

Key Points
Hypoxia increases expression of TET3 via an enhancer located in intron 2, which is required for hypoxia-mediated upregulation of TET3.
Loss of HIF-1α–binding sites in intron 2 of TET3 leads to decreased erythroid differentiation and loss of viability under hypoxia.

Abstract
In mammalian cells, cytosines found within cytosine guanine dinucleotides can be methylated to 5-methylcytosine (5-mC) by DNA methyltransferases and further oxidized by the Ten-eleven translocation dioxygenase (TET) enzymes to 5-hydroxymethylcytosine (5-hmC). We have previously shown that hematopoietic stem and progenitor cells (HSPCs) with TET2 mutations have aberrant 5-hmC distribution and less erythroid differentiation potential. However, these experiments were performed under standard tissue culture conditions with 21% oxygen (O2), whereas HSPCs in human bone marrow reside in ∼1% O2. Therefore, to model human erythropoiesis more accurately, we compared 5-hmC distribution and gene expression in hypoxic vs normoxic conditions. Despite TET enzymes having limited O2 as a substrate in hypoxia, 5-hmC peaks were more numerous and pronounced than in normoxia. Among the TET genes, TET3 was upregulated specifically in hypoxia. We identified 2 HIF-1 binding sites in TET3 by chromatin immunoprecipitation of HIF-1α followed by sequencing, and TET3 upregulation was abrogated with deletion of both sites, indicating that TET3 is a direct HIF-1 target. Finally, we showed that loss of one or both of these HIF-1 binding sites in K562 cells disrupted erythroid differentiation in hypoxia and lowered cell viability. This work provides a molecular link between O2 availability, epigenetic modification of chromatin, and erythroid differentiation.
Introduction
5-Hydroxymethylcytosine (5-hmC) is an epigenetic mark that regulates chromosome structure and promotes transcription.1-3 The Ten-eleven translocation dioxygenases (TETs) convert 5-methylcytosine (5-mC) to 5-hmC in a reaction that requires oxygen (O2), Fe(II), and α-ketoglutarate and is facilitated by ascorbate as a cofactor. The human genome contains 3 TET genes (TET1-3) expressed at different tissue and developmental stages. In human hematopoietic cells, both TET2 and TET3 are expressed.4 Moreover, TET2 is one of the most frequently somatically mutated genes in a condition now commonly referred to as clonal hematopoiesis, as well as myeloid malignancies, T-cell lymphomas, melanomas, and gliomas.3,5-9 Previously, we reported that TET2 is the predominant TET enzyme in erythropoiesis under normoxic conditions, and its activity is augmented by JAK2-mediated phosphorylation.10,11 These studies highlight the importance of 5-hmC regulation for erythroid lineage differentiation.
Hematopoietic stem and progenitor cells (HSPCs) reside within the bone marrow niche, which is poorly oxygenated.12,13 In addition, environmental hypoxia is a strong driver for erythropoiesis through stimulating erythropoietin (EPO) production in renal cells, which then stimulate erythroid differentiation of HSPCs.14 We therefore undertook a study to understand how hypoxia affects 5-hmC distribution and gene expression during erythropoiesis in HSPCs. We expected that hypoxia would lead to decreased global 5-hmC levels and attenuation of 5-hmC peaks compared with normoxia. We expected that changes in 5-hmC distribution together with gene expression changes directed by HIF transcription factors would promote erythroid differentiation of HSPCs.
Materials and methods
Full details of the methods used in this study are given in the supplemental Materials and methods.
In vitro human erythroid differentiation in normoxia and hypoxia
5-hmC pull-down, HIF-1α chromatin immunoprecipitation, and sequencing data processing
5-hmC pull-down and sequencing were performed as previously described.16 HIF-1α chromatin immunoprecipitation (ChIP) was performed on sonicated chromatin with rabbit anti–HIF-1α antibody (Abcam; ab2185). Raw sequences in fastq format were aligned to the hg19 reference genome by Burrows-Wheeler Aligner.17 Peaks were called by MACS218 with input sequences as control.
RNA-sequencing and data processing
CRISPR-Cas9 targeted deletion
CRISPR guide sequences were inserted to the lentiCRISPR v2 plasmid (#52961; Addgene) according to the associated protocol.21 Single-cell clones were isolated from the transduced population, and the targeted site was sequenced from each clone. Double deletion clones were made from validated single deletion clones by targeting the intact binding site using CRISPR-Cas9.
Results
Hypoxia promotes 5-hmC accumulation during erythropoiesis
To investigate the effects of hypoxia on 5-hmC distribution and gene expression during erythropoiesis, we performed our established erythroid differentiation protocol on normal human CD34+ HSPCs under parallel normoxic (21% O2) vs hypoxic (1% O2) conditions. Samples were collected for DNA and RNA extraction at days 0, 3, 7, and 10 of the differentiation assay. We measured total levels of 5-mC and 5-hmC in genomic DNA using mass spectrometry. No significant differences were found between normoxic and hypoxic samples in 5-mC or 5-hmC levels (Figure 1A-B). This observation was contrary to our expectation that a lack of O2 would lower substrate availability for the 5-mC to 5-hmC conversion and result in a decrease in total 5-hmC levels.

Hypoxia increases overall 5-hmC density during in vitro erythroid differentiation. (A) Mass spectrometry quantification of 5-mC relative to all cytosine species during erythroid differentiation. (B) Mass spectrometry quantification of 5-hmC relative to all cytosine species during erythroid differentiation. Quantification of 5-hmC peaks by peak counts (C) and by total bases covered by peaks (D) in normoxia vs hypoxia at days 3, 7, and 10. Normalized to corresponding measurements at day 0. Day 0 peak count was 473 206 for replicate 1 and 185 665 for replicate 2. (E) Dot plots of numbers of sequencing reads within peaks called by MACS2 per million total aligned reads. Each dot represents a genomic region of 1 Mb. (F) Similar to panel E, global measurement of sequencing reads within peaks, normalized to day 0. (G) Box plot of all 5-hmC peaks FPKM in all time points in normoxia vs hypoxia. Wilcoxon rank-sum test was used to evaluate statistical significance. ‡P < 10−8. Quantification of peaks that gained or lost 5-hmC in hypoxia by peak counts (H) and by total bases covered by peaks (I) as percentages of total peaks. N = 2 for mass spectrometry and 5-hmC pull-downs. *P < .05.
Hypoxia increases overall 5-hmC density during in vitro erythroid differentiation. (A) Mass spectrometry quantification of 5-mC relative to all cytosine species during erythroid differentiation. (B) Mass spectrometry quantification of 5-hmC relative to all cytosine species during erythroid differentiation. Quantification of 5-hmC peaks by peak counts (C) and by total bases covered by peaks (D) in normoxia vs hypoxia at days 3, 7, and 10. Normalized to corresponding measurements at day 0. Day 0 peak count was 473 206 for replicate 1 and 185 665 for replicate 2. (E) Dot plots of numbers of sequencing reads within peaks called by MACS2 per million total aligned reads. Each dot represents a genomic region of 1 Mb. (F) Similar to panel E, global measurement of sequencing reads within peaks, normalized to day 0. (G) Box plot of all 5-hmC peaks FPKM in all time points in normoxia vs hypoxia. Wilcoxon rank-sum test was used to evaluate statistical significance. ‡P < 10−8. Quantification of peaks that gained or lost 5-hmC in hypoxia by peak counts (H) and by total bases covered by peaks (I) as percentages of total peaks. N = 2 for mass spectrometry and 5-hmC pull-downs. *P < .05.
We next examined the genomic distribution of 5-hmC during erythroid differentiation under normoxic vs hypoxic conditions using hMe-SEAL16 (GSE40243 [as published in Madzo et al10 ], GSE142870 [hypoxia data from the current study]). We found that 5-hmC peaks were more prominent in hypoxia than in normoxia in multiple hypoxia-responsive genes (supplemental Figure 1). MACS2 was then used to identify 5-hmC peaks in all samples. Consistent with our previous observations,10 5-hmC distribution in all samples was enriched in promoters, enhancers, and gene bodies but was depleted in intergenic regions (supplemental Figure 2A-C). More peaks were observed in hypoxic samples than in normoxic samples, especially at days 7 and 10 (Figure 1C). Similarly, peak coverage across the genome, defined as total base pairs covered by peaks in a data set, was higher in hypoxia by day 10 (Figure 1D).
We then examined the normalized counts across the genome (Figure 1E-F) and found that although normoxic 5-hmC peaks gradually diminished throughout differentiation, hypoxic 5-hmC peaks remained prominent up to day 10. This trend was observed across the entire genome, resulting in a higher percentage of 5-hmC reads located inside peaks in hypoxic samples at later time points compared with normoxic samples. Consistently, overall 5-hmC peak density (FPKM [fragments per kilobase of transcript per million mapped reads]) at days 7 and 10 was higher in hypoxia compared with normoxia (Figure 1G), suggesting enhanced maintenance and/or de novo synthesis of 5-hmC. Lastly, we quantified the percentage of 5-hmC peaks that were gained or lost in hypoxia compared with those in normoxia (Figure 1H-I). To reduce noise in the data, a peak was classified as a differential peak if it had FPKM >1 in both normoxia and hypoxia and if the difference in FPKM between normoxia and hypoxia was greater than twofold. With this definition, nearly 40% of all 5-hmC peaks had higher density in hypoxia (5-hmC gain), whereas <10% of the peaks had higher 5-hmC density in normoxia (5-hmC loss). 5-hmC gains were enriched in promoters, gene bodies, CpG islands and shores, and especially in enhancers throughout differentiation, whereas 5-hmC losses were not enriched in any genomic elements at days 7 and 10 (supplemental Figure 2A-C). Based on these observations, we hypothesized that TET activity in hypoxia increases to reorganize 5-hmC distribution across the genome to promote hypoxic gene expression.
TET3 is upregulated in hypoxia in erythropoietic cells
We next performed RNA-sequencing in normoxic vs hypoxic CD34+ HSPCs during erythroid differentiation (GSE40243 [as published in Madzo et al10 ], GSE142870 [hypoxia data from the current study]). We examined whether 5-hmC changes correlated with changes in gene expression by calculating the enrichment of 5-hmC gain or loss in or near upregulated or downregulated genes. We defined 5-hmC peaks to be near a gene if they were located within 2 kb flanking the gene. Although 5-hmC peaks were enriched in genes generally, a particularly strong correlation was noted between 5-hmC gains and upregulated genes at days 7 and 10 (Figure 2A-C; supplemental Figure 2D-F). Gene ontology enrichment analysis22,23 was used to show that the upregulated genes containing 5-hmC gains at day 7 were involved in glycolysis, hypoxic response, signal transduction, and cytoskeleton remodeling (Figure 2D). The upregulated genes containing 5-hmC gain regions at day 10 were enriched in glycolysis (26.5-fold) and positive regulation of cell projection organization (Figure 2E). In contrast, gene ontology analysis on all upregulated genes at days 7 and 10, regardless of associated 5-hmC changes, returned notably different lists (supplemental Figure 3). Glycolysis and hypoxic response genes were less enriched, whereas a greater number of terms related to ion homeostasis and small molecule metabolism were more enriched. These results suggest that 5-hmC may have an important function in promoting the expression of hypoxia-inducible genes and may contribute to cellular response to hypoxia.

The 5-hmC gain is enriched in hypoxia-induced genes. Enrichment of all 5-hmC (A), 5-hmC gain (B), and 5-hmC loss (C) in upregulated genes at day 3, day 7, and day 10 of in vitro erythropoiesis. (D) Gene ontology term enrichment of day 7 upregulated genes containing regions with 5-hmC gain (green bar in panel B). All terms shown have a false discovery rate <0.05. (E) Gene ontology term enrichment of day 10 upregulated genes containing regions with 5-hmC gain (dark green bar in panel B). All terms shown have a false discovery rate <0.05. N = 2 for RNA-sequencing experiments. VEGF, vascular endothelial growth factor.
The 5-hmC gain is enriched in hypoxia-induced genes. Enrichment of all 5-hmC (A), 5-hmC gain (B), and 5-hmC loss (C) in upregulated genes at day 3, day 7, and day 10 of in vitro erythropoiesis. (D) Gene ontology term enrichment of day 7 upregulated genes containing regions with 5-hmC gain (green bar in panel B). All terms shown have a false discovery rate <0.05. (E) Gene ontology term enrichment of day 10 upregulated genes containing regions with 5-hmC gain (dark green bar in panel B). All terms shown have a false discovery rate <0.05. N = 2 for RNA-sequencing experiments. VEGF, vascular endothelial growth factor.
We further examined the RNA-sequencing data to find candidate genes that could explain the increased 5-hmC in hypoxia. Hypoxic conditions were confirmed by observing the upregulation of canonical hypoxia responsive genes as well as the downregulation of mitochondrial genes (supplemental Figure 4A). Both TET2 and TET3 were expressed throughout erythropoiesis (Figure 3A-C). In particular, TET3 expression was upregulated as erythropoiesis progressed, whereas TET2 expression remained relatively stable. TET1 expression was very low compared with TET2 or TET3 (Figure 3A-C). The expression pattern of TET3 found here was measured by RNA-sequencing, which we consider to be more reliable than previous methods10 and is validated by independent work4 as well as consensus bone marrow expression data from the Human Protein Atlas (supplemental Figure 7E).24,25 Notably, we found that TET3, but not TET2, was upregulated in hypoxia during erythroid differentiation up to day 7 (Figure 3A-C). This scenario indicated that TET3 could be upregulated by hypoxia and facilitate 5-hmC accumulation and gene expression. DNMT1, which encodes the main maintenance DNA methyltransferase, was suppressed at days 7 and 10, whereas expression of DNMT3A, encoding a de novoDNAmethyltransferase, was increased at these time points (Figure 3D-F).
Lastly, we examined the expression of genes involved in erythropoiesis in normoxia vs hypoxia. We found that hypoxic cells express more EPO receptor (EPOR), glycophorin A (GYPA), and transferrin receptor 2 (TFR2) at days 3 and/or 7 (Figure 3G-I), suggesting a higher sensitivity to EPO stimulation and a more robust early erythroid phenotype. Erythroferrone (ERFE), which encodes a secreted protein that inhibits hepcidin production by the liver to increase blood iron levels, was upregulated at day 3 (Figure 3J). We then examined the genes involved in iron homeostasis in erythrocytes: SLC40A1 encodes the only known iron exporter (ferroportin)26 ; NCOA4 encodes a receptor for ferritin endocytosis27,28 ; SLC11A2 encodes the proton-coupled divalent metal ion transporter responsible for transporting Fe(II) from endosomes to the cytosol26 ; and SLC25A37 encodes the erythroid-specific mitochondrial iron transporter (Mitoferrin1) that is crucial for heme biosynthesis.26,29 All 4 genes were significantly upregulated by hypoxia at day 3, and all but SLC11A2 were significantly upregulated by hypoxia (up to threefold) at day 7 (Figure 3K-N). By day 10, ferroportin remained significantly upregulated in hypoxia (1.9-fold) (Figure 3K). The genes encoding the copper transporter SLC31A1 and zinc transporters SLC30A5 and SLC39A3 were not upregulated by hypoxia (supplemental Figure 4H-J). Together, the RNA-sequencing data suggest that HSPCs in hypoxia are more sensitive to EPO stimulation, exhibit a more robust activation of erythroid lineage genes, and have increased iron homeostatic activities, consistent with previous studies.30-32 Finally, expression of erythroid transcription factors KLF1, GATA1/2, and RUNX1 was not significantly different in normoxia vs hypoxia (supplemental Figure 4B-D). This finding suggests that the differences in expression described here were not mediated by transcriptional changes of these erythroid transcription factor genes.

Hypoxia upregulates TET3 expression and enhances early erythroid differentiation.TET1-3 expression in normoxia vs hypoxia at day 3 (A), day 7 (B), and day 10 (C) of in vitro erythropoiesis. DNMT1, DNMT3A, and DNMT3B expression in normoxia vs hypoxia at day 3 (D), day 7 (E), and day 10 (F) of in vitro erythropoiesis. (G-N) Expression of EPOR, GYPA, TFR2, ERFE, SLC40A1, NCOA4, SLC11A2, and SLC25A37 in normoxia vs hypoxia at day 3, 7, and 10 of in vitro erythropoiesis. N = 2 for RNA-sequencing experiments. *P < .05; **P < .01.
Hypoxia upregulates TET3 expression and enhances early erythroid differentiation.TET1-3 expression in normoxia vs hypoxia at day 3 (A), day 7 (B), and day 10 (C) of in vitro erythropoiesis. DNMT1, DNMT3A, and DNMT3B expression in normoxia vs hypoxia at day 3 (D), day 7 (E), and day 10 (F) of in vitro erythropoiesis. (G-N) Expression of EPOR, GYPA, TFR2, ERFE, SLC40A1, NCOA4, SLC11A2, and SLC25A37 in normoxia vs hypoxia at day 3, 7, and 10 of in vitro erythropoiesis. N = 2 for RNA-sequencing experiments. *P < .05; **P < .01.
HIF-1α binds to enhancers within TET3 intron 2
We then asked whether TET3 is a direct target of HIF-1. We found that K562 cells tolerate hypoxia treatment well and that TET3 is upregulated in hypoxia in these cells (Figure 4A; supplemental Figure 7E).24,25 We therefore chose to use this cell line as a model to investigate the molecular mechanisms controlling the hypoxia-induced TET3 upregulation. HIF-1α ChIP-sequencing was performed in K562 cells cultured under normoxic vs hypoxic conditions (GSE142870). We found HIF-1α binding at 2 sites (Sites 1 and 2) within intron 2 (Figure 4B). In contrast, HIF-1α did not bind either TET1 or TET2 (supplemental Figure 5). K562 Hi-C data showed no strong interactions between TET3 and other nearby HIF-1–binding sites, suggesting that Sites 1 and 2 are the main binding sites responsible for TET3 upregulation33,34 (supplemental Figure 6).
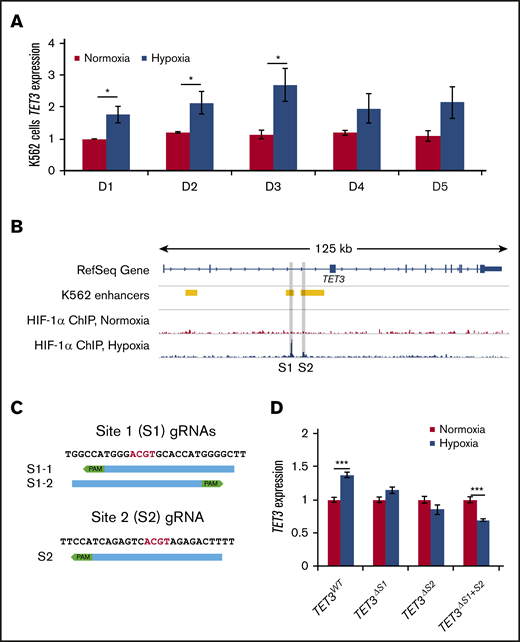
HIF-1 binding is required for TET3 upregulation in hypoxia. (A) TET3 expression in K562 cells in normoxia vs hypoxia, up to 5 days. (B) Overlay of K562 enhancers with HIF-1α ChIP in normoxia and hypoxia in TET3 genes. The binding sites are highlighted in gray (Sites 1 and 2). (C) Sequences of binding Site 1 and Site 2, as well as positions of CRISPR guides used to delete the binding sites. Core E-box motif (ACGT) is indicated by red letters. PAM sequences (NGG) are indicated by green boxes. (D) TET3 expression in normoxia vs hypoxia in TET3WT, TET3ΔS1, TET3ΔS2, and TET3ΔS1+S2 clones. N = 2 for HIF-1α ChIP. N = 3 for hypoxia treatment of CRISPRed K562 clones. *P < .05; ***P < .001.
HIF-1 binding is required for TET3 upregulation in hypoxia. (A) TET3 expression in K562 cells in normoxia vs hypoxia, up to 5 days. (B) Overlay of K562 enhancers with HIF-1α ChIP in normoxia and hypoxia in TET3 genes. The binding sites are highlighted in gray (Sites 1 and 2). (C) Sequences of binding Site 1 and Site 2, as well as positions of CRISPR guides used to delete the binding sites. Core E-box motif (ACGT) is indicated by red letters. PAM sequences (NGG) are indicated by green boxes. (D) TET3 expression in normoxia vs hypoxia in TET3WT, TET3ΔS1, TET3ΔS2, and TET3ΔS1+S2 clones. N = 2 for HIF-1α ChIP. N = 3 for hypoxia treatment of CRISPRed K562 clones. *P < .05; ***P < .001.
HIF-1α binding is required for TET3 upregulation in hypoxia
To assess the effects of HIF-1α binding at these sites, CRISPR/Cas9 guides were designed to delete one or both of the binding sites (Figure 4C). In total, we identified 3 clones with Site 1 deletions (TET3ΔS1), 2 with Site 2 deletions (TET3ΔS2), and 2 with double-deletions (TET3ΔS1+S2), as well as 3 wild-type clones from vector-only transfection (TET3WT) (supplemental Figure 7A-C). We then treated these clones in normoxia vs hypoxia for 72 hours and measured TET3 expression (Figure 4D; supplemental Figure 7D). TET3WT cells exhibited the expected upregulation of TET3 in hypoxia (∼1.4-fold), whereas neither TET3ΔS1 nor TET3ΔS2 cells upregulated TET3 (Figure 4D). Strikingly, TET3ΔS1+S2 cells exhibited significant TET3 suppression (0.7-fold) in hypoxia, showing an additive effect of these sites. These results suggest that both HIF-binding sites are key sites that dictate TET3 expression in response to hypoxia in erythropoiesis.
Loss of TET3 induction impairs K562 cell differentiation potential and survival in hypoxia
To investigate the physiological significance of the 2 HIF-1α–binding sites in erythropoiesis, we subjected the parental and CRISPR ΔS1/ΔS2 cell lines to erythroid differentiation in normoxia and hypoxia. We used 1 mM sodium butyrate to drive erythropoiesis in K562 cells under normoxic or hypoxic conditions.35 After 3 days of treatment, samples were collected to make cytospin slides, which were stained with hematoxylin and benzidine to quantify hemoglobin production in each cell (Figure 5A). Interestingly, parental K562 cells spontaneously differentiated in hypoxia without sodium butyrate treatment, resulting in a higher percentage of hemoglobin-producing cells (Figure 5B; supplemental Figure 8A). However, this phenomenon was not observed in any of the cell lines containing deletions of the HIF-1–binding sites. We quantified the percentage of hemoglobin-producing cells that accumulated high levels of hemoglobin (Figure 5C; supplemental Figure 8B-F). We found that cells with Site 2 deleted were less capable of accumulating hemoglobin in hypoxia, which can be seen as a shift in benzidine staining intensity from normoxia to hypoxia (supplemental Figure 8E-F). Together, these results show that loss of either Site 1 or Site 2 impairs the initiation of erythropoiesis in response to hypoxia and that the loss of Site 2 further inhibits hemoglobin production.

Loss of TET3 negatively affects K562 erythroid differentiation potential and chromatin stability. (A) Representative images of benzidine-hematoxylin staining of K562 TET3WT and TET3ΔS1+S2 cells. Benzidine staining (brown) differentiates hemoglobin-positive (Hb+) and hemoglobin-negative (Hb−) cells. Scale bars indicate 50 μm. (B) Quantification of Hb+ cells under normoxia vs hypoxia in parental K562 cells and TET3ΔS1/ΔS2 lines. (C) Quantification of cells with high hemoglobin levels under normoxia vs hypoxia in parental K562 cells and TET3ΔS1/ΔS2 lines. Normoxic butyrate-treated cells were used to establish the range of benzidine staining intensity in each replicate. High hemoglobin cells were defined as having higher benzidine staining intensity than 50% of Hb+ cells in the normoxic butyrate-treated cells. (D) Quantification of cells with pyknotic nuclei in normoxia vs hypoxia, with or without sodium butyrate, in parental K562 cells and TET3ΔS1/ΔS2 lines. (E) Quantification of cells with karyorrhectic nuclei in normoxia vs hypoxia, with or without sodium butyrate, in parental K562 cells and TET3ΔS1/ΔS2 lines. N = 3 for all hypoxia/sodium butyrate treatments. A minimum of 500 cells were counted in each replicate for each treatment combination. *P < .05; **P < .01; ***P < .001.
Loss of TET3 negatively affects K562 erythroid differentiation potential and chromatin stability. (A) Representative images of benzidine-hematoxylin staining of K562 TET3WT and TET3ΔS1+S2 cells. Benzidine staining (brown) differentiates hemoglobin-positive (Hb+) and hemoglobin-negative (Hb−) cells. Scale bars indicate 50 μm. (B) Quantification of Hb+ cells under normoxia vs hypoxia in parental K562 cells and TET3ΔS1/ΔS2 lines. (C) Quantification of cells with high hemoglobin levels under normoxia vs hypoxia in parental K562 cells and TET3ΔS1/ΔS2 lines. Normoxic butyrate-treated cells were used to establish the range of benzidine staining intensity in each replicate. High hemoglobin cells were defined as having higher benzidine staining intensity than 50% of Hb+ cells in the normoxic butyrate-treated cells. (D) Quantification of cells with pyknotic nuclei in normoxia vs hypoxia, with or without sodium butyrate, in parental K562 cells and TET3ΔS1/ΔS2 lines. (E) Quantification of cells with karyorrhectic nuclei in normoxia vs hypoxia, with or without sodium butyrate, in parental K562 cells and TET3ΔS1/ΔS2 lines. N = 3 for all hypoxia/sodium butyrate treatments. A minimum of 500 cells were counted in each replicate for each treatment combination. *P < .05; **P < .01; ***P < .001.
In addition to hemoglobin production, we examined the nuclear morphology of the non–hemoglobin-producing cells under all treatment conditions. Notably, compared with any other cells in any treatment condition, the TET3ΔS1+ΔS2 double deletion cells contained more pyknotic nuclei after sodium butyrate treatment and under hypoxic conditions (Figure 5D), suggesting higher rates of cell death. Likewise, TET3ΔS1+ΔS2 cells were the only ones with higher rates of karyorrhectic (fragmented) nuclei after sodium butyrate treatment and under hypoxic conditions (Figure 5E). This outcome indicates that loss of both binding sites and the subsequent loss of TET3 expression (Figure 4D) decrease cell survivability in hypoxia, especially under differentiating conditions.
Discussion
Erythropoiesis in humans is intricately regulated by O2 availability. Low O2 tension, as occurs at high altitude, leads to HIF-α stabilization in renal cells, which then induce production of EPO to stimulate erythropoiesis in the bone marrow.14 Changes in the HIF-EPO axis have been shown to have major effects on erythropoiesis. For example, patients with congenital VHL mutations or VHL-mutated renal cell carcinoma cells do not ubiquitinate HIF-α subunits and constitutively upregulate the EPO gene, leading to polycythemia.36-40 Genetic adaptations (eg, a gain-of-function variant of the PHD2 protein that increases O2 affinity) have been identified in Tibetan populations that attenuate erythropoiesis in hypoxia to avoid polycythemia.41-44 These studies highlight the O2-sensitive control of EPO production in renal cells but did not examine the effects of hypoxia on HSPCs.
HIF stabilization alters histone modifications by recruiting p300 histone acetyltransferase in VHL-deficient clear cell renal cell carcinoma, which in turn activates gene transcription.39,40 In addition, HIF binding promotes chromatin accessibility by remodeling local histone occupancy.45 Our study suggests that hypoxia sensitizes and promotes erythroid differentiation of HSPCs by inducing EPOR expression as well as by changing the epigenomes via TET3 and possibly DNMT3A. Zhang et al46 have shown that DNMT3A and TET2 exhibit both competitive and cooperative activities in HSPCs to suppress the expression of lineage-specific genes. It is possible that DNMT3A and TET3 have similar interactions in the erythroid lineage under hypoxic conditions. We also showed that hypoxia increases expression of genes involved in transporting iron ions, which is both required for heme biosynthesis and a crucial cofactor for dioxygenases such as TETs. The effects of hypoxia on HSPCs could amplify the renal cell response and contribute to rapid acclimation to hypoxic environments.
Available ENCODE (Encyclopedia of DNA Elements) ChIP data in K562 revealed that Site 1 and Site 2 in TET3 are in fact very different with respect to transcription factor binding in general (supplemental Figure 9). Although the 2 HIF-1 binding sites have similar effects on TET3 expression under hypoxia, Site 2, but not Site 1, is bound by a number of erythroid transcription factors or complexes, including GATA1, GATA2, TAL1, EP300, NCOR1, REST, and NCOA4. Two binding motifs of GATA1/2 (GATA-box) are upstream (–60 bp and −105 bp) of Site 2, suggesting that this site is likely also important in regulating erythropoiesis-specific TET3 expression. This hypothesis is supported, in part, by the observation that K562 cells with Site 2 deletion were less capable of accumulating hemoglobin in hypoxic conditions (Figure 5C). Notably, despite the motif similarity of MYC and MAX to HIF-1, no strong MYC/MAX binding was found at either Site 1 or Site 2 (supplemental Figure 9B), which implies that these sites are HIF specific. We thus conclude that Sites 1 and 2 are the main hypoxia-responsive regulatory sites of TET3 in erythropoiesis.
Despite having similar core catalytic domains, TET3 harbors a CXXC domain that is not present in TET2.1,3 In contrast, TET2 is uniquely phosphorylated by JAK2 at 2 tyrosine residues near the C terminus, which increases its enzymatic activity.11 Yan et al4 have shown that TET3-knockdown HSPCs have slower growth and a higher apoptosis rate, and fail to enucleate properly, whereas TET2-knockdown HSPCs have increased growth and delayed differentiation. Adding to the differences between TET2 and TET3, our data show that TET3 expression is responsive to hypoxia, whereas TET2 expression is insensitive to O2 availability (Figure 3A-C). These results show that TET2 and TET3 respond to different environmental signals and have distinct functions during erythropoiesis.
Further differences between the 2 TET genes have been observed in clinical studies. Although TET2 mutations are common in patients with hematopoietic malignancies,3,5-9 TET3 mutations are relatively rare. Two studies have described several TET3 somatic exonic mutations in chronic myelomonocytic leukemia.8,47 One of these mutations resulted in a truncated protein (TET3Y473) lacking the catalytic domain, and another missense mutation (TET3R1548H) was shown to severely impair TET3 catalytic function. Other variants identified in TET3 are of unknown significance. Another study on chronic myeloid leukemia also identified 1 patient with a TET3 mutation from a cohort of 24 patients.48 The significance of this mutation (TET3A128T) is also unknown. The results from these studies seem to suggest that TET3 mutations in hematopoietic cells may predispose patients to chronic leukemias. Considering the remarkable effects of hypoxia on the epigenome, future studies regarding erythropoiesis and especially relating to TET functions should incorporate hypoxia to better simulate the physiological environment of the HSPCs.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE142870).
Acknowledgments
The authors thank the ENCODE Consortium for the K562 ChIP data.
This work was supported by an EvansMDS grant from the Edward P. Evans Foundation (A.W. and L.A.G.). J.Z.C. was supported by funding from the University of Chicago Biological Sciences Division Dean’s Office, the University of Chicago Comprehensive Cancer Center Women’s Board, and the Goldblatt Scholarship.
Authorship
Contribution: J.Z.C. designed and performed the experiments, analyzed the data, and wrote the manuscript; H.L. performed the differentiation experiments; L.A.G. conceived of the study and provided insights in experimental design and data interpretation; and A.W. provided primary HSPCs for the differentiation experiments and additional input for experimental design and data interpretation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lucy A. Godley, The University of Chicago, 5841 S Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: lgodley@medicine.bsd.uchicago.edu.

