

Key Points
Myeloid-specific Morrbid is hyperactivated in a mouse model of JMML (Shp2E76K) and regulates the number of leukemic cells.
Low expression of Morrbid/MORRBID is associated with improved survival in mice and humans.

Abstract
Mutations in PTPN11, which encodes the protein tyrosine phosphatase SHP2, contribute to ∼35% of cases of juvenile myelomonocytic leukemia (JMML). A common clinical picture in children with JMML is that it presents as a constitutive hyperinflammatory syndrome, partially reminiscent of chronic myelomonocytic leukemia in adults. Thus, a component of JMML is associated with a hyperinflammatory state and abundant innate immune cells such as neutrophils and monocytes. Recently, we showed that the evolutionarily conserved mouse lncRNA Morrbid is specifically expressed in myeloid cells and uniquely represses the expression of the proapoptotic gene Bim to regulate the lifespan of myeloid cells. However, its role in JMML has not been investigated. In this study, we characterized the role of Morrbid and its target Bim, which are significantly dysregulated in Shp2E76K/+-bearing myeloid cells, in driving JMML. Loss of Morrbid in a mouse model of JMML driven by the Shp2E76K/+ mutation resulted in a significant correction of myeloid and erythroid cell abnormalities associated with JMML, including overall survival. Consistently, patients with JMML who had PTPN11, KRAS, and NRAS mutations and high expression of MORRBID manifested poor overall survival. Our results suggest that Morrbid contributes to JMML pathogenesis.
Introduction
Juvenile myelomonocytic leukemia (JMML) is the most common myeloproliferative neoplasm (MPN) in childhood. JMML is characterized by mutations in NF1, CBL, KRAS, NRAS, or PTPN11, and cells from JMML patients show hypersensitivity to GM-CSF in vitro.1-3 Traditional cytotoxic chemotherapeutic agents are ineffective in JMML, and the only curative modality is allogeneic hematopoietic stem cell transplantation.4 As the most prevalent mutation in children with MPN, the activated gain-of-function mutation in PTPN11, which encodes the protein tyrosine phosphatase SHP2, contributes to ∼35% of cases of JMML, and leads to hyperactive Ras-MAPK signaling.5
A common clinical picture in JMML is that it presents as a hyperinflammatory syndrome and is often indistinguishable from viral infections.6 Unlike acute myeloid leukemia (AML) but similar to CMMLchronic myelomonocytic leukemia, JMML rarely progresses to a blast crisis; rather, death is caused by extramedullary myeloid cell expansion and enhanced survival, leading to organ failure, respiratory failure, bleeding, and infection.1 SHP2 mutations such as SHP2E76K/+ and SHP2D61Y/+ downregulate the expression of the proapoptotic protein Bim, which is associated with enhanced survival relative to controls.7 Loss of Bim in hematopoietic progenitor cells enhances their survival and shows resistance to apoptosis.8,9
Despite the discovery of transcripts derived from the noncoding genome, how these noncoding RNAs, including long noncoding RNAs (lncRNAs), regulate MPN initiation or development is poorly understood. Although only a few lncRNAs related to myeloid malignancies have been described, their function and the targets that they regulate is largely unknown.10 We have recently shown that, under physiologic conditions, the evolutionarily conserved novel lncRNA Morrbid is uniquely expressed in myeloid cells and represses the expression of the proapoptotic gene Bim to regulate the lifespan of these cells.11 Using murine Shp2E76K-driven JMML models and a JMML patient database, we studied the role of MORRBID in disease initiation and progression. Although genetic loss of Morrbid partially mitigates signatures of JMML, our results show that Morrbid plays a role in the onset and progression of the disease.
Methods
All the animal experiments were approved by the Laboratory Animal Resource Center at Indiana University School of Medicine. All strains were on a C57/B6 background, and wild-type (WT) mice were procured from the Indiana University School of Medicine Core Facility. JMML model mice (LysMCre; Ptpn11E76K/+) and Morrbid-deficient mice (Morrbid−/−) have been described11,12 and were bred to generate the compound mutants. Analysis involving mouse experiments (ie, flow cytometry strategies and intracellular staining for annexin-V and anti-Bim) have been described.13 Unless stated otherwise, statistical analysis in animal experiments was performed with GraphPad Prism 6 for Kaplan-Meier survival curve comparison or using Student t test or 1-way analysis of variance for group comparison.
The bioinformatics analysis of MORRBID in human JMML was conducted as follows. To identify which lncRNAs potentially associate with JMML, we downloaded raw microarray data for JMML samples, with or without PTPN11, KRAS, and NRAS mutation from the GEO database (GSE71935),14 along with the pertinent clinical data on the patients. The downloaded data were normalized by the MAS5 method. Survival analysis was performed over the expression of lncRNA MORRBID and clinical data, using the log-rank test encoded in the survival R package (https://github.com/therneau/survival). Considering that the data of very young patients (<18 months) were not representative of the disease progression, only older patients with JMML (>18 months at the last follow-up) were included for survival analysis in the study.
Results and discussion
Shp2 is indispensable for hematopoiesis.15,16 Hematopoietic cells expressing the gain-of-function isoform of Shp2, Shp2E76K, which mimics the human PTPN11 mutation in JMML, confer cell survival advantage and alterations in cell proliferation and differentiation.12 The murine model bearing Shp2E76K (Ptpn11E76K/+) driven by lysosome-cre (LysMCre; Ptpn11E76K/+, indicated as Shp2* mice hereafter) is often used to study JMML, because it induces acute alterations in hematopoiesis similar to human JMML.12,15 We have shown that, in inflammatory conditions, the expression of lncRNA Morrbid is elevated, whereas the level of Bim is downregulated.11 Consistently, we have shown that Morrbid is upregulated, and Bim is downregulated in myeloid cells derived from Shp2* mice.13 To test the functional requirement of Morrbid in the development of JMML, we generated a mouse model of JMML that lacked Morrbid: LysMCre; Shp2E76K/+; Morrbid−/− (indicated herein as Shp2*M). We compared the disease progression between Shp2* and Shp2*M mice, along with WT and Morrbid−/− (M) controls (Figure 1A).
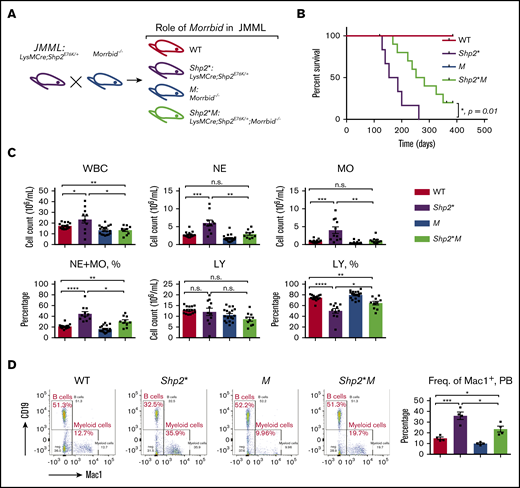
Loss of Morrbid enhances the survival of Shp2E76K-driven JMML mice. (A) Generation of JMML mice (Shp2*: LysMCre; Shp2E76K/+) and JMML mice lacking Morrbid (Shp2*M: LysMCre; Shp2E76K/+; Morrbid−/−) along with WT control and Morrbid−/− (M). (B) Kaplan-Meier survival curve of Shp2* and Shp2*M mice and control animals. (C) Analysis of hematopoietic cells in PB by the automatic blood cell counter Element HT5 Analyzer. (D) Analysis of WBCs from PB by flow cytometry. Mac1+ staining for myeloid cells and CD19+ staining for B cells (n = 4-18 mice). *P < .05; **P < .01; ***P < .001; ****P < .0001. See supplemental Figure 1 for improvement of the anemia phenotype in Shp2*M mice compared with Shp2* mice. LY, lymphocytes; n.s., not significant.
Loss of Morrbid enhances the survival of Shp2E76K-driven JMML mice. (A) Generation of JMML mice (Shp2*: LysMCre; Shp2E76K/+) and JMML mice lacking Morrbid (Shp2*M: LysMCre; Shp2E76K/+; Morrbid−/−) along with WT control and Morrbid−/− (M). (B) Kaplan-Meier survival curve of Shp2* and Shp2*M mice and control animals. (C) Analysis of hematopoietic cells in PB by the automatic blood cell counter Element HT5 Analyzer. (D) Analysis of WBCs from PB by flow cytometry. Mac1+ staining for myeloid cells and CD19+ staining for B cells (n = 4-18 mice). *P < .05; **P < .01; ***P < .001; ****P < .0001. See supplemental Figure 1 for improvement of the anemia phenotype in Shp2*M mice compared with Shp2* mice. LY, lymphocytes; n.s., not significant.
We assessed overall survival of Shp2*M mice relative to Shp2* controls up to a year (Figure 1A). Although both WT and M mice showed a normal lifespan, the median survival for Shp2* mice was 170 days, which was extended to 270 days for Shp2*M mice (n = 4-10; *P = .01), indicating that Shp2*M mice show an improved overall lifespan (Figure 1B). We next compared the hematological parameters in peripheral blood (PB), bone marrow (BM), and spleen of 4 experimental groups. At the moribund or semimoribund stage (typically, 5-6 months for Shp2* and 8-9 months for Shp2*M), both Shp2* and Shp2*M mice showed comparable high overall white blood cell (WBC) counts and neutrophil (NE) and monocyte (MO) counts in PB (∼10-fold higher than WT; data not shown). At 1 to 4 months of age, compared with WT mice, Shp2* mice developed JMML-like symptoms, including higher WBC, NE, and MO counts, which were noted in animals as young as 3 to 4 weeks. At the same age, however, Shp2*M mice showed significantly lower WBC, NE, and MO counts compared with Shp2* mice (Figure 1C). Furthermore, the percentage of NEs and MOs in WBCs was also reduced in Shp2*M mice compared with that in Shp2* mice, but was still higher than that in WT (Figure 1C). Consistent with these findings, flow cytometry showed a significant correction in the frequency of Mac-1+ cells in Shp2*M mice compared with Shp2* mice (Figure 1D).
To determine whether the loss of Morrbid alters the hematopoietic abnormalities associated with Shp2* mice in the BM and spleen, we again analyzed all 4 groups of mice at 3 to 4 months of age. A significant difference in the frequency of granulocyte-macrophage progenitors (GMPs) was observed in the BM of WT and Shp2* mice (Shp2* vs WT, P < .01; Figure 2A). In contrast, although the increased frequency of GMPs was not modulated in Shp2*M mice, the BM cellularity in Shp2*M mice was significantly reduced compared with that in Shp2* mice (Shp2* vs Shp2*M, P < .05; Figure 2C). Thus, the absolute number of GMPs in Shp2*M mice was significantly reduced compared with the number in Shp2* mice (Shp2* vs Shp2*M, P < .01; Figure 2C). With regard to mature myeloid cells in the BM, both the frequency and the absolute number of Mac1+ cells were significantly reduced in Shp2*M mice compared with Shp2* mice (Figure 2B-C). Consistent with previous reports, Shp2* mice manifested an obvious splenomegaly compared with WT mice as young as 1 month. Although, spleen weight and size were comparable between Shp2* and Shp2*M mice at all time points examined (data not shown), Shp2*M mice showed a correction in the frequency of both GMPs and Mac1+ cells compared with Shp2* mice (Figure 2D-E). Furthermore, although Shp2*M mice rapidly developed anemia at the moribund stage, similar to Shp2* mice, the onset of the anemia was much later and milder in young Shp2*M mice than in Shp2* mice. At 2 to 4 months of age, Shp2*M mice showed normal frequency of erythroid progenitors, red blood cell counts, and hemoglobin concentration, similar to WT mice, whereas Shp2* mice showed impaired maturation of erythroid progenitors (supplemental Figure 1).
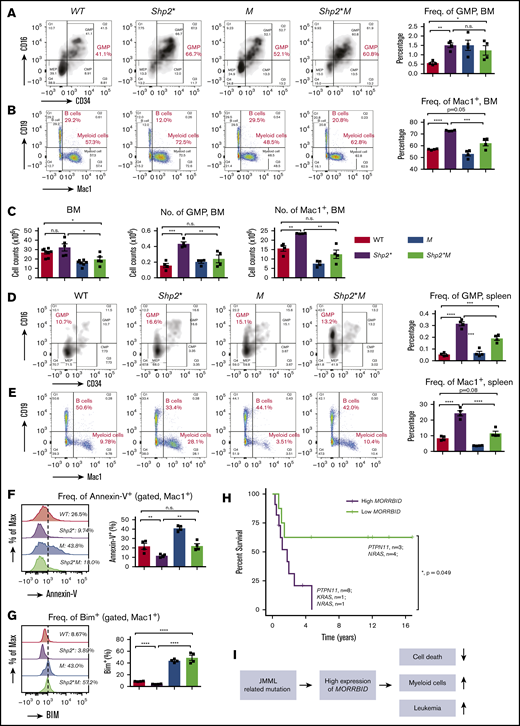
Loss of Morrbid rescues mature and immature myeloid cells in the BM and spleen of Shp2* mice. Mononuclear cells were collected from the BM (A-C) and spleens (D-E) of the 4 experimental groups (WT, Shp2*, M, and Shp2*M; age, 3-4 months) for analyzing hematopoiesis. Changes in myeloid progenitors, GMPs, and mature myeloid cells were analyzed. Lin−Sca1−cKit+ cells were pregated for GMP flow cytometry profiles (CD34+CD16+). In BM, both the frequency and number of GMPs and mature myeloid cells were quantified. In spleen, only the frequency of GMPs and mature myeloid cells was quantified. (F) The level of apoptosis in mature myeloid cells was determined by flow cytometry using annexin-V staining, a marker for proapoptosis. (G) The level of Bim expression in mature myeloid cells was determined by intracellular flow cytometry with an anti-Bim antibody. (H) Kaplan-Meier survival plots of JMML patients with high or low expression of MORRBID. The purple line indicates the survival curve of children with JMML who had high expression of MORRBID (n = 10 patient samples: 8 with mutations in PTPN11, 1 in KRAS, and 1 in NRAS; age >18 months), and the green line indicates that of JMML children with low expression of MORRBID (n = 7 patient samples; 3 with mutations in PTPN11 and 4 in NRAS; age >18 months). The high expression of MORRBID was associated with poor overall survival of these patients (P = 0.049). See supplemental Figure 2 for the survival curve of JMML patients without mutations in PTPN11, KRAS, and NRAS. (I) Model for Morrbid regulation of myeloid cell survival in JMML. For mouse samples, n = 4 mice; for human samples, n = 7-10. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Loss of Morrbid rescues mature and immature myeloid cells in the BM and spleen of Shp2* mice. Mononuclear cells were collected from the BM (A-C) and spleens (D-E) of the 4 experimental groups (WT, Shp2*, M, and Shp2*M; age, 3-4 months) for analyzing hematopoiesis. Changes in myeloid progenitors, GMPs, and mature myeloid cells were analyzed. Lin−Sca1−cKit+ cells were pregated for GMP flow cytometry profiles (CD34+CD16+). In BM, both the frequency and number of GMPs and mature myeloid cells were quantified. In spleen, only the frequency of GMPs and mature myeloid cells was quantified. (F) The level of apoptosis in mature myeloid cells was determined by flow cytometry using annexin-V staining, a marker for proapoptosis. (G) The level of Bim expression in mature myeloid cells was determined by intracellular flow cytometry with an anti-Bim antibody. (H) Kaplan-Meier survival plots of JMML patients with high or low expression of MORRBID. The purple line indicates the survival curve of children with JMML who had high expression of MORRBID (n = 10 patient samples: 8 with mutations in PTPN11, 1 in KRAS, and 1 in NRAS; age >18 months), and the green line indicates that of JMML children with low expression of MORRBID (n = 7 patient samples; 3 with mutations in PTPN11 and 4 in NRAS; age >18 months). The high expression of MORRBID was associated with poor overall survival of these patients (P = 0.049). See supplemental Figure 2 for the survival curve of JMML patients without mutations in PTPN11, KRAS, and NRAS. (I) Model for Morrbid regulation of myeloid cell survival in JMML. For mouse samples, n = 4 mice; for human samples, n = 7-10. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Given the correction in the frequency and absolute number of Mac-1+ cells in Shp2*M mice compared with Shp2* mice, we sought to determine the extent to which loss of Morrbid in these cells impacts their survival. Reduced apoptosis observed in Mac-1+Shp2* cells was normalized to WT levels in Shp2*M Mac-1+ cells (Figure 2F-G), which was associated with increased expression of Bim compared with controls (Figure 2G). These results show that targeting Morrbid in Shp2* Mac-1+ cells induces apoptosis and significantly corrects the absolute number of mature myeloid cells in the BM, spleen, and PB (Figure 2I).
To assess whether MORRBID also plays a role in human JMML, we conducted data mining studies to assess the correlation between expression of MORRBID and JMML prognosis. In a limited number of human JMML samples with PTPN11, KRAS, and NRAS mutations, we found that children with JMML who showed increased expression of MORRBID were associated with poor overall survival compared with patients who had low levels of MORRBID (Figure 2H; n = 7-10; P = .049; patients aged >18 months were included for survival analysis, whereas those aged <18 months were excluded). These data are consistent with our survival studies using the murine model of JMML shown in Figure 1B. Moreover, although it was not statistically significant because of the small sample size, we observed a similar trend between MORRBID expression and survival in patients with JMML lacking mutations in PTPN11, KRAS, and NRAS (n = 9 for each subgroup; P = .227; supplemental Figure 2).
In summary, our results demonstrate an essential role for Morrbid in regulating the pathogenesis of Shp2E76K mice, including hematologic abnormalities and mortality seen in this model of human JMML. Future studies will determine how inhibition of MORRBID in JMML patient-derived cells affects their growth and engraftment in mice. In addition, it would also be prudent to assess the impact of Morrbid deletion in a model of Shp2E76K in which Shp2E76K is active in hematopoietic stem cells. Although Morrbid is expressed in myeloid cells, use of a conditional knockout model of Morrbid to assess its function specifically in hematopoietic stem cells would shed new light on how Morrbid contributes to the JMML phenotypes driven by Shp2E76K in stem cells.
Original data are available by e-mail request to either of the corresponding authors.
Acknowledgments
The authors thank their colleagues for technical support and for critically reading the manuscript and making suggestions to improve it and Tracy Winkle for administrative support.
This work was supported in part by grants from the National Institutes of Health, National Cancer Institute (R01-CA134777 and R01-CA173852) (R.K.) and National Heart, Lung, and Blood Institute (R01-HL140961 and R01-HL146137 [R.K.]; and T32HL007910 [Z.C.]), and by funds from Riley Children’s Foundation (R.K.).
Authorship
Contribution: Z.C. and R.K. conceived and designed the experiments, analyzed the data, and wrote the manuscript; Z.C. performed most of the experiments and acquired the data; C.Z. performed the bioinformatics analysis; and J.J.K., A.W., and J.H.-M. contributed reagents.
Conflict of interest: The authors declare no competing financial interests.
Correspondence: Zhigang Cai, School of Medicine, Indiana University, 1044 W Walnut St, R4-169, Indianapolis, IN 46202; e-mail: zcai@iupui.edu; and Reuben Kapur, School of Medicine, Indiana University, 1044 W Walnut St, R4-168, Indianapolis, IN 46202; e-mail: rkapur@iupui.edu.

