

Key Points
GPVI regulates aggregation and PS exposure on collagen and noncollagen surfaces under flow.
The estimated frequency of the (c.711_712insA) variant in GP6 in Chile is 2.9%.

Abstract
The role of glycoprotein VI (GPVI) in platelets was investigated in 3 families bearing an insertion within the GP6 gene that introduces a premature stop codon prior to the transmembrane domain, leading to expression of a truncated protein in the cytoplasm devoid of the transmembrane region. Western blotting and flow cytometry of GP6hom (homozygous) platelets confirmed loss of the full protein. The level of the Fc receptor γ-chain, which associates with GPVI in the membrane, was partially reduced, but expression of other receptors and signaling proteins was not altered. Spreading of platelets on collagen and von Willebrand factor (which supports partial spreading) was abolished in GP6hom platelets, and spreading on uncoated glass was reduced. Anticoagulated whole blood flowed over immobilized collagen or a mixture of von Willebrand factor, laminin, and rhodocytin (noncollagen surface) generated stable platelet aggregates that express phosphatidylserine (PS). Both responses were blocked on the 2 surfaces in GP6hom individuals, but adhesion was not altered. Thrombin generation was partially reduced in GP6hom blood. The frequency of the GP6het (heterozygous) variant in a representative sample of the Chilean population (1212 donors) is 2.9%, indicating that there are ∼4000 GP6hom individuals in Chile. These results demonstrate that GPVI supports aggregation and PS exposure under flow on collagen and noncollagen surfaces, but not adhesion. The retention of adhesion may contribute to the mild bleeding diathesis of GP6hom patients and account for why so few of the estimated 4000 GP6hom individuals in Chile have been identified.
Introduction
Glycoprotein VI (GPVI) is a member of the immunoglobulin receptor superfamily and a major signaling receptor in platelets for collagen, fibrin, and fibrinogen.1 GPVI is associated with the Fc receptor (FcR) γ-chain in the membrane. Clustering of GPVI leads to Src family kinase–mediated phosphorylation of a conserved immunoreceptor tyrosine-based activation motif in the FcR γ-chain and binding of the tyrosine kinase Syk through its tandem SH2 domains. This initiates a signaling cascade that culminates in activation of phospholipase Cγ2 (PLCγ2) and Ca2+ mobilization, integrin αIIbβ3 activation, granule secretion, and phosphatidylserine (PS) exposure, providing a surface for thrombin generation.2
Several unrelated patients homozygous for an adenine insertion (c.711_712insA) in exon 6 of the GP6 gene have been identified in Chile. The insertion generates a premature stop codon prior to the transmembrane region, leading to expression of a truncated protein of ∼50 kDa that is retained in the cytosol. In 2013, Matus et al3 described the phenotype of 5 GP6hom individuals, 3 females (aged 22, 11, and 5 years) and 2 males (aged 23 and 12 years). All of the individuals had a normal platelet count, and 4 had a mild bleeding diathesis, with the fifth, the 5-year-old girl, being asymptomatic. Symptoms started in early childhood and consisted of mild mucosal and skin bleeding. The Ivy bleeding time, recorded for 2 individuals, was 9 and 13 minutes, thus similar or above the normal level (9.5 min) and consistent with a mild bleeding diathesis. All hemostatic parameters and plasma von Willebrand factor (VWF) activity were within the normal range.3
The lack of GPVI surface expression on GP6hom platelets was shown by flow cytometry using an antibody against the extracellular region of GPVI.3 The expression of the truncated protein in the cytosol was shown using immunofluorescence in permeabilized platelets and by western blotting. GP6hom platelets failed to aggregate or secrete 14C-serotonin in response to the GPVI agonists collagen, convulxin, and C-reactive protein (CRP), whereas their response to arachidonic acid and adenosine 5′-diphosphate was unaltered.3 GP6het relatives did not exhibit signs of bleeding3 and displayed normal aggregation and secretion upon stimulation with collagen and convulxin.3 In 2 GP6het individuals, the response to CRP was reduced, which is in accordance with a previously reported effect in heterozygous mouse platelets.4 The loss of response to CRP in the GP6het individuals is likely due to loss of avidity, whereas for collagen, this is masked by the presence of integrin α2β1.
Since 2013, additional GP6hom individuals have been identified in Chile, bringing the total to 9 from 8 unrelated families. With no consanguinity and the locations of the families being geographically disperse over several hundred kilometers, the heterozygous (carrier) frequency for c.711_712insA may be relatively high in Chile. These are the only identified individuals worldwide with a homozygous variant that prevents surface expression of GPVI and as such represent a unique resource to study the role of the immunoglobulin receptor in platelet activation.
In this study, we have measured platelet activation on collagen and noncollagen (a mixture of rhodocytin, laminin, and VWF) surfaces under static and flow conditions in 3 GP6het and 4 GP6hom individuals from 3 unrelated families. The results show that GPVI is critical for spreading under static conditions on a variety of surfaces and for aggregation and PS exposure, but not for adhesion on collagen and noncollagen surfaces under flow. Given the unique appearance of GP6hom patients in the Chilean population, we have sequenced exon 6 of 1212 DNA samples representative of the Chilean population5 and determined a carrier frequency of 2.9% for this variant.
Methods
Ethical statement
Experiments involving blood samples and DNA from Chilean patients were approved by the Ethical Scientific Committee at the Pontificia Universidad Catolica de Chile and were conducted in accordance with the guidelines of the National Commission on Science and Technology of Chile. Informed consent was obtained according to the guidelines of the local ethics committee and complied with the ethical principles according to the Declaration of Helsinki. The experiments on mice were performed in line with UK Home Office approval (see supplemental Materials and methods).
Patients
The major patient bleeding features, including a Bleeding Assessment Tool,6 are reported in supplemental Table 1.
Reagents and samples
The list of reagents including the control and GPVI-null mouse platelets are reported in supplemental Materials and methods.
Blood withdrawal and platelet preparation
Blood was taken with 3.2% sodium citrate (ratio 1:9) as the anticoagulant. Blood was taken from 3 families with a total of 4 GP6hom and 3 GP6het individuals. In 2 individuals, blood was drawn on 2 occasions separated by 9 months. All family members had been genotyped for a predicted adenine insertion between positions 711 and 712 of exon 6. Blood was taken from 4 healthy, unrelated individuals on the same experimental days. Washed platelets were separated from plasma by centrifugation, resuspended in a modified Tyrode’s buffer, and recentrifuged before final suspension (see supplemental Materials and methods).
Western blotting
Platelets were mixed with sodium dodecyl sulfate reducing sample buffer and proteins separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The antibodies and concentrations are described in supplemental Materials and methods.
Platelet spreading
Glass coverslips were coated with collagen (10 μg/mL) and VWF (100 μg/mL) overnight at 4°C and blocked with 1% bovine serum albumin in phosphate-buffered saline (PBS) for 60 min at room temperature before washing with PBS. Washed platelets were spread on coated and uncoated coverslips for 45 minutes at 37°C. The coverslips were washed in PBS, and adherent platelets were fixed in 10% formalin, permeabilized with 0.1% Triton X-100 (in PBS), and stained with Alexa Fluor 488-Phalloidin. Platelets were imaged on a Zeiss LSM7 microscope.
Platelet measurements including area were calculated using a semiautomated machine learning–based workflow7 and implemented using the open source software KNIME8 and ilastik.9 First, a pixel classifier was trained within ilastik to produce binary segmentations. The classifier was then run on the full data set within KNIME and touching platelets were identified manually by clicking on their center. A watershed transformation was then used to extract cellular boundaries facilitating the calculation of per-platelet measurements including area and circularity. Any objects <1 μm2 were discarded. The measurements for each platelet were then used to train a random forest classifier to automatically group platelets into predefined subtypes, specifically nonspread, partially spread, and fully spread.
Flow studies
Thrombus formation was assessed in whole blood.10 Glass coverslips were coated with collagen (50 µg/mL) or a combination of human VWF (50 μg/mL), laminin (100 μg/mL), and rhodocytin (250 μg/mL) before blocking with 1% bovine serum albumin in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer (pH 7.45). Citrated whole blood, recalcified with CaCl2 (7.5 mM) and MgCl2 (3.75 mM), in the presence of PPACK was perfused for 4 minutes at 1000 s−1 and then labeled by perfusion with AF647-Annexin-A5 (1:200) in a modified N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer (pH 7.45) supplemented with CaCl2 (2 mM) and heparin (1 U/mL) for 1.5 minutes. Phase-contrast and fluorescence images were taken for analysis of surface area coverage of adherent platelets and PS exposure. For studies under coagulating conditions,11 citrated whole blood without PPACK was perfused over slides coated with collagen (50 μg/mL) and recombinant tissue factor (500 pM), and simultaneously, PS exposure was labeled with AF647-Annexin-A5. Image analysis was performed by using predefined scripts12,13 in the open source software Fiji,14 and contraction was assessed by visual inspection compared with representative images.12
Thrombin generation
Thrombin generation in platelet-rich plasma (PRP) was assessed by continuous assessment of thrombin generation using a calibrated automated thrombogram as described.15 In brief, the PRP platelet count was adjusted to 150 × 109/L with autologous platelet-poor plasma. PRP (80 µL) was supplemented with tissue factor (20 µL) and fluorogenic substrate plus CaCl2 (20 µL) to trigger thrombin generation. The resultant curves were analyzed, and results are expressed as endogenous thrombin potential (ETP).
Exon 6 genotyping
Genomic DNA was extracted from the whole blood of 1235 individuals, representative of the Chilean population as previously described.5 PCR amplification was performed across the GP6 exon 6 variant region using the following primers: 3′-CTCAAAAGGGGAATGGAGATA-5′ and 5′-AAGAGAGAGCTCCGTCCTCAC-3′ (as used in Matus et al3 ). DNA sequencing was performed using the BigDye Terminator sequencing kit v3.1 and run on the 3730 DNA analyzer. Sequencing results were analyzed using SnapGene Software.
Statistics
Data are represented as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism v8 software (San Diego, CA). Significance was determined using unpaired Student t test or a 1-way ANOVA with Tukey posttest, as appropriate. Differences with P < .05 were considered significant.
Results
GP6hom donors have a reduced level of the FcR γ-chain
The presence of a premature stop in GP6 results in the formation of a truncated protein of ∼50 kDa, which lacks the transmembrane and is retained in the cytosol.3 Consistent with this, the expression of full-length GPVI was reduced and abolished in GP6het and GP6hom individuals, respectively, as shown by western blotting with an antibody against the cytoplasmic tail of GPVI (Figure 1A). Flow cytometry revealed that surface expression of GPVI was reduced and abolished in GP6het and GP6hom individuals, respectively (not shown), as previously described.3 The level of the FcR γ-chain was reduced by 40% to 65% relative to controls in 3 GP6hom individuals (Figure 1B). The stimulation of whole tyrosine phosphorylation, including Syk (525/526), LAT (200), and PLCγ2 (1217), by collagen was abolished in GP6hom platelets (Figure 1C). The levels of various membrane proteins (GPIbα, GPIIb, and CLEC-2), signaling proteins (Syk, LAT, Btk, RhoA, and PLCγ2), and α-tubulin were similar among control, GP6het, and GP6hom individuals (Figure 1A-E).
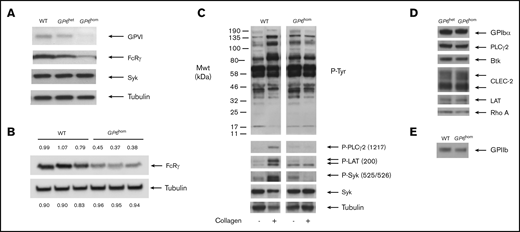
Protein expression and tyrosine phosphorylation in control and GP6homplatelets. (A) Western blot showing levels of GPVI, FcRγ, Syk, and tubulin (loading control) in control, GP6het, and GP6hom individuals. (B) Densitometry reading of the level of FcRγ and tubulin (loading control) in a GP6hom individual relative to a control (the GP6hom individual is not related to the donor in panel A). (C) Collagen (30 μg/mL) stimulation of tyrosine phosphorylation in a control and GP6hom individual (90-second stimulation). (D) Western blot of representative proteins from GP6het and GP6hom donors. (E) Western blot of GPIIb from control and GP6hom donors. Results are representative of 3 (A-C) or 4 (D-E) unrelated GP6hom individuals. Mwt, molecular weight; WT, wild type.
Protein expression and tyrosine phosphorylation in control and GP6homplatelets. (A) Western blot showing levels of GPVI, FcRγ, Syk, and tubulin (loading control) in control, GP6het, and GP6hom individuals. (B) Densitometry reading of the level of FcRγ and tubulin (loading control) in a GP6hom individual relative to a control (the GP6hom individual is not related to the donor in panel A). (C) Collagen (30 μg/mL) stimulation of tyrosine phosphorylation in a control and GP6hom individual (90-second stimulation). (D) Western blot of representative proteins from GP6het and GP6hom donors. (E) Western blot of GPIIb from control and GP6hom donors. Results are representative of 3 (A-C) or 4 (D-E) unrelated GP6hom individuals. Mwt, molecular weight; WT, wild type.
These results confirm the previous reports of reduction and loss of full-length GPVI in GP6het and GP6hom platelets and demonstrate that the level of the FcR γ-chain is also reduced. This decrease was not anticipated, as the level has been reported to be unchanged in GP6hom mouse platelets.16 However, on repeating these studies, we observed a small reduction (∼15%) in GP6hom mice platelets, which may be due to increased degradation (supplemental Figure 1). It is unclear to what extent the decrease in the FcR γ-chain contributes to the clinical phenotype of the patients.
Spreading on collagen, VWF, and glass
Adhesion and spreading were investigated under static conditions on surfaces coated with collagen, VWF, and glass (uncoated), with the latter serving as a representative charged surface. The majority of platelets from controls generated filopodia and lamellipodia on collagen and on glass (Figure 2A-B). The small number of partially and nonspread platelets on both surfaces is likely to represent cells that have recently adhered (Figure 2A). In contrast, only one-third of platelets underwent partial spreading on VWF (Figure 2A). Spreading of GP6hom platelets on collagen and VWF was abolished and markedly reduced on uncoated glass, whereas adhesion was not altered on all 3 surfaces (Figure 2A-B), indicating involvement of additional receptors, such as integrin α2β1 for collagen and GPIb-IX-V and integrin αIIbβ3 for VWF. Spreading, but not adhesion, was reduced in GP6het platelets on all 3 surfaces, although for collagen, this was less marked than for GP6hom platelets (Figure 2B). These results demonstrate that GPVI is essential for spreading, but not adhesion, of platelets on collagen, VWF, and a charged surface.
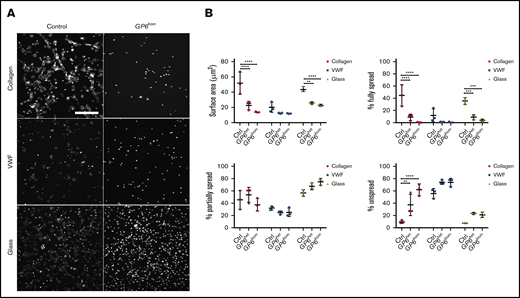
Platelet spreading on collagen, VWF, and glass. (A) Human platelets (2 × 107/mL) were spread on coated and uncoated (not blocked) coverslips as described in “Methods.” The images are representative of 3 control and 3 GP6hom patients. (B) The graphs illustrate quantification of surface area and the percentage of fully, partially spread, or nonspread platelets (control = 3, GP6het = 2, GP6hom = 3). Measurements are the mean of 48 882 platelets in a replicate (7 fields of view per replicate), and the figures are representative of part of a field of view. Significance was measured using 2-way ANOVA. **P < .01, ***P < .001; ****P < .0001. The surface area in the field of view is 213 × 213 μm. Scale bar, 50 μm.
Platelet spreading on collagen, VWF, and glass. (A) Human platelets (2 × 107/mL) were spread on coated and uncoated (not blocked) coverslips as described in “Methods.” The images are representative of 3 control and 3 GP6hom patients. (B) The graphs illustrate quantification of surface area and the percentage of fully, partially spread, or nonspread platelets (control = 3, GP6het = 2, GP6hom = 3). Measurements are the mean of 48 882 platelets in a replicate (7 fields of view per replicate), and the figures are representative of part of a field of view. Significance was measured using 2-way ANOVA. **P < .01, ***P < .001; ****P < .0001. The surface area in the field of view is 213 × 213 μm. Scale bar, 50 μm.
Platelet aggregation under noncoagulating flow conditions
Experiments were undertaken to evaluate the role of GPVI in adhesion and aggregation of noncoagulated blood under arterial shear. Coagulation was inhibited by citrate and PPACK. Whole blood was flowed over collagen and noncollagen (mixture of VWF, laminin, and rhodocytin) surfaces at a shear rate of 1000 s−1, and platelet adhesion and aggregation were analyzed along with contraction score. The contraction score was determined based on a predefined set of example images and depicts the tightness of the aggregate.10,17
Blood from GP6het donors form robust, large aggregates on collagen with a similar surface area coverage and contraction score to that of controls, although PS exposure is reduced by ∼50% (Figure 3A). In contrast, only single platelets adhere and small aggregates form on collagen in GP6hom blood (Figure 3A). This is accompanied by a reduction in abrogation of PS exposure, although adhesion is not altered (Figure 3A). This demonstrates a critical role of GPVI in aggregation and PS exposure, but not in adhesion, on a collagen surface.
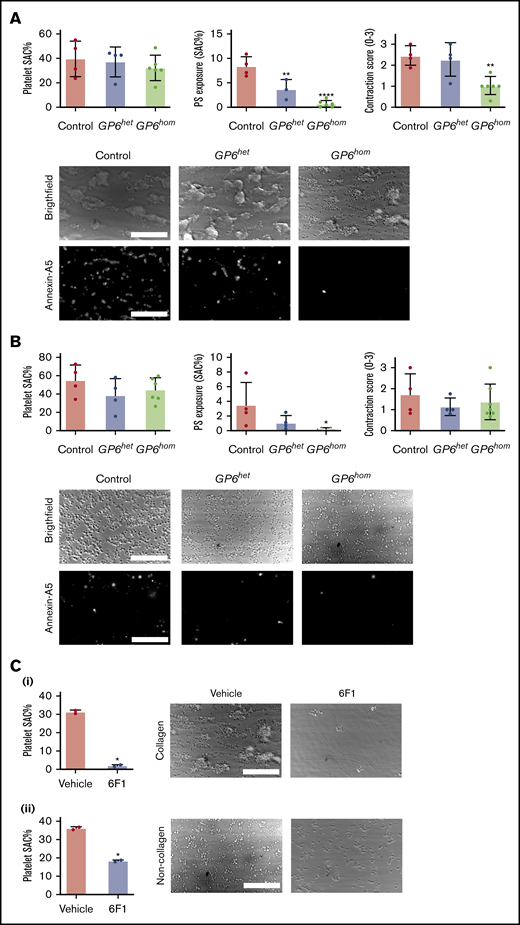
GPVI deficiency leads to abolished PS exposure under flow in the absence of coagulation. Whole blood from control, GP6hom, and GP6het subjects was recalcified in presence of PPACK and perfused over collagen (A) and noncollagen (B) (laminin, VWF, and rhodocytin) surfaces. Surface area coverage (SAC), PS exposure, and contraction score are presented as mean ± SD; control = 4, GP6het = 4, and GP6hom = 6. ****P < .0001, **P < .01, *P < .05. Representative bright-field and Alexa Fluor 647-Annexin A5 images are shown. Images were taken at the end point (8 min) after labeling was performed. (C) Whole blood from GP6hom patients treated with the monoclonal antibody 6F1 (10 μg/mL) was recalcified and perfused over collagen (i) and noncollagen (ii) surfaces. Quantification of surface area coverage is presented as mean ± SD. GP6hom= 2. Representative bright-field images are shown. Scale bars, 50 μm.
GPVI deficiency leads to abolished PS exposure under flow in the absence of coagulation. Whole blood from control, GP6hom, and GP6het subjects was recalcified in presence of PPACK and perfused over collagen (A) and noncollagen (B) (laminin, VWF, and rhodocytin) surfaces. Surface area coverage (SAC), PS exposure, and contraction score are presented as mean ± SD; control = 4, GP6het = 4, and GP6hom = 6. ****P < .0001, **P < .01, *P < .05. Representative bright-field and Alexa Fluor 647-Annexin A5 images are shown. Images were taken at the end point (8 min) after labeling was performed. (C) Whole blood from GP6hom patients treated with the monoclonal antibody 6F1 (10 μg/mL) was recalcified and perfused over collagen (i) and noncollagen (ii) surfaces. Quantification of surface area coverage is presented as mean ± SD. GP6hom= 2. Representative bright-field images are shown. Scale bars, 50 μm.
A similar degree of surface coverage to that on collagen was observed for whole blood on a mixture of VWF, laminin, and rhodocytin (Figure 3B). However, in contrast to the collagen surface, coverage consisted of smaller aggregates and single platelets, resulting in a lower contraction score. The proportion of PS-positive platelets was ∼50% of that observed on collagen (Figure 3B). The surface coverage of blood from GP6het and GP6hom donors was similar to that of controls on the noncollagen surface, although the number of single platelets was increased, notably in GP6hom blood (Figure 3B). The exposure of PS was abrogated on both surfaces (Figure 3B). Thus, GPVI is also important for aggregation and PS exposure, but not adhesion, on a noncollagen surface.
To investigate whether the adhesion of GP6hom platelets on collagen is through the second platelet receptor for collagen, integrin α2β1, experiments were performed using the blocking monoclonal antibody (mAb) 6F1.18 mAb 6F1 abrogated adhesion of GP6hom blood on collagen (Figure 3Ci), but also partially reduced adhesion on the noncollagen surface (Figure 3Cii). Since there are no reports of binding of VWF, laminin, or rhodocytin to integrin α2β1, this may be due to steric hindrance.
These results demonstrate a critical role for GPVI in supporting platelet aggregation and PS exposure in the absence of coagulation on collagen and noncollagen surfaces, with the adhesion of GP6hom platelets on collagen being mediated by integrin α2β1.
Platelet aggregation under coagulating flow conditions
Additional flow studies were performed at the same arterial shear rate (1000 s−1) in the presence of recombinant tissue factor and Ca2+ (without PPACK) to determine the role of GPVI in the presence of thrombin and fibrin. Under these conditions, fibrin strands were seen to develop in control blood from platelet aggregates on collagen (Figure 4), suggesting that they are catalyzed by platelet-dependent coagulation, as described previously.11 In contrast, there was no detectable fibrin formation on the noncollagen surface (not shown), and further studies were not performed on this surface.
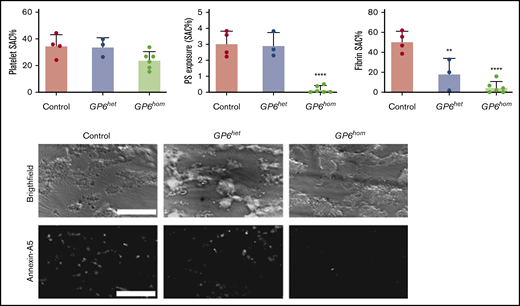
GPVI deficiency leads to abolished PS exposure under flow. Whole blood from control, GP6hom, and GP6het individuals was recalcified and perfused over a collagen surface cocoated with tissue factor. Quantification of surface area coverage (SAC), PS exposure, and fibrin surface area coverage is shown. Data are shown as mean ± SD; control = 4, GP6het = 3, and GP6hom = 6. ****P < .0001, **P < .01. Representative bright-field and Alexa Fluor 647-Annexin A5 images taken after 6 minutes of blood perfusion are shown from a control and a GP6hom subject on a collagen surface. Scale bars, 50 μm.
GPVI deficiency leads to abolished PS exposure under flow. Whole blood from control, GP6hom, and GP6het individuals was recalcified and perfused over a collagen surface cocoated with tissue factor. Quantification of surface area coverage (SAC), PS exposure, and fibrin surface area coverage is shown. Data are shown as mean ± SD; control = 4, GP6het = 3, and GP6hom = 6. ****P < .0001, **P < .01. Representative bright-field and Alexa Fluor 647-Annexin A5 images taken after 6 minutes of blood perfusion are shown from a control and a GP6hom subject on a collagen surface. Scale bars, 50 μm.
Large platelet aggregates/thrombi are formed when control blood is flowed over collagen under coagulating conditions (Figure 4). PS exposure is also observed and was similar in magnitude to that on collagen in the absence of coagulation (compare Figures 3A and 4). A similar degree of thrombus formation was observed with GP6het and GP6hom blood, but fibrin formation was reduced and abolished, respectively (Figure 4). PS was not altered in GP6het blood but was abrogated in GP6hom blood (Figure 4).
These results demonstrate a critical role for GPVI in supporting PS exposure and fibrin formation under coagulating conditions, but not in thrombus formation on collagen at arterial shear.
Thrombin generation is reduced in GP6-deficient platelets
Using the calibrated automated thrombogram (also known as the CAT assay), we observed a trend toward a reduction in thrombin generation in PRP isolated from GP6hom individuals, depicted as a reduced peak height and ETP (Figure 5A-C). However, this trend was not seen in other parameters (eg, time to peak), and the ETP was not significantly different, suggesting that there is only a slight decrease in the total amount of thrombin formed in GP6hom platelets. This partially resembles the results using GPVI-deficient platelets (3 patients with immune thrombocytopenia and 1 compound heterozygote with 2 GPVI variants) and healthy PRP pretreated with GPVI-blocking Fab 9012. Mammadova-Bach et al19 reported a significant decrease in peak height in GPVI-deficient patients, while our results only show a trend toward a decrease. This difference is likely to due to the partial nature of the decrease and the low number of patient samples and the variation in response between donors. A similar level of reduction in thrombin generation was seen in PRP from GP6het relative to GP6hom individuals.
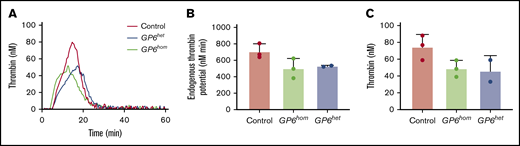
GPVI deficiency leads to a partial reduction in thrombin generation. Thrombin generation assay was performed in platelets from control, GP6het, and GP6hom patients. (A) Thrombin generation curves from a control, GP6het, and GP6hom individual are shown. (B) ETP is shown. (C) Peak height is shown. Data are resented as mean ± SD; control = 3, GP6het = 2, and GP6hom = 3.
GPVI deficiency leads to a partial reduction in thrombin generation. Thrombin generation assay was performed in platelets from control, GP6het, and GP6hom patients. (A) Thrombin generation curves from a control, GP6het, and GP6hom individual are shown. (B) ETP is shown. (C) Peak height is shown. Data are resented as mean ± SD; control = 3, GP6het = 2, and GP6hom = 3.
Frequency of the GP6 c.711_712insA variant in the Chilean population
To investigate the frequency of the c.711_712insA GP6 variant in the Chilean population, sequencing of GP6 exon 6 was performed on 1235 DNA samples. Samples represent mixed Chilean Latinos with Mapuche Native American ancestry, as described previously.5 Of these, 23 samples gave sequencing traces that were of too poor quality to analyze and were excluded. Of the remaining 1212 samples, a total of 36 were found to be heterozygous for the c.711_712insA variant (supplemental Figure 2), equivalent to 2.9% of the total analyzed. No GP6hom samples were identified.
Discussion
This study demonstrates (1) a critical role for GPVI in aggregation and PS exposure, but not adhesion, in human platelets flowed at arteriolar shear over collagen and noncollagen surfaces under noncoagulating conditions; and (2) a critical role for GPVI in PS exposure, but not thrombus formation, on collagen under coagulating conditions. In addition, this study shows that spreading of platelets on collagen, VWF, and uncoated glass, as well as thrombin formation, is reduced or abolished in GP6hom platelets. Together, these results show that the role of GPVI in supporting aggregation at arteriolar shear extends to noncollagen surfaces and that GPVI is critical for PS exposure and supporting thrombin formation.
GPVI has long been recognized as a collagen receptor, but more recently, it has been shown to serve as a receptor for further ligands in the vasculature and vessel wall, including fibrinogen, fibrin, fibronectin, vitronectin, and laminin.1,19-21 In addition, GPVI has been shown to associate with GPIbα in the membrane22 and support thrombus formation and platelet adhesion to immobilized VWF at arterial and venous shear.23 VWF stimulates tyrosine phosphorylation of the FcR γ-chain,24,25 and treatment with an anti-GPVI antibody reduces FcR γ-chain and Syk phosphorylation upon ristocetin stimulation.23 One or more of these interactions could explain the reduction in spreading on VWF and platelet aggregation and PS exposure on a mixture of VWF, laminin, and rhodocytin. Alternatively, the reduction in these responses could be due to the ∼50% reduction in the FcR γ-chain that is seen in GP6hom platelets, and this could be tested in transgenic mice that are heterozygous for the FcR γ-chain. Aggregation to botracetin/VWF is abolished in mouse platelets deficient in FcR γ-chain-deficient but not GPVI-deficient mouse platelets.26
The present study also reports that spreading of GP6hom platelets is reduced on uncoated glass, suggesting that the GPVI is activated by a charge interaction. Previously, we have shown that GPVI can be activated by a diverse group of charged ligands, including diesel exhaust particles, polysulfated sugars such as fucoidan and dextran sulfate, and histones.20 These ligands are structurally distinct and have no resemblance to endogenous ligands, consistent with crosslinking of GPVI and platelet activation being mediated by a charge interaction. The role of GPVI in spreading on glass raises the possibility that other endogenous ligands may activate GPVI through a charge interaction, including ligands from the platelet releasate, and this could potentially also contribute to the reduction in aggregation and PS exposure.
The role of GPVI in supporting platelet aggregation on collagen in the absence of coagulation is well documented.27,28 However, it is only recently that conditions have been developed that permit the study of platelet aggregation on collagen in the absence of thrombin inhibition.11 The observation therefore that GPVI is critical for platelet aggregation and PS exposure under both coagulating and noncoagulating conditions highlights the critical role of the glycoprotein receptor in thrombus formation. The retention of adhesion and formation of small aggregates on the 2 surfaces is explained by the presence of a second receptor for collagen, integrin α2β1, and by receptors for rhodocytin (CLEC-2), laminin (α6β1), and VWF (GPIb-IX-V). The observation that adhesion of GP6hom platelets is retained on collagen is consistent with previous reports of integrin α2β1 supporting platelet adhesion to collagen in human platelets under flow.18,29-33 The retention of adhesion to collagen by integrin α2β1, in combination with the vascular wall damage that leads to the exposure of negatively-charged phospholipid on the cell surface thus leading to tissue factor driven thrombin formation, could account for the relatively minor bleeding phenotype of GP6hom individuals. The role of integrin α2β1 in supporting adhesion of human platelets in the absence of GPVI is in contrast to mice, in which adhesion to collagen is abolished in the absence of GPVI.34 This difference could be due to the affinity of nonactivated integrin α2β1 for collagen, the relative density of GPVI and α2β1 on the platelet surface, or the difference in size of mouse and human platelets.
The present results demonstrate that the degree of platelet PS exposure is governed by the level of GPVI expression, as a marked defect was seen in GP6het and GP6hom individuals under coagulating and noncoagulating conditions. The reduction in platelet procoagulant activity was associated with a slight decrease in thrombin formation. This is in agreement with the results of Mammadova-Bach et al using GPVI-deficient platelets and a GPVI-blocking Fab.19 GPVI signals synergize with Gq family G protein–coupled receptors to induce PS exposure and a procoagulant surface.2,35,36
The potential significance of the present findings should be considered in light of the frequency of the GP6 c.711_712insA in the Chilean population. The prevalence of the variant in the Chilean cohort was estimated with a carrier status probability of 0.0297 (36/1212). If we consider this observation, and that the allele frequency for the c.711_712insA variant is in Hardy-Weinberg equilibrium, then the theoretical prevalence of GP6hom in Chile could be 1/4534 (ie, the product of 0.0297 × 0.0297 with division by 4) or greater. This result, combined with population data from the last national census, suggests that 4079 individuals with GP6hom could be currently living in Chile, which is more than double the number of registered hemophilia patients. With such a high predicted prevalence, the question remains as to why so few patients have been identified. This may due to the limited genetic testing that has been performed in patients with a bleeding disorder and the relatively mild bleeding diathesis caused by loss of GPVI. A bleeding assessment tool (BAT) in GP6hom individuals is shown in supplemental Table 1 and includes an asymptomatic girl (age 12 years) who is the sister of one of the index cases. This suggests that some homozygous carriers of GP6 c.711_712insA do not present with bleeding issues. The majority of GP6het individuals do not have signs of excessive bleeding. This further underscores the interest in targeting GPVI as an antiplatelet therapy.
An important consideration is whether GP6hom individuals are protected from thrombosis. As yet, the number and age of the GP6hom individuals is too few and too low to ascertain whether this is the case. With the relatively high carrier frequency (2.9%), there is a chance that the heterozygous variant is being selected for in the Chilean population. This is unlikely to have conferred a selection advantage in the context of atherosclerosis, as this is a relatively modern disease and relatively late in onset. Whether this has conferred an advantage in other thrombotic-related or other conditions is not known. Larger, longer-term screening of GP6het and GP6hom individuals and their cardiovascular clinical manifestations will help to inform their propensity for bleeding and thrombosis.
For data sharing, e-mail the corresponding author, Steve P. Watson (s.p.watson@bham.ac.uk).
Acknowledgments
The authors thank Barry Coller for the gift of mAb 6F1.
M.N. and J.W.M.H. thank the Interreg V Euregio Meuse-Rhine Program (Poly-Valve) for financial support. S.P.W. holds a British Heart Foundation Chair (CH03/003). E.E.G. receives funding from the National Health and Medical Research Council of Australia and the Australian Research Council. This work was supported by a Birmingham-Maastricht Studentship (G.P.) and by the BHF Accelerator Award (AA/18/2/34218).
Authorship
Contribution: M.N. and G.P. designed and performed experiments, analyzed and interpreted data, and wrote the manuscript; A.D. performed experiments and wrote the manuscript; L.G.Q. performed experiments; J.A.P. designed and performed the spreading analysis and edited the manuscript; N.V.M. designed the genetic analysis and edited the manuscript; E.E.G. provided essential reagents and edited the manuscript; J.W.M.H. edited the manuscript; D.M. diagnosed and recruited patients and edited the manuscript; S.P.W. designed experiments, supervised research, interpreted data, and wrote the manuscript; M.F.B. organized the patient recruitment, coordinated the blood extraction, performed the thrombin generation assay, and participated in the flow cytometry experiments; and J.F.M. and L.A. selected the subjects representative of the Chilean population, drew blood, and extracted leukocyte DNA.
Conflict-of-interest disclosure: J.W.M.H. is a cofounder and shareholder of FlowChamber. The remaining authors declare no competing financial interests.
Correspondence: Steve P. Watson, Institute of Cardiovascular Sciences, IBR Building, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, United Kingdom; e-mail: s.p.watson@bham.ac.uk.

