

Abstract
The surge of human genetic information, enabled by increasingly facile and economically feasible genomic technologies, has accelerated discoveries on the relationship of germline genetic variation to hematologic diseases. For example, germline variation in GATA2, encoding a vital transcriptional regulator of multilineage hematopoiesis, creates a predisposition to bone marrow failure and acute myeloid leukemia termed GATA2 deficiency syndrome. More than 300 GATA2 variants representing missense, truncating, and noncoding enhancer mutations have been documented. Although these variants can diminish GATA2 expression and/or function, the functional ramifications of many variants are unknown. Studies using genetic rescue and knockin mouse systems have established that GATA2 mutations differentially affect molecular processes in distinct target genes and within a single target cell. Considering that target genes for a transcription factor can differ in sensitivity to altered levels of the factor, and transcriptional mechanisms are often cell type specific, the context-dependent consequences of GATA2 mutations in experimental systems portend the complex phenotypes and interindividual variation of GATA2 deficiency syndrome. This review documents GATA2 human genetics and the state of efforts to traverse from physiological insights to pathogenic mechanisms.
Introduction
Linking GATA2 dysregulation to human hematologic disease
The discoveries of master regulatory transcription factors that control hematopoiesis have been rapidly followed, or even preceded, by evidence for causative roles in human blood diseases. Transcriptional circuitry governing hematopoietic system development and function generates almost infinite opportunities for hematopoiesis to go awry. This is exemplified by GATA1,1-4 Runx1,5,6 Etv6,7,8 and Scl/TAL1,9,10 as their dysregulation causes human diseases, including anemia, thrombocytopenia, bone marrow failure, and leukemia. After more than a decade of intensive study, strong evidence has emerged from human genetic analyses that GATA2 alterations also cause hematologic disease.
After discovery of the founding member of the GATA transcription factor family GATA1,1,2 GATA2 was shown by Orkin and colleagues in 1994 to be a vital determinant of multilineage hematopoiesis.11 Although high GATA2 expression correlates with disease severity in pediatric and adult acute myeloid leukemia (AML),12,13 analyses of GATA2 germline mutations definitively established its role in causing a complex disorder termed GATA2 deficiency syndrome.14-17 This syndrome involves immunodeficiency with monocytopenia; B cell, natural killer cell, and dendritic cell deficiencies; and common Mycobacterium, fungal, and viral infections. Patients with GATA2 deficiency syndrome may also exhibit lymphedema or monosomy 7.17-20 GATA2 mutations create an myelodysplastic syndrome/AML predisposition, and physiological GATA2 levels suppress bone marrow failure and leukemogenesis.13,21 The only potentially curative therapy is bone marrow transplantation.22,23 Chronic myelogenous leukemia and AML patients with somatic GATA2 mutations reportedly share select phenotypes with germline GATA2 deficiency syndrome,24,25 , albeit the differences between GATA2 deficiency syndrome and these leukemias are considerable.
GATA2 deficiency syndrome has a highly variable penetrance, with unpredictable presentations in children and adults.18,19,26,27 The enigmatic penetrance has profound therapeutic implications. GATA2 mutations in children and adults can be asymptomatic, despite having family members with GATA2 deficiency syndrome. Somatic mutations in a host of genes (RUNX1, ETV6, CEBPA, ASXL1, SETBP1, and STAG2) occur commonly with germline GATA2 mutations and may constitute pathogenic triggers.21,28-37 However, whether different constellations of somatic mutations, in concert with the GATA2 germline mutation, qualitatively or quantitatively influence pathogenesis is unknown. Infection or environmental stresses may establish or influence epigenetic mechanisms to trigger pathogenesis and dictate penetrance.37-39 Epigenetic plasticity is a double-edged sword, as it may promote disease progression40 or mitigate disease phenotypes.41
Mechanistic basis of GATA2-instigated pathogenesis
Understanding GATA2 deficiency syndrome requires knowledge of GATA2-dependent mechanisms that sustain hematopoiesis and permit the hematopoietic system to adapt to diverse stresses. GATA2 promotes hematopoietic stem and progenitor cell (HSPC) generation and function11,42-48 and regulates erythroid and megakaryocytic precursors.13,49,50 In human embryonic stem cell systems, GATA2 overexpression promotes the conversion of hemogenic endothelium to hematopoietic precursors51 and maintains multipotent precursors.52 In addition to HSPCs, GATA2 is expressed in select differentiated cell types, including macrophages, endothelial, neuronal, and endocrine cells.49,51,53-57 Importantly, GATA2 regulation can vary greatly in different biological systems. For example, targeted ablation of the murine Gata2 +9.5 intronic enhancer strongly reduces Gata2 expression in HSPCs without affecting expression in brain.58 Targeted ablation of the far upstream Gata2 −77 enhancer abrogates the multilineage differentiation potential of fetal liver progenitor cells without affecting hematopoietic stem cell emergence,44 which is abrogated by the +9.5 enhancer deletion.43,45 Targeted ablation of the Gata2 −1.8 “GATA switch site” disrupts the mechanism that maintains Gata2 repression during erythroid differentiation, without affecting the initiation of Gata2 repression and Gata2 expression before repression.59 GATA2-instigated genetic networks vary in different cellular contexts and in the steady state vs stress.39,44-46,60 In progenitor cells, GATA2 activates expression of a cadre of mechanistically linked and diverse genes, including those encoding c-Kit receptor tyrosine kinase, c-Kit signaling facilitator Samd14, erythroid and megakaryocytic differentiation inducer GATA1, and histidine decarboxylase mediating histamine biosynthesis.13,61-64 During mouse embryogenesis, GATA2 suppresses expression of innate immune genes, endowing progenitors with multilineage differentiation potential.46 During erythrocyte development, a subset of GATA2-activated genes, including Gata2 itself, are repressed as GATA1 replaces GATA2 on chromatin, a process termed a GATA switch.49,53,65,66 Thus, in certain contexts, GATA1 and GATA2 function distinctly.
The foundation described here raises the question of whether GATA2 germline mutations uniformly disrupt GATA2 expression and function in all cellular contexts, or if the mutations compromise processes in select cellular contexts and/or a subset of GATA2-regulated processes within a single cell. In addition, does the phenotypic complexity of GATA2 deficiency syndrome reflect an amalgamation of hematopoietic cell–intrinsic and non–cell-autonomous defects arising solely from insufficient GATA2, or do certain GATA2 mutants exert ectopic activities? GATA2 germline and acquired mutations include coding and noncoding sequence deletions, frameshifts, and missense mutations. Of 343 GATA2 coding mutations designated “pathogenic,” “likely pathogenic,” and “uncertain significance” by ClinGen/ClinVar (Figure 1), 148 are deemed pathogenic, with 61% (90) altering sequences within the zinc finger domain. Although GATA2 N-finger function is unresolved, the C-finger mediates GATA motif binding.49 In GATA1, the N-finger mediates binding to the coregulator Friend of GATA167,68 and modulates DNA binding at complex sites containing composite GATA motifs.69 Pathogenic mutations external to the GATA2 zinc fingers (41) are predominantly frameshift or nonsense (28% of pathogenic mutations; 41 of 148). GATA2 zinc-finger domain mutations and external frameshift and nonsense mutations amount to 88% of the pathogenic mutations (131 of 148). Apart from nonsynonymous coding mutations, synonymous GATA2 coding mutations have also been described in a small number of patients. Despite unaltered protein composition, nucleotide changes in coding regions induced selective loss of messenger RNA expression and/or splicing errors.70 Phenotypic consequences resembled that of nonsynonymous mutations, and a single variant of synonymous germline mutation generated intrafamilial phenotypic variability.71
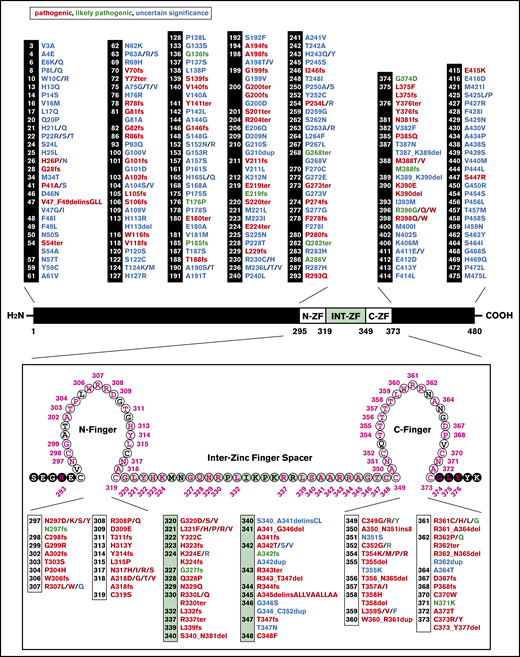
Human GATA2 coding mutations. GATA2 mutations that are designated “pathogenic,” “likely pathogenic,” and “uncertain significance” and their corresponding protein sequences. Top, GATA2 protein sequence depicting mutations external to the zinc finger domain. Bottom, enlargement of GATA2 zinc finger domain illustrating amino acids affected by mutations. Reported mutations are indicated in pink. N-ZF, N-finger; INT-ZF, inter-zinc finger spacer; C-ZF, C-finger. N- and C-fingers are also referred to as Zf1 and Zf2, respectively.
Human GATA2 coding mutations. GATA2 mutations that are designated “pathogenic,” “likely pathogenic,” and “uncertain significance” and their corresponding protein sequences. Top, GATA2 protein sequence depicting mutations external to the zinc finger domain. Bottom, enlargement of GATA2 zinc finger domain illustrating amino acids affected by mutations. Reported mutations are indicated in pink. N-ZF, N-finger; INT-ZF, inter-zinc finger spacer; C-ZF, C-finger. N- and C-fingers are also referred to as Zf1 and Zf2, respectively.
To develop principles underlying GATA2-dependent pathogenesis, it is instructive to consider the relationship between human mutations and posttranslational modifications that regulate GATA2 function. The MAPKs p38α and extracellular signal–regulated kinase can phosphorylate murine GATA2 at multiple sites (S73, S119, S192, S290, and S340), which are conserved between mouse and man (Figure 2A).62 The Ras-MAPK pathway mediates GATA2 multisite phosphorylation in a mechanism requiring S192 and promoted by amino acids 171-174,72 which conform to an extracellular signal–regulated kinase docking sequence termed a “DEF motif” (FXFP) (Figure 2).73,74 GATA2 multisite phosphorylation amplifies its activity to regulate select target genes. In Kasumi-1 AML cells, these genes include IL1β and CXCL2, which generate positive-feedback regulation of the RAS/MAPK-GATA2-IL1β/CXCL2 axis.72 CXCL2 promotes Kasumi-1 cell proliferation, and high CXCL2 expression correlates with poor prognosis of patients with AML. The DEF motif is also implicated in GATA2-mediated megakaryocyte generation from murine hemogenic endothelial cells ex vivo.75 In HEK293 and 3T3-F442A preadipocyte cells, insulin-dependent activation of the Akt kinase induces phosphorylation of GATA2 S401,76 and GRB10, a receptor tyrosine kinase–binding adapter protein, interferes with Akt-mediated GATA2 phosphorylation.77 Whether this signal-dependent mechanism operates in hematopoietic cells is unclear. CDK1 phosphorylates GATA2 at T176 and promotes GATA2 degradation by Fbw7.78 In vitro studies revealed that p300 and GCN5 acetylate GATA2 at K102, K281, K285, K334, K336, K389, K390, K399, K403, K405, K406, K408, and K409 residues (Figure 2B),79 and GATA1 was known to be acetylated.80 Functional analyses revealed that these residues enhance DNA binding in vitro and transactivation activity, and studies in Xenopus eggs implicated GATA2 acetylation in regulating primitive erythropoiesis.81 Two missense mutations reported in ClinGen/ClinVar occur at phosphorylation sites (T176P and S192F), one missense mutation is immediately at the 3′-side of the DEF motif (P175S), and 2 deletion mutations and 2 missense mutations occur at acetylation sites (K389_K390del, K390del, K390E, and K406M).
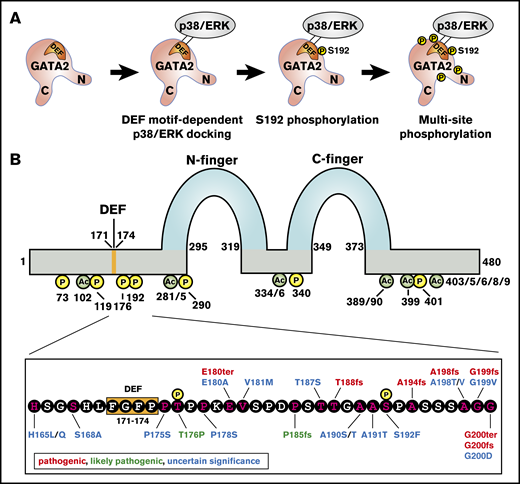
Relationship of human clinical genetic mutations and GATA2 posttranslational modification sites. (A) Multisite GATA2 phosphorylation model. Extracellular signal–regulated kinase (ERK)/p38α docks on the GATA2 DEF motif, S192 is phosphorylated, and other serine residues (S73, S119, S290, and S340) are phosphorylated subsequently. (B) Top, GATA2 protein structure. N- and C-zinc fingers, DEF motif, phosphorylated residues, and acetylated residues are indicated. Bottom, amino acid sequence from 165 aa to 200 aa of GATA2 protein. Phosphorylated residues, DEF motif, and mutations reported by ClinVar/ClinGen are indicated.
Relationship of human clinical genetic mutations and GATA2 posttranslational modification sites. (A) Multisite GATA2 phosphorylation model. Extracellular signal–regulated kinase (ERK)/p38α docks on the GATA2 DEF motif, S192 is phosphorylated, and other serine residues (S73, S119, S290, and S340) are phosphorylated subsequently. (B) Top, GATA2 protein structure. N- and C-zinc fingers, DEF motif, phosphorylated residues, and acetylated residues are indicated. Bottom, amino acid sequence from 165 aa to 200 aa of GATA2 protein. Phosphorylated residues, DEF motif, and mutations reported by ClinVar/ClinGen are indicated.
Among the essential GATA2 noncoding sequences, GATA2 intron 5 (+9.5) enhancer can be mutated in GATA2 deficiency syndrome. These mutations disrupt +9.5 enhancer function, thus reducing GATA2 expression.13,38,39,58,82 The +9.5 enhancer,83-86 which is highly homologous to the human enhancer, is essential for hematopoiesis, HSPC generation/function, and hematopoietic regeneration upon myeloablative stress.39,43,45,58 The most commonly reported germline aberration in the +9.5 enhancer is a c.1017+572C>T transition mutation38,82 that destroys a motif binding members of the ETS transcription factor family.87,88 Although mice harboring this mutation develop into adults with largely normal steady-state hematopoiesis, the mutants are hypersensitive to 5-fluorouracil–mediated myeloablation.39 This phenotype reflects a hematopoietic cell–intrinsic defect in HSPC regeneration.38
In aggregate, the frameshift, nonsense, and +9.5 enhancer mutations suggest a haploinsufficiency mechanism of pathogenesis. Certain missense mutations can generate proteins defective in naked DNA binding, transfection-based transactivation, and/or chromatin occupancy, also consistent with haploinsufficiency. However, transcription factor mutations can generate ectopic activities,89 which may not be revealed without comprehensive multi-omic analyses. Taken together with the granulopoiesis-inducing activity of GATA2 R307W90,91 and the many unanswered questions regarding GATA2 function at the genomic level, it is attractive to consider a model in which mutations inhibit certain, but not all, GATA2 activities, and mutation-induced ectopic activities further corrupt GATA2 mechanisms and cellular physiology. However, much more research is required to elucidate the molecular and cellular consequences of GATA2 disease mutations and, importantly, to determine if firm genotype–phenotype correlations can be established.
Experimental strategies to elucidate GATA2-dependent pathogenic mechanisms
What are the most insightful assays to inform how human GATA2 mutations compromise GATA2 mechanisms to yield pathogenesis? Naked DNA binding has limitations, as affinities may or may not extrapolate to chromatin binding, based on influences from protein–protein interactions and protein posttranslational modifications. GATA factors assemble and integrate into multiprotein complexes on chromatin that may enable tethering of a DNA binding–defective mutant into the complex. Chromatin immunoprecipitation assays reveal whether a mutant loses capacity to occupy all or a subset of sites or acquires nonphysiological sites. Human GATA1 mutations can impair friend of GATA1 (FOG1) binding,4 and FOG1 facilitates chromatin occupancy in a locus-specific manner.49,92 Genome-wide analysis indicates that GATA1 mutations can impair occupancy at certain sites, concomitant with ectopic site acquisition.93 Some GATA2 disease mutants retain chromatin occupancy at certain sites.90 No evidence exists to suggest that transfection-based transactivation assays predict GATA factor–mediated activation or repression of target genes.
To surmount these complexities and limitations, a genetic rescue assay was developed in Gata2 −77 enhancer-mutant murine progenitor cells, in which GATA2 levels are ∼80% lower than wild-type progenitor cells.90,91 The −77 enhancer confers GATA2 expression in progenitors,44,46 and a 3q21q26 inversion in AML allows the −77 to induce MECOM1 as a leukemogenic mechanism.13,94,95 Using a retroviral-based expression strategy, GATA2 or a GATA2 disease mutant can be replaced at a near-physiological level, and the impact on target genes, differentiation, or any process of interest can be quantified. It is essential to compare wild-type and mutant proteins at normal expression levels, as transcription factor target genes can differ in sensitivity to reduced levels of the factor.96 In addition, in humans, there are multiple examples of disease phenotypes resulting from transcription factor haploinsufficiency.97 The C-finger germline T354M mutant can be stably expressed in cells, retains measurable naked DNA binding capacity15,98 and activity to regulate select target genes and to promote granulocytic differentiation in the rescue assay, albeit at reduced levels.90,91 An R307W N-finger mutation from patients with AML also can be stably expressed in cells and retains activity to regulate certain, but not all, GATA2 target genes. Although this mutant does not support erythroid progenitor function in the rescue assay, it retains the capacity to promote granulocytic differentiation at least as effectively as wild-type GATA2. A familial nine amino acid insertion between the zinc fingers abrogates target gene regulation and differentiation.91 The differential impact of GATA2 mutations on target gene expression is consistent with the existing paradigm that GATA factors function through multiple regulatory modes.49,99 Based on this mechanistic diversity, in which a cohort of GATA factor target genes may be regulated differently from another target gene cohort, a molecular aberration would not be expected to disrupt all mechanisms equivalently. This epitomizes the problem: how can a single assay inform pathogenic mechanisms that involve multiple genes controlled by distinct mechanisms? The differential functional consequences of GATA2 mutations illustrate the need to rigorously analyze relationships between GATA2 function in vivo with ex vivo and in vitro assay readouts; the most instructive assay readouts are unknown, and it is certainly possible that multiple assays will be required.
Although in vivo approaches are intrinsically low-throughput, they have considerable potential for modeling human GATA2-linked disease phenotypes, unveiling disease mechanisms, and potentially providing screening systems to achieve clinical/translational outcomes. The first Gata2 knockin mouse disease model was reported recently. This strain harbors a G320D mutant allele and biallelic Cebpa mutation, which recapitulates that detected in patients with acute erythroid leukemia, albeit the human mutations were somatic and not germline.100 Approximately 40% of the mice developed acute erythroid leukemia, illustrating the power of combining mutations to model disease phenotypes.
GATA2 disease mutations: one or more paths to pathogenesis?
The aggregate data raise the question as to whether GATA2 disease mutations cause pathogenesis by attenuation of all GATA2-dependent processes, a subset of processes, acquisition of ectopic GATA2 activities, or a combination of these mechanisms. A mutation-sensitive process may be inextricably linked to a process not affected directly by the mutation, thus generating composite defects that can be difficult to deconvolute experimentally and therefore to fully understand. It will be crucial to systematically analyze a cohort of disease mutants with different assays and systems, including rescue assays ex vivo46,90,91 and knockin mice,101 to unveil GATA2 deficiency syndrome mechanisms. Furthermore, as mutations analogous to GATA2 disease mutations have been reported in genes encoding other GATA factors, and these mutations can be associated with human disease, insights from GATA2 mutations will almost certainly inform biology and pathology linked to other GATA factors.49 For example, the GATA1 R216Q mutation, detected in a patient with congenital erythropoietic porphyria, is analogous to the GATA2 R307W mutation.102
Considering the thousands of GATA2 chromatin occupancy sites, cell type–specific GATA2 target genes, and context-dependent transcriptional regulatory modes, abrogating function at certain, but not all, target genes destroys stoichiometric relationships within the delicately balanced, GATA2-regulated genetic and protein networks. The aberrant networks may generate ectopic mechanisms that are deleterious to cellular physiology and not characteristic of normal hematopoiesis. Networks can be corrupted via multiple means, and the quantitative and qualitative impact of distinct lesions in network components may differ, creating phenotypic diversity. Alternatively, any aberration that creates a network imbalance might yield dominant phenotypes that are largely invariant across distinct molecular lesions. The impact of this problem extends beyond mechanism, as this may guide therapies.
From a therapeutic perspective, the objective to restore inadequate levels of GATA2 may be exceedingly challenging, without exacerbating pathological phenotypes. As noted earlier, high GATA2 correlates with poor-prognosis leukemia,12,103 and elevating GATA2 in murine bone marrow suppressed hematopoiesis in a transplant model.104 As an alternative or complement to bone marrow transplantation, it may be necessary to destroy the GATA2 mutant, repair the GATA2 mutation, supplant ectopically elevated mechanisms, or reinstate attenuated mechanisms. These promising approaches need to be intensively evaluated, yet each presents unique challenges that may or may not be surmountable.
Conclusions
While the depth of knowledge of GATA2 mechanisms and biological functions continues to increase, the phenotypic complexity and highly variable penetrance of GATA2 deficiency syndrome remains enigmatic. A simple explanation for this complexity is different GATA2 disease-linked mutations may create diverse molecular and cellular aberrations through quantitative differences in the severity of aberrations and/or qualitative differences. For example, certain aberrations may generate ectopic activity, whereas others diminish physiological activity, and there may be cases in which a given aberration elicits both consequences. Thus, multiple molecular paths to pathogenesis may exist. A common thread of distinct aberrations is their contribution to deconstruction of a finely balanced network, which reinforces the vital need to elucidate how such networks are established and maintained. Ascertaining whether GATA2 deficiency syndrome involves a predominant path or multiple paths to pathogenesis is an exceptionally high priority, as this mechanistic framework will guide the development of the most appropriate therapeutic options, and the outcomes will guide patient-specific precision medicine strategies.
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (DK68634 and DK50107) (E.H.B.), the Carbone Cancer Center (P30CA014520), and the Edward P. Evans Foundation.
Authorship
Contribution: All authors wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emery H. Bresnick, Department of Cell and Regenerative Biology, Carbone Cancer Center, University of Wisconsin School of Medicine and Public Health, 1111 Highland Ave, Madison, WI 53705; e-mail: ehbresni@wisc.edu.

