blasts in the spleens and livers of birinapant/tariquidar-treated mice compared with the Ara-c–only group (Figure 6G; supplemental Figure 9B). This finding correlated with a reduction in MLL-AF9-GFP cells in the bone marrow and strongly reduced hepato- and splenomegaly (Figure 6H-J; supplemental Figures 9C-D and 10A). Furthermore, red blood cell counts were unaffected, whereas white blood cell and platelet counts were, respectively, significantly decreased and increased in the combination-treated groups compared with those treated with birinapant alone (supplemental Figure 10B-D), thus confirming the effectiveness of the combination treatment.
Combination birinapant/tariquidar therapy kills human primary leukemias
To further explore the safety of such a combination therapy in human primary cells, we determined the impact of birinapant and birinapant/tariquidar treatment on healthy human CD34+ HSPCs in vitro. Although human CD34+ cells have MDR1 activity (Figure 7A), no increase in birinapant-mediated cell killing was observed in the presence of an MDR1i (Figure 7B). In contrast, VP16/tariquidar treatment was toxic to these cells (Figure 7B). Finally, we determined the efficacy of birinapant/MDR1i treatment in patient-derived primary AML cells with a different cytogenic and treatment history (supplemental Table 3). Similar to what we observed in murine AMLs and cell lines (Figures 2 and 3), MDR1 activity and response to SM treatment was heterogeneous in primary leukemias (Figure 7C-L). In AML with functional MDR1 (patients 1-4), birinapant/tariquidar treatment was more effective at inducing cell death than birinapant alone, and the most pronounced differences were observed in leukemias with the highest levels of MDR1 (patients 2 and 3). Moreover, in patients 2 and 4, addition of TNF increased cell survival in the presence of birinapant, whereas in patient 3, it had no effect. Nevertheless, addition of tariquidar once again dramatically sensitized patients 2 and 4 AML cells to killing by birinapant (Figure 7C-F). AML cells with no detectable MDR1 activity (patients 5-9), when compared with MDR1H AML cells, were more sensitive to SM single-agent treatment, with no enhanced killing induced by addition of tariquidar (Figure 7G-K). Lastly, patient 10 MDR1L AML was unresponsive to all treatments tested (Figure 7L). These results support the notion that high MDR1 activity can impair birinapant bioavailability and reduce SM treatment efficacy.

Combination birinapant/tariquidar therapy kills human primary leukemias. (A) MDR1 activity was determined in primary human CD34+ HSPCs through a Rho-123–retention assay. Cells were pretreated with Rho-123 (250 nM), with or without Tari (1 μM), and fluorescence was determined by flow cytometry. (B) Human CD34+ HSPCs were treated with Bir or VP16 (0.4, 2, and 10 μM), with or without Tari (1 μM), for 24 to 48 hours (n = 2-7 independent samples, error bars SD). (C-L) MDR1 activity was determined through a Rho-123–retention assay and cell death analysis of samples from patients 1 to 10. Cells from the patients were treated with Bir (0.016, 0.08, 0.4, 2, and 10 μM), with or without Tari (1 μM) and with or without TNF (25 ng/mL), for 24 to 48 hours (1 independent experiment for each sample). Cell survival was measured by flow cytometry analysis of AxV exclusion and is represented relative to control.
Combination birinapant/tariquidar therapy kills human primary leukemias. (A) MDR1 activity was determined in primary human CD34+ HSPCs through a Rho-123–retention assay. Cells were pretreated with Rho-123 (250 nM), with or without Tari (1 μM), and fluorescence was determined by flow cytometry. (B) Human CD34+ HSPCs were treated with Bir or VP16 (0.4, 2, and 10 μM), with or without Tari (1 μM), for 24 to 48 hours (n = 2-7 independent samples, error bars SD). (C-L) MDR1 activity was determined through a Rho-123–retention assay and cell death analysis of samples from patients 1 to 10. Cells from the patients were treated with Bir (0.016, 0.08, 0.4, 2, and 10 μM), with or without Tari (1 μM) and with or without TNF (25 ng/mL), for 24 to 48 hours (1 independent experiment for each sample). Cell survival was measured by flow cytometry analysis of AxV exclusion and is represented relative to control.
Discussion
Chemoresistance is a common feature of AML and one of the main causes of disease relapse and poor overall survival (<30% at 5 years). Clinical trial therapies, such as venetoclax, cytarabine, and hypomethylating agent combinations, have been effective in newly diagnosed AML but have shown modest and short-lived responses in chemoresistant patients; thus, new therapies are needed for this group. High levels of IAPs have been linked to drug resistance in AML, and SM drugs that target IAPs, such as birinapant, have entered late-phase clinical trials. Unfortunately, despite initial promising results, a good safety profile, and the ability to elicit an anticancer immune response, SMs, used as single-agent therapies, have not been overwhelmingly successful in myelodysplastic syndrome (MDS) and relapsed AML.7-9 Using an unbiased screen of preclinical and clinical compounds, we identified MDR1i as a class of drugs that can enhance killing of AML cells by a SM. Expression of MDR1 has extensively been reported to be a predictor of treatment outcome in AML, with high levels of MDR1 being identified in >50% of patients with relapse or secondary disease, such as MDS, chronic myeloproliferative neoplasia, and therapy-related AML.24,26,53,54 Therefore, we suggest that a drug regimen of SM plus MDR1i can be used in chemotherapy for relapsed or refractory disease to improve outcomes for these patients.
MDR1 actively expels diverse cytotoxic agents from cells, including chemotherapies, maintaining the levels of anticancer drugs below toxic levels.22,55 Therefore, cancers that express high levels of MDR1 are less responsive to chemotherapies. Clinical trials of new therapies are frequently performed in patients with cancer, in whom conventional therapies have failed, and thus their tumors are likely to have high MDR1 activity. This finding is important because birinapant and other SMs have been unsuccessfully trialed in patients with relapse of MDS or AML. In this study, we showed that the combination of birinapant with third-generation MDR1-specific inhibitors is well tolerated, efficiently kills murine MLL-AF9 LSCs, and prolongs survival in MDR1H leukemia models. Thus, screening for MDR1 expression and activity may enable birinapant/MDR1i therapy to be targeted to MDR1H cancers.
That MDR1 is a member of the large ABC transporter family and is implicated in chemoresistance25 has prompted both the U.S. Food and Drug Administration and the European Medicines Agency to mandate that new anticancer drugs be assessed for interactions with MDR1 and other drug transporters.56 However, older anticancer agents have not been selected to meet this requirement. An outstanding question in the field is how ABC transporters select their substrates, given that they are structurally diverse.22 We were intrigued that birinapant and other chemically distinct SMs synergized with MDR1i57,58 ; however, our tests with ABT-199 and JQ1 indicated that not all peptide-mimetic drugs are substrates.
Regulation of cellular mechanisms other than drug efflux, including differentiation and cell survival, have been proposed as possible mechanisms by which inhibition of MDR1 may sensitize cells to chemotherapy.25 Using MS, we showed that cotreatment with the MDR1i tariquidar tripled the intracellular levels of birinapant in MDR1H AML cells, indicating that regulation of birinapant efflux is the main mechanism by which these drugs enhance the killing of tumor cells by birinapant.
Unfortunately, although MDR1i are well tolerated by patients, their combination with cytotoxic drugs has led to increased toxicity toward healthy cells, limiting the clinical use of such combination treatments.22,25,27,28,39 Moreover, given that HSPCs express higher levels of MDR1 than their differentiated progeny, we were concerned that combining birinapant with an MDR1i would cause hematopoietic toxicity. However, in addition to observing no adverse effects of the birinapant/MDR1i combination in immunocompetent mice, we found that this treatment did not diminish the survival of mouse or human HSPCs in culture, whereas the current standard-of-care chemotherapy Ara-c or the combination VP16/MDR1i was toxic, as previously observed.11 We speculate that this differential toxicity may be explained by the fact that birinapant is not inherently toxic and therefore, increasing doses of this SM in healthy cells has no discernible effect on their viability. Collectively, these results suggest that repurposing of MDR1 inhibitors in combination with SMs may be a safe and efficacious new approach for cancer therapy.
The reason that birinapant preferentially kills transformed cells while leaving normal cells intact remains unclear. One explanation put forward is that, contrary to chemotherapeutic drugs that alter multiple cellular pathways,59,60 SMs specifically affect IAP signaling pathways. Another explanation may be that abnormal expression of IAPs and/or TNF by the tumor or stromal cells in their environment leads to dependency on the NF-κB and/or the TNFR-1 signaling pathways. Consistent with the latter idea, our in vitro assays with murine and human AMLs revealed that tumors must be able to produce TNF in response to birinapant to be killed efficiently by SMs.
For novel therapies to cure AML, they must eradicate LSCs, yet these cells are highly resistant to many anticancer drugs and therefore are often responsible for relapse.29-32 Our discoveries suggest that MDR1 expression is the Achilles heel of LSCs, which birinapant can exploit if combined with an MDR1i, and thereby prevent disease relapse and prolong survival.
Although further investigation of combination therapy in patient-derived xenograft models would be valuable before clinical trials, availability and inexpensive screening tools for MDR1 activity as a biomarker for SM-related therapies could improve patient stratification. Furthermore, the safety and efficacy of birinapant/MDR1i treatment in preclinical models of AML suggests a new therapeutic approach for patients with AML.
The data sets generated in this study are available from the corresponding authors on request (silke@wehi.edu.au and brumatti@wehi.edu.au).
Acknowledgments
The authors thank the staff of the Walter and Eliza Hall Institute (WEHI) Bioservices facilities for technical assistance, and all members of the Silke Laboratory, Andreas Strasser, Paul Ekert, and George Kiossoglou for discussions.
This work was supported by Leukemia & Lymphoma Society (SCOR [Specialized Centre of Research]) grant 7015-18 (J.S.); National Health and Medical Research Council (NHMRC) grants 1025594, 1046010, and 1081376; Cancer Australia and Leukaemia Foundation Australia priority grant PdCCRS 1162023 (G.B.); NHMRC fellowship 1107149 (J.S.); Victoria Cancer Agency (VCA) mid-career fellowship MCRF 15027 (G.B.); and Australian Cancer Research Foundation and Victorian State Government Operational Infrastructure Support and Australian Government NHMRC Independent Research Institutes Infrastructure Support Scheme (IRIISS) grant 9000433.
Authorship
Contribution: E.M. performed most of the experiments; G.B., A.C., N.S., A.L., G.E., L.M., G.P., K.C.F., J.A.B., and E.L.C. performed the remaining experiments; J.J.S. performed the MS analyses; K.E.J. performed the initial drug screening; D.M.M., A.H.W., J.A.P., A.C.L., and S.M.P., provided expertise, processing, and analysis of primary leukemia samples; L.M. and M.A.D. provided MLL-AF9 leukemic stem cells and blasts; E.M., J.S., and G.B. planned the project, designed the experiments, analyzed the data, and wrote the manuscript; and all authors contributed to discussion of the results and review of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John Silke, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052, Australia; e-mail: silke@wehi.edu.au; and Gabriela Brumatti, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, VIC 3052 Australia; e-mail: brumatti@wehi.edu.au.

