

Key Points
NEO2734 binds to both BET proteins and CBP/EP300.
NEO2734 has preclinical antitumor activity, especially in lymphomas and leukemias.

Abstract
Bromodomain and extra-terminal domain (BET) proteins, cyclic adenosine monophosphate response element-binding protein (CBP), and the E1A-binding protein of p300 (EP300) are important players in histone acetylation. Preclinical evidence supports the notion that small molecules targeting these proteins individually or in combination can elicit antitumor activity. Here, we characterize the antitumor activity of the pan BET/CBP/EP300 inhibitor NEO2734 and provide insights into its mechanism of action through bromodomain-binding assays, in vitro and in vivo treatments of cancer cell lines, immunoblotting, and transcriptome analyses. In a panel of 60 models derived from different tumor types, NEO2734 exhibited antiproliferative activity in multiple cell lines, with the most potent activity observed in hematologic and prostate cancers. Focusing on lymphoma cell lines, NEO2374 exhibited a pattern of response and transcriptional changes similar to lymphoma cells exposed to either BET or CBP/EP300 inhibitors alone. However, NEO2734 was more potent than single-agent BET or CBP/EP300 inhibitors alone. In conclusion, NEO2734 is a novel antitumor compound that shows preferential activity in lymphomas, leukemias, and prostate cancers.
Introduction
Epigenetic mechanisms are essential for maintaining homeostatic gene expression of normal cells. Deregulation of the epigenetic apparatus is frequently observed in tumors, leading to the overexpression of oncogenes and silencing of tumor suppressor genes.1 An important epigenetic mediator of transcription is histone acetylation. This covalent epigenetic mark is mainly regulated by 3 classes of proteins: histone acetyltransferases, also called “writers,” which covalently introduce acetyl groups; histone deacetylases, called “erasers,” which remove these chemical modifications; and bromodomain (BRD) proteins, called “readers,” which bind to acetylated histones.2 BRD and extra-terminal domain (BET) proteins associate with acetylated histone tails and are critical regulators of transcription. In cancer cells, they drive the expression of key oncogenic networks. Thus, BET proteins are considered relevant therapeutic targets in various tumors, and this has driven the development of numerous BET inhibitors.3 Many preclinical studies substantiate the rationale to pharmacologically target BET proteins in human cancers, and these are supported by clinical studies reporting the efficacy of this class of compounds.3-14
The acetyltransferase cyclic AMP response element-binding protein (CBP) and the E1A-interacting protein of 300 kDa (EP300) are highly homologous BRD-containing transcriptional coactivators that are often mutated in human cancers, including diffuse large B-cell lymphoma (DLBCL).15,16 In DLBCL, CREBBP mutations are predominantly mono-allelic (80% of CREBBP mutated cases), suggesting that some EP300 and CBP activity is needed by cancer cells. A basis for targeting both proteins in cancer came from a synthetic-lethal screen revealing that CBP-deficient cancer cells were killed by suppression of EP300.17 Moreover, it has been shown that EP300/CBP inhibitors are significantly more toxic in CBP−/− than CBP+/+ DLBCL cell lines.8 Compared with BET inhibitors, few data are available for inhibitors of CBP and EP300. However, there is preclinical evidence supporting the notion that small molecules targeting both proteins elicit antitumor activity,8,9,18-24 and early clinical trials are ongoing (NCT04068597, NCT03568656). Furthermore, the combination of a BET inhibitor and a CBP/EP300 inhibitor is synergistic in acute myeloid leukemia (AML) cell lines.18
Together, these observations provide a logical rationale for targeting both epigenetic writers (CBP/EP300) and readers (BET proteins). Due to the homology of the BRDs present in CBP/EP300 and BET proteins, small molecules with such dual inhibitory capacity can be designed.25-28 NEO2734 and NEO1132 are 2 lead clinical candidates from a series of dual inhibitors of BET and CBP/EP300.29 Here, we present their biological characterization as antitumor compounds and their mechanism of action by using preclinical DLBCL models.
Methods
Screening of BRD binding
Screening of BRD binding was performed at DiscoverX Corporation (Fremont, CA). T7 phage strains displaying BRDs were grown in parallel in 24-well blocks in an Escherichia coli host derived from the BL21 strain. E coli were grown to log-phase and infected with T7 phage from a frozen stock (multiplicity of infection = 0.4) and incubated with shaking at 32°C until lysis (90-150 minutes). The lysates were centrifuged (5000g) and filtered (0.2 μm) to remove cell debris. Streptavidin-coated magnetic beads were treated with biotinylated small molecule or acetylated peptide ligands for 30 minutes at room temperature to generate affinity resins for BRD assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock, Thermo Fisher Scientific, Waltham, MA; 1% bovine serum albumin, 0.05% Tween 20, 1 mM dithiothreitol) to remove unbound ligand and to reduce nonspecific phage binding. Binding reactions were assembled by combining BRDs, liganded affinity beads, and test compounds in 1× binding buffer (17% SeaBlock, 0.33× phosphate-buffered saline [PBS], 0.04% Tween 20, 0.02% bovine serum albumin, 0.004% sodium azide, 7.4 mM dithiothreitol). Test compounds were prepared as 1000× stocks in 100% dimethyl sulfoxide (DMSO). Dissociation constants (Kds) were determined by using an 11-point threefold compound dilution series with 1 DMSO control point. All compounds for Kd measurements were distributed by acoustic transfer (noncontact dispensing) in 100% DMSO. The compounds were then diluted directly into the assays such that the final concentration of DMSO was 0.09%. All reactions were performed in polypropylene 384-well plates, each at a final volume of 0.02 mL. The assay plates were incubated at room temperature with shaking for 1 hour, and the affinity beads were washed with wash buffer (1× PBS, 0.05% Tween 20). The beads were then resuspended in elution buffer (1× PBS, 0.05% Tween 20, 2 μM non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. The BRD concentration in the eluates was measured by using quantitative polymerase chain reaction (PCR).
Proliferation and apoptosis assays
The screening of 60 cell lines was performed at Crown Bioscience facilities (Beijing, People’s Republic of China). All the cell lines, short tandem repeat verified for their cell identity, were cultured in media supplemented with 10% to 15% FBS. Cells (4000 in each well) were seeded in 96-well plates and treated for 72 hours. Viability of cells was assessed with CellTiter-Glo (Promega, Madison, WI) and luminescence, recorded by using an EnVision Multilabel Reader (PerkinElmer, Waltham, MA). Antiproliferative activity in DLBCL cell lines was assessed as previously described.6 The identity of DLBCL cell lines was validated by short tandem repeat DNA fingerprinting.30 Briefly, cells were manually seeded in 96-well plates at a concentration of 50 000 cells/mL (10 000 cells in each well). Treatments were performed by using the Tecan D300e Digital Dispenser (Tecan, Männedorf, Switzerland). After 72 hours, cell viability was determined by using 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide; 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide and the reaction stopped after 4 hours with sodium dodecyl sulfate lysis buffer. Cell cycle analysis was performed as previously described.31 Combination studies and calculation of the Chou-Talalay combination index (CI) were performed as previously described.32,33 NEO2734 (EP31670) and NEO1132 were provided by NEOMED Therapeutics 1 Inc., birabresib and SGC-CBP30 were puchased from MedKoo Biosciences Inc. (Morrisville, NC), and venetoclax from Selleckchem (Selleckchem, Houston, TX).
In DLBCL cell lines, BCL2, MYC, TP53, and MYD88 status was defined as previously described.34 CREBBP and EP300 mutational status was specifically obtained from the cell lines by using a recently reported targeted gene panel.35 Sequenced reads were demultiplexed and converted from the Illumina binary format into FASTQ format. Adaptor sequences were trimmed by using Cutadapt (https://pypi.python.org/pypi/cutadapt). Sequenced reads were then aligned against the human genome reference sequence (hg19) with Burrows-Wheeler Aligner.36 Subsequent local insertion/deletion realignment and base quality score recalibration were performed by using the Genome Analysis Toolkit (GATK; https://www.broadinstitute.org/gatk). Genetic variants were called with GATK Unified Genotyper using the default parameters except –minIndelFrac (set to 0.05).37 Variants were annotated using Ensembl Variant Effect Predictor.38 The effect of an amino acid substitution induced by a mutation was predicted with SIFT39 and PolyPhen240 algorithms. Nonsense, frameshift, and canonical splice site variants (positions −2 and −1 upstream of an exon start and +1 and+2 downstream of an exon end) were considered for further analysis.
Immunoblotting
Protein extraction was performed by using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific). Proteins were resolved on 4% to 15% Mini-PROTEAN TGX Precast Gel (Bio-Rad, Hercules, CA). Membranes were incubated with anti-MYC (D3N8F, CST, Cell Signaling Technology, Danvers, MA) and anti–glyceraldehyde-3-phosphate dehydrogenase (14-9523-80, Thermo Fisher Scientific). Glyceraldehyde-3-phosphate dehydrogenase was used as loading control.
Transcriptome profiling
Total RNA was extracted and processed for RNA-sequencing (stranded, single-ended 75bp-long sequencing reads) using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs Inc., Ipswich, MA) on a NextSeq 500 (Illumina, San Diego, CA) as previously described.33 Data mining was performed as previously described.41 Significantly differentially expressed transcripts had absolute log fold-change >0.25 and Benjamini-Hochberg multiple test-corrected P values (false discovery rate) <0.05. Functional annotation was done using Gene Set Enrichment Analysis on fold-change preranked lists with gene sets from the MSigDB collection (c2cp, hallmarks)42 and from different publications,32 applying as thresholds P and false discovery rate values <0.05. Expression data are available at the National Center for Biotechnology Information Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) database (GSE152497).
Real-time PCR
Cell lines were seeded at a density of 500 000 cells/mL and treated for 6 hours with 100 nM of NEO2734. Total RNA was extracted with TRIzol Reagent (Thermo Fisher Scientific). Reverse transcription and quantitative real-time PCR were performed with the QuantiFast SYBR Green PCR Kit (Qiagen, Hilden, Germany).
Animal studies
For the leukemia xenograft experiments, mice maintenance and animal experiments were performed under Amplia’s animal use protocol PRN008, which was approved by Neomed’s animal use ethics committee. SCID (CB17) mice from Charles River Laboratories (Wilmington, MA) were injected with 3 × 106 MV-4-11 cells in their lower left flanks. When tumor size reached ∼150 mm3, mice were randomized (n = 6 mice in each group) for treatment.
For the lymphoma xenograft experiments, mice maintenance and animal experiments were performed under the institutional guidelines established for the animal facility and with study protocols approved by the local Cantonal Veterinary Authority (license TI-49-2018). NOD-SCID mice were obtained from The Harlan Laboratory (S. Pietro al Natisone, Udine, Italy). Xenografts were established by injecting lymphoma cells (15 × 106 cells/mouse, 200 µL of PBS) into the left flanks of female NOD-SCID mice (6 weeks of age, ∼20 g of body weight). Tumor size was measured on a regular basis and until tumors reached ∼5 mm in diameter (100 mm3). Tumor volumes were calculated as previously described.43 In both experiments, mice were euthanized when tumor volumes reached ethical limits. Differences in tumor volumes between treatment groups were calculated by using the Mann-Whitney U test (GraphPad Prism version 7.0d, GraphPad Software, La Jolla, CA) as previously performed.33 The P value for significance was <.05.
Data mining
Associations in 2-way tables were tested for statistical significance by using either the χ2 test or Fisher’s exact test, as appropriate. Binomial exact 95% confidence intervals (CIs) were calculated for median percentages. Differences in 50% inhibitory concentration (IC50) values among subtypes were calculated by using the Mann-Whitney U test. The degree of correlation among antiproliferative activities was calculated by standard Pearson correlation coefficients. Statistical significance was defined by P ≤ .05. Statistical analyses were conducted with Stata/SE 12.1 for Mac (Stata Corp, College Station, TX) and Prism 7 for Mac.
Results
NEO2734 binds to both BET proteins and CBP/EP300
The binding potency of NEO2734 was determined against a panel of BRDs using a ligand-binding, site-directed competition assay. NEO2734 bound both BET and CBP/EP300 proteins (supplemental Table 1; supplemental Figure 1) with Kds in the nanomolar range. In particular, the Kds for BRD2, BRD3, BRD4, and BRDT were in the single nanomolar range. The values for CBP and EP300 were 19 nM and 31 nM, respectively. The pattern shown by a BET inhibitor, molibresib (iBET-762, GSK525762), clearly differed in its complete lack of binding to CBP and EP300. The affinity for BET proteins was also stronger for NEO2734 than for molibresib.
NEO2734 has antiproliferative activity in cancer cell lines with a preference for blood and prostate cancers
The in vitro antitumor activity of NEO2734 was tested against a panel of 60 cell lines (Figure 1; supplemental Table 2) derived from different tumor types. After an exposure time of 72 hours, the compound showed antiproliferative activity in many cell lines, with a stronger activity observed in cell lines derived from leukemia (median IC50 = 280 nM), prostate cancer (median IC50 = 460 nM), and lymphomas (median IC50 = 300 nM) (P < .0001, P = .075, and P < .001, respectively), which also comprised a higher percentage of sensitive cells, defined as an IC50 < 500 nM (supplemental Figures 2 and 3). Concurrently, cell lines were also exposed to molibresib. Overall, NEO2734 showed significantly more antiproliferative activity than molibresib (P < .0001) (supplemental Table 2).

Distribution of NEO2734 IC50 values across cell lines derived from different tumor tissues. IC50 values calculated after 72 hours of treatment. Each dot represents a cell line and its respective IC50. Cell lines are divided according to tumor type. Numbers below the boxes show the median IC50 for the corresponding tumor type.
Distribution of NEO2734 IC50 values across cell lines derived from different tumor tissues. IC50 values calculated after 72 hours of treatment. Each dot represents a cell line and its respective IC50. Cell lines are divided according to tumor type. Numbers below the boxes show the median IC50 for the corresponding tumor type.
NEO2734 has in vitro antitumor activity in DLBCL
Because lymphoma exhibited the highest degree of sensitivity to NEO2734, 27 DLBCL cell lines were exposed to increasing concentrations of the inhibitor for 72 hours (supplemental Table 3). NEO2734 showed antitumor activity with a median IC50 of 157 nM (95% CI, 129-225) (Figure 2A). Cell lines derived from activated B-cell–like DLBCL (ABC-DLBCL) (n = 8) were more sensitive than those derived from germinal center B-cell (GCB-DLBCL) (n = 19) in terms of IC50 (P < .01) (Figure 2B) but not 90% inhibitory concentration (IC90) values. No differences were observed based on MYC gene status, BCL2 gene status, TP53 gene status, double-hit MYC/BCL2, CREBBP gene status, or EP300 gene status. A significant difference in sensitivity was observed based on MYD88 mutation (mutated, n = 4; wild type, n = 11; P = .017), in agreement with the association with the DLBCL cell of origin (3 of 4 mutated cell lines are ABC-DLBCL).
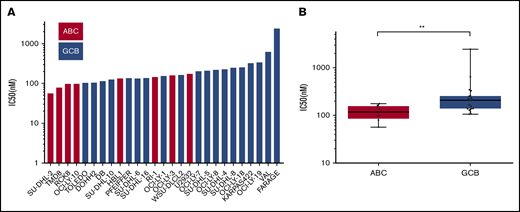
Distribution of NEO2734 IC50 values in 27 DLBCL cell lines showing higher antitumor activity in ABC-DLBCLs than GCB-DLBCLs. (A) IC50 values in the 27 DLBCL cell lines measured after 72 hours of exposure to NEO2734. (B) Differences in IC50 values between ABC-DLBCL and GCB-DLBCL cell lines. The Mann-Whitney U test was used to compare groups. **P < .01.
Distribution of NEO2734 IC50 values in 27 DLBCL cell lines showing higher antitumor activity in ABC-DLBCLs than GCB-DLBCLs. (A) IC50 values in the 27 DLBCL cell lines measured after 72 hours of exposure to NEO2734. (B) Differences in IC50 values between ABC-DLBCL and GCB-DLBCL cell lines. The Mann-Whitney U test was used to compare groups. **P < .01.
The antiproliferative activity of NEO2734 seemed to be largely cytotoxic based on cell cycle analysis in 3 of 4 cell lines (2 GCB-DLBCL and 2 ABC-DLBCL) exposed to 300 nM NEO2734 for 72 hours (Figure 3A). Similar data were obtained treating with single BET (300 nM) or single CBP (10 μM) inhibitors. Immunoblotting analyses showed that NEO2734 (250 nM, 24 hours) reduced MYC protein levels (Figure 3B-C).
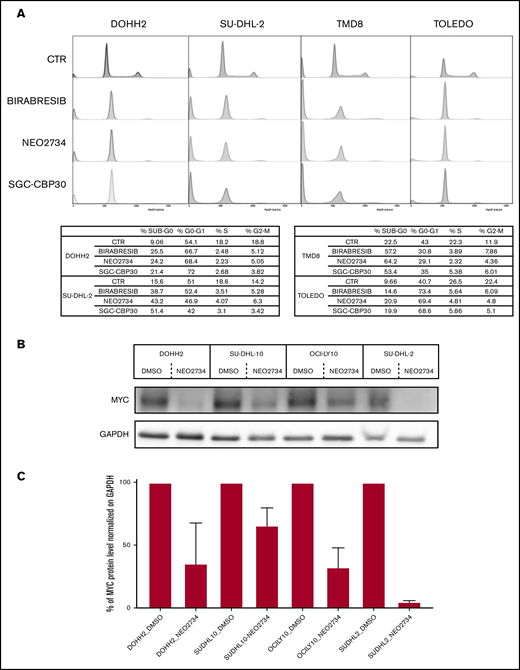
Effect of NEO2734 on cell cycle and MYC protein level in DLBCL cell lines. (A) Representative cell cycle profiles of 2 replicates with the respective quantification; 2 GCB-DLBCL (TOLEDO, DOHH2) and 2 ABC-DLBCL (SU-DHL-2, TMD8) cell lines were treated with DMSO or birabresib (300 nM) or NEO2734 (300 nM) or SGC-CBP30 (10 μM) for 72 hours. (B) Representative immunoblot of 2 replicates. Two GCB-DLBCLs and 2 ABC-DLBCLs were exposed to NEO2734 (250 nM) for 24 hours. (C) Immunoblot quantification.
Effect of NEO2734 on cell cycle and MYC protein level in DLBCL cell lines. (A) Representative cell cycle profiles of 2 replicates with the respective quantification; 2 GCB-DLBCL (TOLEDO, DOHH2) and 2 ABC-DLBCL (SU-DHL-2, TMD8) cell lines were treated with DMSO or birabresib (300 nM) or NEO2734 (300 nM) or SGC-CBP30 (10 μM) for 72 hours. (B) Representative immunoblot of 2 replicates. Two GCB-DLBCLs and 2 ABC-DLBCLs were exposed to NEO2734 (250 nM) for 24 hours. (C) Immunoblot quantification.
To identify features responsible for the sensitivity or resistance to NEO2734, we divided DLBCL cells based on IC90, and we took advantage of the baseline gene expression profiles we had previously obtained in the cell lines.32 Sensitive cell lines (IC90 < 1 μM) vs resistant cell lines (IC90 > 10 μM) revealed BCL2 as the top gene with higher expression in resistant cell lines compared with sensitive ones (supplemental Figure 4A). Based on these data, we combined NEO2734 with the BCL2 inhibitor venetoclax. The addition of venetoclax was able to overcome resistance to NEO2734, as shown by synergism of the combination in resistant cell lines but not in sensitive cell lines, which had low levels of BCL2 (supplemental Figure 4B,D). The observed synergism was independent of venetoclax sensitivity of the cells (supplemental Figure 4C).
In addition to BCL2 levels, another feature associated with sensitivity to NEO2734 was a higher expression of MYC targets, mirrored by a higher expression of MYC-repressed genes in resistant cell lines (supplemental Table 4). In addition, transcripts involved in oxidative phosphorylation, trichloroacetic acid cycle activity, and cell cycle were enriched in sensitive cells.
NEO2734 differs from the other lead clinical candidate NEO1132
We also compared NEO2734 with NEO1132, the other lead clinical candidate from the same series of dual inhibitors of BET and CBP/EP300.29 NEO1132 exhibited a median IC50 (400 nM; 95% CI, 316-622), which was higher than that achieved with NEO2734 (Figure 4A; supplemental Table 3) but with a similar pattern of antiproliferative activity (r = 0.99; P < .001) (supplemental Figure 5). NEO1132 also presented a slightly different pattern of binding to BRDs when tested with the ligand binding site-directed competition assay, with reduced affinity for CBP/EP300, as well as for a subset of BET BRDs (supplemental Table 1).
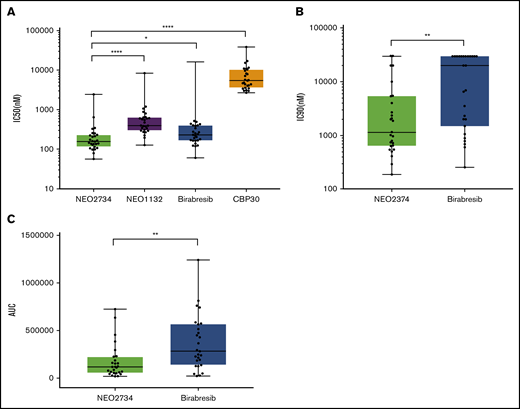
NEO2734 is more active than birabresib, CBP30, and NEO1132 in DLBCL cell lines. IC50 and IC90 values calculated for DLBCL cell lines exposed for 72 hours to the compounds. (A) IC50 value comparison between NEO2734, NEO1132, birabresib, and CBP30. (B) IC90 value comparison between NEO2734 and birabresib. (C) AUC comparison between NEO2734 and birabresib. Each point represents 1 cell line with the respective IC50/IC90/AUC. The line in the middle of the boxes represents the median IC50/IC90/AUC with 95% of CI. IC90 values >20 μM were arbitrarily set as 30 μM. Statistics were calculated with the Mann-Whitney U test. *P < .05, **P < .01, ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
NEO2734 is more active than birabresib, CBP30, and NEO1132 in DLBCL cell lines. IC50 and IC90 values calculated for DLBCL cell lines exposed for 72 hours to the compounds. (A) IC50 value comparison between NEO2734, NEO1132, birabresib, and CBP30. (B) IC90 value comparison between NEO2734 and birabresib. (C) AUC comparison between NEO2734 and birabresib. Each point represents 1 cell line with the respective IC50/IC90/AUC. The line in the middle of the boxes represents the median IC50/IC90/AUC with 95% of CI. IC90 values >20 μM were arbitrarily set as 30 μM. Statistics were calculated with the Mann-Whitney U test. *P < .05, **P < .01, ****P < .0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
NEO2734 has stronger in vitro antilymphoma activity than inhibitors targeting either BET or CBP/EP300 alone
DLBCL cell lines were also exposed to a BET inhibitor and to a CBP/EP300 inhibitor. The BET inhibitor birabresib (OTX015) is a drug with proven preclinical and early clinical antilymphoma activity6,7 and with comparable in vitro activity to molibresib.44 The CBP/EP300 inhibitor was SGC-CBP30 (CBP30), with Kd values of 21 nM for CBP and 38 nM for EP300,45 comparable to those seen for NEO2734. The median IC50 values of these 2 molecules were 240 nM (95% CI, 171-344) and 5.5 μM (95% CI, 4.2-8.3), respectively (supplemental Table 3).
NEO2734 presented a similar pattern of antiproliferative activity across all the cell lines to birabresib (r = 0.97; P < .001), and SGC-CBP30 (r = 0.87; P < .001). Similar behavior was also seen for birabresib vs SGC-CBP30 (r = 0.85; P < .001) (supplemental Figure 5). With respect to IC50 values, NEO2734 had a significantly stronger antiproliferative activity than birabresib (P = .0182) and SGC-CBP30 (P < .001) (Figure 4A). The difference between NEO2734 and birabresib became more pronounced when we considered IC90 values (P = .0026): median values were 1.1 μM (95% CI, 646 nM-4 μM) and 20 μM (95% CI, 2-30) for the dual inhibitor and for the BET inhibitor, respectively (Figure 4B). This was also reflected in a significantly lower area under the curve (AUC) for NEO2734 compared with birabresib-treated cells (Figure 4C).
The stronger antilymphoma activity of NEO2734 was also evident when DLBCL cells underwent a long exposure to birabresib or the novel compound. In this case, the difference between NEO2734 and birabresib increased more with time in the DLBCL cell lines bearing CBP/EP300 mutations than in the cell lines harboring wild-type chromatin-remodeling genes (supplemental Figure 6A). Differences between NEO2734 and birabresib also increased with time in cell lines expressing low BCL2 levels compared with those with high expression of the antiapoptotic gene (supplemental Figure 6B).
NEO2734 shows in vivo antitumor activity in DLBCL
To validate the in vivo relevance of our observations, we performed an in vivo xenograft experiment with the ABC-DLBCL TMD8 model. Cohorts of NOD-SCID mice were treated with vehicle control, NEO2734 (10 mg/kg once daily [QD], 6 days per week, by mouth [PO]), and the BET inhibitor birabresib (50 mg/kg BID, 6 days per week PO). Treatment with NEO2734 repressed tumor growth (P < .001, day 7; P = .008, day 11) compared with vehicle-treated animals (Figure 5). Moreover, the effect was significantly higher (P < .05, day 7) than what was achieved by using the BET inhibitor even when administered at a higher dose and more intensive schedule. Treatment was well tolerated, with no significant loss in body weight.
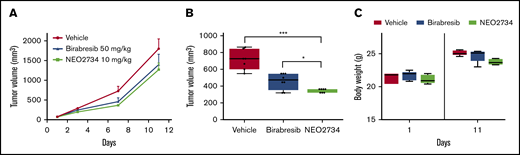
In vivo antitumor activity of NEO2734 in a DLBCL xenograft model. (A) Treatment of TMD8 xenograft (ABC-DLBCL) with NEO2734, birabresib, or vehicle. Lines show median values per time point with the corresponding interquartile range. (B) The boxplot represents the tumor volumes at the best vehicle per treatment day (day 7). (C) Body weight at the first and last day of treatment. Seven mice were treated in each group. The Mann-Whitney U test was used to compare groups. *P < .05, ***P < .001.
In vivo antitumor activity of NEO2734 in a DLBCL xenograft model. (A) Treatment of TMD8 xenograft (ABC-DLBCL) with NEO2734, birabresib, or vehicle. Lines show median values per time point with the corresponding interquartile range. (B) The boxplot represents the tumor volumes at the best vehicle per treatment day (day 7). (C) Body weight at the first and last day of treatment. Seven mice were treated in each group. The Mann-Whitney U test was used to compare groups. *P < .05, ***P < .001.
NEO2734 shows in vivo antitumor activity in AML
Based on the in vitro data of the initial screening, we also validated the activity of NEO2734 in an in vivo AML model. The human leukemic MV-4-11 cell line was injected in the left lower flanks of SCID mice. Mice were treated with vehicle control, NEO2734 (1, 3, 10 mg/kg QD, 14 days, PO) or the BET inhibitor molibresib (10, 30 mg/kg QD, 14 days, PO). NEO2734-treated mice already exhibited reduced tumor growth at only 1 mg/kg, which was enhanced by 3 mg/kg and was significantly more pronounced than molibresib at 10 mg/kg (P < .05, starting from day 8) (Figure 6). Treatment was well tolerated, with no significant loss in body weight.
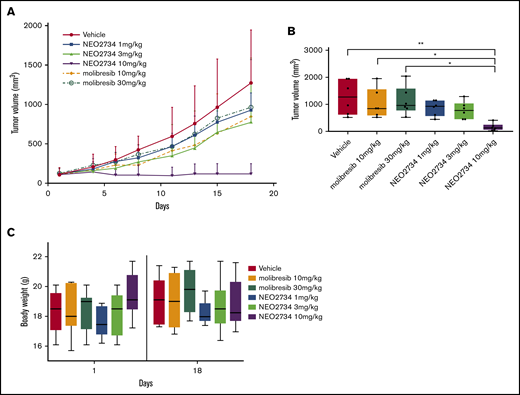
In vivo antitumor activity of NEO2734 in an AML xenograft model. (A) Antitumor activity in the AML MV-4-11 xenograft model treated with NEO2734, molibresib, or vehicle. Lines show median values per time point with the corresponding interquartile range. On the y-axis, the tumor volume is given in mm3; x-axis, days of treatment. (B) Tumor volume at the best vehicle per treatment day (day 18). (C) Body weight at the first and last day of treatment. Treatments started when tumors became visible (150 mm3). Six mice were treated in each group. The Mann-Whitney U test was used to compare groups. *P < .05, **P < .01.
In vivo antitumor activity of NEO2734 in an AML xenograft model. (A) Antitumor activity in the AML MV-4-11 xenograft model treated with NEO2734, molibresib, or vehicle. Lines show median values per time point with the corresponding interquartile range. On the y-axis, the tumor volume is given in mm3; x-axis, days of treatment. (B) Tumor volume at the best vehicle per treatment day (day 18). (C) Body weight at the first and last day of treatment. Treatments started when tumors became visible (150 mm3). Six mice were treated in each group. The Mann-Whitney U test was used to compare groups. *P < .05, **P < .01.
NEO2734 affects the transcriptome of DLBCL cells
To better understand the mechanism of action of NEO2734, we performed transcriptome analysis on RNA extracted from 2 GCB-DLBCL (Toledo, DOHH2) and 2 ABC-DLBCL (TMD8, OCI-Ly-10) cell lines after 6 hours exposure to DMSO, NEO2734, or birabresib (100 nM) (supplemental Tables 5 and 6). NEO2734 altered the transcriptome at this early time point, with downregulated transcripts enriched for MYC targets and genes involved in inflammation, and chemokine signaling. Upregulated transcripts appeared enriched for genes encoding histones but also for ribosomal proteins and members of phosphatidylinositol 3-kinase signaling.
NEO2734 exposure led to a higher number of differentially expressed transcripts than birabresib (P = .044, 1-tailed Fisher’s exact test) (supplemental Figure 7A), but the general effect was very similar in terms of individual transcripts (supplemental Figure 7B) or gene sets. In particular, in both GCB-DLBCL and ABC-DLBCL, the genes affected by birabresib were also regulated by exposure to NEO2734 and vice versa (supplemental Figure 7C). The changes induced by both compounds overlapped with published signatures for BET inhibitors, histone deacetylase inhibitors, CB/EP300 inhibition, and phosphatidylinositol 3-kinase inhibitors (supplemental Tables 4 and 5).
To validate RNA-sequencing data, quantitative polymerase chain reaction was performed in 2 resistant (FARAGE, U2932) and 2 sensitive (TMD8, SU-DHL10) cell lines. Among the downregulated genes, MYC, BCL2, and ASB2 were validated in 4 of 4 cell lines tested and CD180 and USP18 in 3 of 4 cell lines. Among the upregulated genes, CDK9, HIST2H2BE, and HEXIM1 were validated in all 4 cell lines and CCNT1 in 3 of 4 cell lines. None of the validated genes was differently modulated between sensitive and resistant cell lines (supplemental Figure 8).
NEO2734 shows in vitro antitumor activity in canine DLBCL
Because dogs with spontaneous lymphomas are considered excellent effective animal models for drug development, we compared all the compounds in a canine DLBCL cell line (CLBL-1) in which we had previously shown activity of birabresib.46 All the small molecules presented dose-dependent antiproliferative activity, with the BET inhibitor birabresib as the most active followed by NEO2734 (supplemental Figure 9).
Discussion
Our data show that NEO2734 is a novel compound able to bind with high affinity to BET BRD proteins and also to the BRDs of CBP and EP300. NEO2734 exhibited in vitro antitumor activity in a variety of cancer cell lines derived from different tumor types, especially from prostate cancer, leukemias, and lymphomas. Focusing on the latter tumor type, NEO2734 showed clear in vitro antitumor activity in DLBCL and also appeared superior to other epigenetic compounds in its ability to kill tumor cells.
In an initial large-scale screening of cell lines derived from a variety of cancers, leukemias, lymphomas, and prostate cancers seemed to be the tumor types most sensitive to NEO2734, both in terms of IC50 values and of the frequency of sensitive models. The observed activity in prostate cancer is consistent with data recently reported in prostate cancer patient–derived xenografts,47 in which NEO2734 is able to bypass BET inhibitor resistance due to SPOP mutations.9,48
Due to our previous experience with BET inhibitors,6,49,50 we focused on analyzing NEO2734 activity in DLBCL. NEO2734 showed a clear in vitro antitumor activity in DLBCL, which was more potent than either a BET inhibitor (birabresib) or a CBP/EP300 inhibitor (SGC-CBP30). Importantly, NEO2734 antitumor activity was largely cytotoxic. NEO2734 showed stronger in vitro antitumor activity in ABC-DLBCL than in GCB-DLBCL. This observation could be explained by the possible impact of CBP inhibition on the NF-κB pathway due to the reported interaction between CBP/EP300 and the RelA subunit of the NF-κB transcription factor complex.51 There was also an association between the presence of MYD88 mutation and higher sensitivity to NEO2734, which was in agreement with promising preclinical and clinical results with BET inhibitors in this lymphoma subtype.4,6,7,49,52
The in vitro antitumor activity of NEO2734 was confirmed in an ABC-DLBCL xenograft model, in which NEO2734 exhibited moderately superior activity to a BET inhibitor given at a much higher dose. We also confirmed the in vitro activity of NEO2734 in leukemia in an in vivo AML xenograft experiment. NEO2734 showed much stronger antitumor activity compared with vehicle-treated or BET inhibitor–treated mice, especially at the highest concentration. Our data are supported by preliminary findings obtained with NEO2734 in primary AML cells and patient-derived xenografts.53 Although the in vivo activity of NEO2734 in the lymphoma model was lower than in the AML model, it was in line with reported activities of BET inhibitors in models derived from this tumor type.5,6,54 Relevantly, it is possible that low doses of NEO2734 necessary to have an activity similar to the BET inhibitor may ameliorate some of the class-specific toxicities of this class of agents, which have been observed during their clinical development.
NEO2734 regulated the transcriptome of DLBCL cells affecting important expected biological pathways associated with its epigenetic mechanism of action. NEO2734 profiles were similar to those induced by BET inhibitors, but the latter more strongly affected the transcriptome of DLBCL cell lines.
To the best of our knowledge, the compounds we studied are the only potential clinical candidate molecules with the ability to target BET proteins and CBP and EP300, maintaining a high specificity for these vs other BRD-containing proteins. Until now, most efforts had been made to obtain compounds that are selective for the 2 individual classes of proteins instead of targeting both of them.25-28 The data presented here show that the dual inhibitor NEO2734 has a stronger antitumor activity than BET inhibitors in both solid tumor and hematologic models. NEO1132, the other lead clinical candidate from the same series of dual inhibitors, was less active than NEO2734 and was characterized by weaker CBP/EP300–binding properties and equal BET-binding affinity, suggesting an important role for CBP inhibition in NEO2734 activity. However, it is difficult to pinpoint the contribution given by adding CBP/EP300 targeting to BET inhibition. The tumor types with the highest sensitivity to BET or to CBP/EP300 inhibitors overlap.9,21,23,55 Here, at short exposure time (72 hours), despite lower IC50 values and higher cytotoxicity, cell lines that were sensitive or resistant to BET inhibitors, or to a specific CBP/BET inhibitor and/or to the dual inhibition, highly overlapped. In lymphoma, the genetic events affecting CBP and EP300 genes loci are usually mono-allelic, suggesting that the enzymatic activity of EP300 and CBP is still needed by cancer cells.15,16,41 Thus, tumors with a single remaining allele would be more sensitive to CBP/EP300 inhibitors. When we treated DLBCL cell lines bearing different CBP/EP300 mutational states, longer exposure to BET inhibitor or NEO2734 increased the difference in activity between the 2 compounds in cell lines with mutated EP300 or CBP, favoring stronger activity of NEO2734 with increasing time. On the contrary, the difference between NEO2734 and BET inhibitor activities decreased with time in wild-type cell lines. These data suggest that patients with mutated tumors might benefit even more in a prolonged regimen treatment from dual inhibitors than patients with tumors that are wild type for CBP/EP300. However, the complex genetic landscape of tumors with multiple lesions leading to potentially similar biological consequences makes it difficult to find robust genetic biomarkers for epigenetic drugs. Indeed, convincing data showing that the genetic loss of CBP confers higher sensitivity to the pharmacologic inhibition of EP300 come from experiments performed using isogenic DLBCL cells.8
We identified BCL2 as the most differentially expressed gene between cell lines with different sensitivities to NEO2734, with resistant cells expressing higher BCL2, in line with previously reported results.56 Importantly, the inhibition of BCL2 was synergistic with NEO2734 in these cell lines.
Although we did not observe any correlation between sensitivity to NEO2734 and the presence of MYC translocation, in accordance with the literature,6,7,57,58 we did note an association between higher NEO2734 activity and higher baseline expression of MYC targets.
Our data, supported by other recent reports,47,53,59-61 provide a strong rationale toward a clinical evaluation of NEO2734 in patients with both hematologic and solid tumors. Reversible thrombocytopenia, anemia, neutropenia, nausea, diarrhea and dysgeusia, fatigue, and bilirubin elevation represent the most common side effects of BET inhibitors.3 No clinical data are available yet with CBP/EP300 inhibitors. Although NEO2734 did not seem more toxic than BET inhibitors in mice, only phase 1 studies will be able to assess the feasibility of dual targeting in humans.
In conclusion, NEO2734 is a potentially novel antitumor compound, worthy of further evaluation, particularly in hematologic cancers such as lymphomas and leukemias.
Acknowledgments
The visual abstract was created by using templates from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported License.
This study was partially supported by research funds from Epigene Therapeutics, from a donation from The Church of Saint Edward The Confessor in memory of Harminda Borioli (F.B.), and an Associazione Italiana per la Ricerca sul Cancro 5X100 grant (no. 21198) (S.P.).
Authorship
Contribution: F.S., F.G., and F.B. were responsible for conception and design: F.S., E.G., A.A.M., F.G., and F.B. developed the methodology; F.S., L.A., S.P., M.W., B.B., C.W., F.G., and F.B. were responsible for acquisition of data (eg, provided animals, acquired and managed patients, provided facilities); F.S., L.C., and F.B. analyzed and interpreted data (eg, statistical analysis, biostatistics, computational analysis); F.S., E.G., A.A.M., L.A., V.P., M.W., B.B., E.Z., F.G., A.S., and F.B. wrote, reviewed, and/or revised the manuscript; F.S., L.C., C.T., M.W., B.B., C.W., F.G., and F.B. provided administrative, technical, or material support (ie, reporting or organizing data, constructing databases); F.B. was responsible for study supervision; and V.P., F.M., G.M., A.R., C.T., E.G., and G.G. performed other experiments.
Conflict-of-interest disclosure: E.Z. received institutional research funds from Celgene, Roche, and Janssen; advisory board fees from Celgene, Roche, Mei Pharma, AstraZeneca, and Celltrion Healthcare; travel grants from AbbVie and Gilead; and provided expert statements to Gilead, Bristol Myers Squibb, and MSD. S.P. received advisory board fees from Celgene, Roche, and NanoString. M.W. received institutional research funds from Admare Bioinnovations. C.W. is a cofounder of Epigenetix Inc. F.G. received a consultancy fee from Epigene Therapeutics; and has equity interest in Epigene Therapeutics. A.S. received institutional research funds from Bayer, ImmunoGen, Merck, Pfizer, Novartis, Roche, MEI Pharma, ADC-Therapeutics; and travel grants from AbbVie and PharmaMar. F.B. received institutional research funds from Acerta, ADC Therapeutics, Bayer AG, Cellestia, CTI Life Sciences, EMD Serono, Helsinn, ImmunoGen, Menarini Ricerche, NEOMED Therapeutics 1, Nordic Nanovector ASA, Oncology Therapeutic Development, and PIQUR Therapeutics AG; consultancy fees from Helsinn and Menarini; provided expert statements to HTG; and received travel grants from Amgen, AstraZeneca, Jazz Pharmaceuticals, and PIQUR Therapeutics AG. The remaining authors declare no competing financial interests.
The current affiliation for B.B. is Paraza Pharma Inc., Saint-Laurent, QC, Canada.
Correspondence: Francesco Bertoni, Institute of Oncology Research, via Vincenzo Vela 6, 6500 Bellinzona, Switzerland; e-mail: francesco.bertoni@ior.usi.ch.

