

Key Points
In-frame VWF deletions have differing impacts on VWF biosynthesis and secretion linked to pseudo-WPB formation and intracellular localization.
Characterization of in-frame deletions highlights new pathogenic mechanisms and an influence of the A3 domain in VWF packaging and processing.

Abstract
Copy number variation (CNV) is known to cause all von Willebrand disease (VWD) types, although the associated pathogenic mechanisms involved have not been extensively studied. Notably, in-frame CNV provides a unique opportunity to investigate how specific von Willebrand factor (VWF) domains influence the processing and packaging of the protein. Using multiplex ligation-dependent probe amplification, this study determined the extent to which CNV contributed to VWD in the Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease cohort, highlighting in-frame deletions of exons 3, 4-5, 32-34, and 33-34. Heterozygous in vitro recombinant VWF expression demonstrated that, although deletion of exons 3, 32-34, and 33-34 all resulted in significant reductions in total VWF (P < .0001, P < .001, and P < .01, respectively), only deletion of exons 3 and 32-34 had a significant impact on VWF secretion (P < .0001). High-resolution microscopy of heterozygous and homozygous deletions confirmed these observations, indicating that deletion of exons 3 and 32-34 severely impaired pseudo-Weibel-Palade body (WPB) formation, whereas deletion of exons 33-34 did not, with this variant still exhibiting pseudo-WPB formation similar to wild-type VWF. In-frame deletions in VWD, therefore, contribute to pathogenesis via moderate or severe defects in VWF biosynthesis and secretion.
Introduction
von Willebrand disease (VWD), a common autosomal inherited bleeding disorder, results from mild or moderate (type 1; VWD1) or severe (type 3; VWD3) quantitative, or functional (type 2; VWD2) deficiency of plasma von Willebrand factor (VWF).1 Accurate identification and characterization of disease-causing variants in VWD are essential to fully understand the molecular mechanisms contributing to pathogenesis.2
Both VWD1 and VWD2 are caused predominantly by heterozygous variants within the VWF gene.3 Historically, the variant screening approaches used in studies of VWD1 and VWD2 were unable to identify heterozygous copy number variation (CNV; large deletions and/or duplications) because polymerase chain reaction (PCR)-based methodologies also amplify the wild-type (WT) allele, masking the presence of heterozygous CNV. Most identified CNV were therefore homozygous and reported in VWD3, resulting in a protein coding frameshift and lack of VWF expression.2,3 Several studies have now used multiplex ligation-dependent probe amplification (MLPA) screening of VWF to identify large homozygous and heterozygous CNV in patients with VWD1-3.4-14 However, to date, the pathogenic mechanisms have only been investigated for 4 of 33 (12%) of identified CNV; deletions of exons 4-5,15,16 26-34,17 and 32-34,18 and an exon 9-10 duplication.12
Using MLPA, this study aimed to determine the prevalence of CNV in a cohort of patients historically diagnosed with VWD1 recruited by the European study Molecular and Clinical Markers for the Diagnosis and Management of Type 1 VWD (MCMDM-1VWD). Subsequently we characterized identified CNV using in vitro expression and high-resolution microscopy, which highlighted that CNV have varying impacts on VWF biosynthesis, storage, and secretion.
Methods
Local ethics review committees at each center approved the study protocol. In accordance with the Declaration of Helsinki, informed consent was obtained from all individuals at recruitment by the individual signing a consent form in their own language.
Study population and phenotypic analysis
Index cases (IC), along with affected and unaffected family members, were recruited as part of the MCMDM-1VWD study as previously described.19 Available phenotypic data for VWF antigen (VWF:Ag), ristocetin cofactor activity (VWF:RCo), collagen binding (VWF:CB), propeptide (VWFpp), factor VIII procoagulant activity (FVIII:C), binding of VWF to FVIII (VWF:FVIIIB), and bleeding score (BS) were measured as previously described.20-24 Multimer analysis of plasma samples was performed using sodium dodecyl sulfate (SDS)–agarose gel electrophoresis as described.25
Generation of VWF CNV expression plasmids
Plasmids expressing VWF CNV (exon 3 deletion, c.56_220del; exon 32-34 deletion, c.5456_5842del; exon 33-34 deletion, c.5621_5842del) were generated in a pCI-neo mammalian expression plasmid (Promega UK Ltd., Southampton, United Kingdom [UK]) containing full-length WT VWF cDNA (pCI-neo-VWF) using a QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies LDA UK Ltd., Stockport, UK). Successful mutagenesis was confirmed via DNA sequence analysis.
In vitro recombinant VWF (rVWF) expression
HEK293 cells were maintained in Dulbecco’s modified Eagle medium containing GlutaMAX supplemented with 10% volume-to-volume ratio (vol/vol) fetal bovine serum (Thermo Fisher Scientific, Paisley, UK). Transient cell transfections with 700 ng/mL WT pCI-neo-VWF or mutant VWF CNV expression plasmid (or 350 ng/mL of both plasmids for 50:50 cotransfections) were performed using either Lipofectamine LTX reagent (Thermo Fisher Scientific) or Xfect (Takara Bio Europe S.A.S, Saint-Germain-en-Laye, France) according to the manufacturer’s recommendations. Cells were maintained at 37°C/5% CO2 for 24 hours before culture media was replaced. Following a further 48-hour incubation, culture media was collected, and the cells were lysed in 300 µL of 1× passive lysis buffer (Promega) containing a protease inhibitor cocktail (Sigma-Aldrich Co. Ltd., Poole, UK).
VWF:Ag levels from cellular supernatants and intracellular lysates were evaluated by enzyme-linked immunosorbent assay (ELISA) using the matched pair VWF:Ag capture and detection antibody kit (Enzyme Research Laboratories Ltd., Swansea, UK). Total VWF:Ag (lysate plus supernatant) was calculated for WT and each mutant and was expressed as a percentage of WT total (WT = 100%). The secreted/retained VWF:Ag ratios (VWF in supernatant/VWF in lysate) were normalized to WT (WT ratio given as 1). VWF multimer analysis on 1.6% medium resolution SDS-agarose gels was performed on cellular supernatants as previously described.25
To determine the ability of the mutants to release VWF after extrinsic stimulation, HEK293 cells were transfected with plasmids as described above, and 72 hours post-transfection the cells were incubated with 160 nM phorbol 12‐myristate 13‐acetate (PMA; Sigma‐Aldrich) or vehicle (dimethyl sulphoxide [DMSO]). The amount of VWF released in 1 hour was determined by the percentage of VWF secreted in medium over the total VWF levels.
Immunocytochemistry and fluorescence microscopy
Microscopy was performed on HEK293 cells 72 hours posttransfection. Cells were fixed using BD Cytofix/Cytoperm (BD Biosciences, Wokingham, UK) at room temperature for 20 minutes, washed 3 times with PBS and 1 time× with 50 mM NH4Cl, and permeabilized with 0.2% vol/vol Triton X-100. Cells were then blocked with 5% vol/vol goat serum (Sigma-Aldrich), washed 3 times with PBS, and incubated at room temperature for 1 hour with primary antibodies at the following concentrations: polyclonal rabbit antihuman VWF at 1:1000 (Dako [UK] Ltd., Ely, UK), mouse antihuman calnexin at 1:500 (Abcam, Cambridge, UK), and sheep antihuman TGN46 at 1:250 (Bio-Rad AbD Serotec Ltd., Kidlington, UK). Following incubation, cells were washed 3 times with 5% vol/vol goat serum and twice with PBS before incubation with 1:500 secondary antibodies (goat antirabbit IgG Alexa Fluor 488, goat antimouse IgG Alexa Fluor 555/568, and donkey antisheep IgG Alexa Fluor 647; Thermo Fisher Scientific) at room temperature for 1 hour. Finally, cells were washed 3 times with 5% vol/vol goat serum and 3 times with PBS before mounting with ProLong Gold Antifade Mountant (Thermo Fisher Scientific).
Widefield images were captured on a Nikon TiE inverted microscope with a 100× oil objective and Andor Zyla scientific complementary metal oxide semiconductor (sCMOS) camera. Image files were processed in Fiji26 to make minor adjustments to contrast and brightness, and are presented as maximum intensity projections.
Structured illumination microscopy (SIM) was carried out using a Deltavision V4 OMX 3D-SIM system fitted with a Blaze module (Applied Precision, GE Healthcare, Issaquah, WA) and a 60 × 1.42 oil planapochromat objective/lens. The system used a standard blue-green-red filter set and used front illuminated sCMOS cameras (2560 × 2160 pixels; pixel size, 6.45 μm; readout speed, 95-286 Mhz; operated at 512 × 512 pixels, 15 bit for 3D-SIM imaging). SIM samples were illuminated using patterned laser illumination at 488 nm. For each z slice, samples were imaged in 5 phase shifts and 3 angles, and z steps were 0.125 nm. Reconstruction and alignment of the subsequent Moiré fringe raw image data were performed in SoftWoRx v6.5.2 (GE Healthcare) using optical transfer functions optimized for the specific wavelength and oil used. All raw images were processed in Fiji.26
Statistical and in silico analysis
Statistical analysis was performed in GraphPad Prism v.7.03 (GraphPad Software Inc., La Jolla, CA). In silico analysis to investigate signal peptide (SP) cleavage was performed using SignalP v4.1.27
Results
Identification of CNV in VWD patients
Of the 150 MCMDM-1VWD IC originally recruited,19 104 were screened for the presence of CNV using MLPA (supplemental Methods), specifically those with no previously identified causative variant or with a variant identified that failed to fully explain the observed clinical phenotype. Analysis identified 6 heterozygous deletions (supplemental Figure 1), specifically of exon 3 (ex3del), exons 4-5 (in 3 IC; ex4-5del), exons 32-34 (ex32-34del), and exons 33-34 (ex33-34del). Table 1 details the phenotypic characteristics of the 6 IC.
Phenotypic details of the 6 IC with identified large VWF deletions
| IC | VWF:RCo, IU/dL | VWF:Ag, IU/dL | RCo/Ag | VWFpp, IU/dL | pp/Ag | VWF:CB, IU/dL | FVIII:C, IU/dL | VWF:FVIIIB | ABO blood group | BS* | Multimers† | Variant | Exons deleted |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1F5III1 | 28 | 28 | 1.00 | 40 | 1.43 | 29 | 70 | 1.12 | O/A | 4 | Normal | c.221-977_532+7059del (p.Asp75_Gly178del) | 4-5 |
| P6F1II1‡ | 23 | 32 | 0.72 | 131 | 4.09 | 15 | 25 | 0.61 | O/O | 4 | 2A(sm) | c.5621-478_5842+2440delins(18)§ (p.Phe1875_Cys1948del) | 33-34 |
| P6F10II1 | 28 | 29 | 0.97 | 58 | 2.00 | 44 | 65 | 1.06 | O/A | 14 | Normal | c.221-977_532+7059del (p.Asp75_Gly178del) | 4-5 |
| P9F3I2 | 11 | 12 | 0.92 | 57 | 4.75 | 12 | 17 | 0.99 | O/A | 6 | 2A(IIE) | c.5455+279_5842+998delins(7)§ (p.Arg1819_Cys1948delinsSer) | 32-34 |
| P9F11II1|| | 20 | 31 | 0.65 | 47 | 1.52 | 31 | 5 | 0.95 | O/O | 10 | Normal | c.56-335_220+1585del (p.Thr20_Gly74del) | 3 |
| P12F9III1 | 27 | 21 | 1.29 | 41 | 1.95 | 30 | 58 | 1.12 | O/O | 8 | Normal | c.221-977_532+7059del (p.Asp75_Gly178del) | 4-5 |
| Normal range¶ | 75-117 | 76-121 | 0.85-1.17 | 104-136 | 1.00-1.50 | 103-150 | 88-133 | 0.68-1.44 | — | — | — | — | — |
| IC | VWF:RCo, IU/dL | VWF:Ag, IU/dL | RCo/Ag | VWFpp, IU/dL | pp/Ag | VWF:CB, IU/dL | FVIII:C, IU/dL | VWF:FVIIIB | ABO blood group | BS* | Multimers† | Variant | Exons deleted |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1F5III1 | 28 | 28 | 1.00 | 40 | 1.43 | 29 | 70 | 1.12 | O/A | 4 | Normal | c.221-977_532+7059del (p.Asp75_Gly178del) | 4-5 |
| P6F1II1‡ | 23 | 32 | 0.72 | 131 | 4.09 | 15 | 25 | 0.61 | O/O | 4 | 2A(sm) | c.5621-478_5842+2440delins(18)§ (p.Phe1875_Cys1948del) | 33-34 |
| P6F10II1 | 28 | 29 | 0.97 | 58 | 2.00 | 44 | 65 | 1.06 | O/A | 14 | Normal | c.221-977_532+7059del (p.Asp75_Gly178del) | 4-5 |
| P9F3I2 | 11 | 12 | 0.92 | 57 | 4.75 | 12 | 17 | 0.99 | O/A | 6 | 2A(IIE) | c.5455+279_5842+998delins(7)§ (p.Arg1819_Cys1948delinsSer) | 32-34 |
| P9F11II1|| | 20 | 31 | 0.65 | 47 | 1.52 | 31 | 5 | 0.95 | O/O | 10 | Normal | c.56-335_220+1585del (p.Thr20_Gly74del) | 3 |
| P12F9III1 | 27 | 21 | 1.29 | 41 | 1.95 | 30 | 58 | 1.12 | O/O | 8 | Normal | c.221-977_532+7059del (p.Asp75_Gly178del) | 4-5 |
| Normal range¶ | 75-117 | 76-121 | 0.85-1.17 | 104-136 | 1.00-1.50 | 103-150 | 88-133 | 0.68-1.44 | — | — | — | — | — |
—, not applicable; pp/Ag, VWFpp/VWF:Ag ratio; RCo/Ag, VWF:RCo/VWF:Ag ratio.
A score ≥4 is indicative of excessive bleeding.23
Based on detailed analysis (P6F1II1 reclassified as VWD2A).25
IC were also heterozygous for c.2561G>A (p.Arg854Gln) in trans.
A detailed description of deletion is provided in supplemental Figure 4.
|IC were also heterozygous for c.8164C>G (p.Pro2722Ala) in cis.
Based on the analysis of healthy control individuals.19,24
The ex4-5del and ex32-34del IC had no previously identified variant. Heterozygous inheritance of c.8164C>G (p.Pro2722Ala) was previously reported in the ex3del IC.19 This variant was originally considered to be pathogenic because it segregated with disease phenotype19 and has a population frequency <0.0001%.28 However in vitro, p.Pro2722Ala had no effect on VWF expression compared with WT (supplemental Methods; supplemental Figure 2). The ex33-34del IC was previously found to be heterozygous for the known VWF variant c.2561G>A (p.Arg854Gln),19 but this variant did not segregate with disease phenotype in the family.
Exact deletion breakpoints (c.221-977_532+7059del) had previously been reported for a characterized ex4-5del shown to occur in VWD1 and VWD3 patients.15,16 The PCR-based assay designed specifically for this deletion15 confirmed that the same ex4-5del occurred in the 3 MCMDM-1VWD IC and segregated with disease phenotype in the respective families (Table 1; supplemental Methods; supplemental Figure 3). The breakpoints for the other identified deletions were mapped within VWF (supplemental Methods), with all 3 deletions shown to be in-frame (Table 1). PCR-based assays for these 3 deletions confirmed that they all segregated with disease phenotype, and additional in silico analysis highlighted the likely mechanisms by which the deletions arose (supplemental Methods; supplemental Figures 3 and 4).
Characterization of heterozygous VWF CNV
The ex4-5del has been characterized extensively in previous in vitro and in vivo studies.15,16 Therefore, in vitro expression (and subsequent high-resolution microscopy) analyses focused on ex3del, ex32-34del, and ex33-34del characterization. ELISA analysis and quantification of supernatants and lysates was used to calculate the total VWF:Ag present. Heterozygous expression of the 3 deletions (mimicking the inheritance pattern observed in the IC) demonstrated that, in comparison with WT, expression of rVWF for ex3del (rVWF3del) and ex32-34del (rVWF32-34del) resulted in a significant decrease in total VWF:Ag (∼60% reduction [P < .0001] and ∼50% reduction [P < .001], respectively; Figure 1A). In comparison, reduction in total VWF observed for the expression of ex33-34del (rVWF33-34del) was milder yet still significant (∼40% reduction [P < .01]; Figure 1A). Despite the reduction in total VWF, the deletions did not result in significantly altered VWF lysate levels compared with WT (P > .05; Figure 1B).
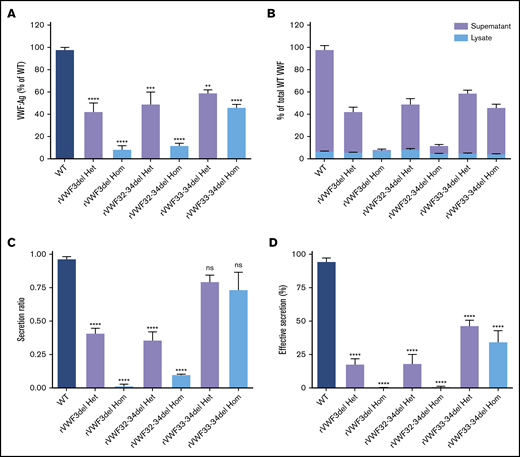
In vitro expression of recombinant in-frame deletions. (A) Quantification of total VWF:Ag as a percentage of WT. (B) Quantification of total VWF:Ag as a percentage of WT (expressed as a proportion of lysate and supernatant). (C) VWF secretion ratios normalized to WT (ratio, 1.0). (D) Effective secretion of VWF normalized to WT (100%). Data were analyzed using 1-way ANOVA and Dunnett’s posttest multiple comparison with WT (**P < .01; ***P < .001; ****P < .0001). Mean values for n = 3 triplicate measurements are shown (bars indicate standard error of the mean). Het, heterozygous; Hom, homozygous; ns, not significant.
In vitro expression of recombinant in-frame deletions. (A) Quantification of total VWF:Ag as a percentage of WT. (B) Quantification of total VWF:Ag as a percentage of WT (expressed as a proportion of lysate and supernatant). (C) VWF secretion ratios normalized to WT (ratio, 1.0). (D) Effective secretion of VWF normalized to WT (100%). Data were analyzed using 1-way ANOVA and Dunnett’s posttest multiple comparison with WT (**P < .01; ***P < .001; ****P < .0001). Mean values for n = 3 triplicate measurements are shown (bars indicate standard error of the mean). Het, heterozygous; Hom, homozygous; ns, not significant.
Quantification of secreted VWF demonstrated that rVWF3del and rVWF32-34del expression also had a profound effect on secretion, resulting in ratios of 0.41 and 0.36, respectively; significant reductions compared with WT (P < .0001; Figure 1C). Additional WT/mutant titration experiments indicated that both affected VWF secretion in a dominant-negative manner (supplemental Figure 5). In contrast, analysis of rVWF33-34del expression revealed that reduction in total VWF was a more significant contributor to reduced VWF levels than a defect in secretion with a ratio of 0.8 (Figure 1C). Given that the expression data indicated an influence of both VWF biosynthesis (total VWF) and secretion, the combined influence of both was considered a more accurate representation of the patient phenotype. Calculation of this effective secretion (secretion ratio × percentage of WT) highlighted significant reductions of ∼80% for both rVWF3del and rVWF32-34del, and a reduction of ∼50% for rVWF33-34del compared with WT (P < .0001; Figure 1D). Secreted multimers displayed a full VWF profile similar to WT for all deletions (supplemental Figure 6), although for rVWF32-34del and rVWF33-34del there was an apparent reduction in intensity of the high molecular weight (HMW) VWF similar to that observed in the respective IC (supplemental Figure 6B,C).
Expression of WT rVWF resulted in the formation of pseudo-Weibel-Palade bodies (WPB) that were clearly visible as elongated VWF-positive structures under widefield fluorescence microscopy, often in close proximity to or emerging from the trans-Golgi network (TGN; Figure 2A-D). Heterozygous rVWF3del expression formed some pseudo-WPB, but these were fewer in number compared with WT, with additional diffuse VWF staining localized to the endoplasmic reticulum (ER; Figure 2E-H). Unlike rVWF3del, rVWF32-34del expression highlighted no diffuse ER localization; however, the pseudo-WPB demonstrated increased clustering around the Golgi/TGN (Figure 2I-L). In contrast to the other deletions, rVWF33-34del expressed pseudo-WPB similar to WT in both appearance and localization (Figure 2M-P).
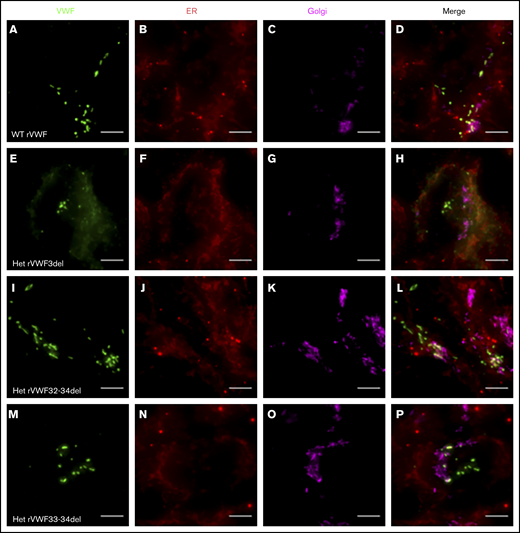
Widefield fluorescence microscopy of recombinant heterozygous in-frame deletions. HEK293 cells were transfected with WT rVWF only (A-D) and WT rVWF with either rVWF3del (E-H), rVWF32-34del (I-L), or rVWF33-34del (M-P) in a 1:1 ratio. Cells were fixed and stained for VWF (green), ER (red), and Golgi (magenta). Scale bars, 5 μm.
Widefield fluorescence microscopy of recombinant heterozygous in-frame deletions. HEK293 cells were transfected with WT rVWF only (A-D) and WT rVWF with either rVWF3del (E-H), rVWF32-34del (I-L), or rVWF33-34del (M-P) in a 1:1 ratio. Cells were fixed and stained for VWF (green), ER (red), and Golgi (magenta). Scale bars, 5 μm.
Characterization of homozygous VWF CNV
Given that the deletions were all in-frame, homozygous expression allowed for their impact on VWF biosynthesis, storage, and secretion to be investigated further in order to gain additional insight regarding potential pathogenic mechanisms. Both rVWF3del and rVWF32-34del expression demonstrated significant effects on total VWF:Ag (>90% and ∼88% reduction, respectively [P < .0001]; Figure 1A) and secreted VWF (ratios 0.016 and 0.1, respectively [P < .0001]; Figure 1C), resulting in a lack of effective secretion (P < .0001; Figure 1D; supplemental Figure 6A-B). Widefield microscopy highlighted that, compared with WT (Figure 3A-D), rVWF3del was unable to form pseudo-WPB and instead produced a diffuse staining pattern highly suggestive of ER retention of VWF (Figure 3E-H). Although rVWF32-34del expression also showed ER-localized VWF staining with no identifiable pseudo-WPB, VWF-positive structures were apparent (Figure 3I-L).
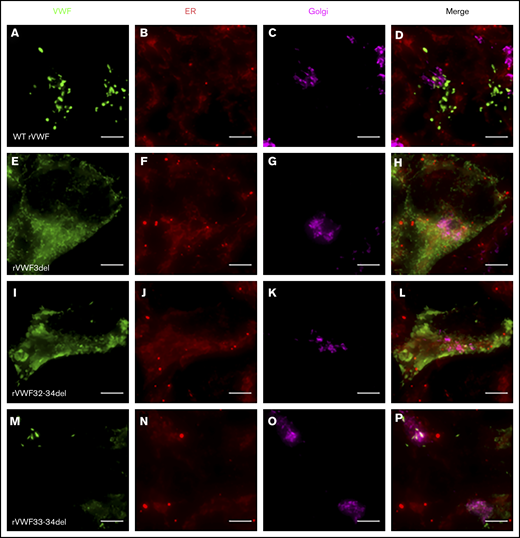
Widefield fluorescence microscopy of recombinant homozygous in-frame deletions. HEK293 cells were transfected with WT rVWF only (A-D) and either rVWF3del (E-H), rVWF32-34del (I-L), or rVWF33-34del (M-P). Cells were fixed and stained for VWF (green), ER (red), and Golgi (magenta). Scale bars, 5 μm.
Widefield fluorescence microscopy of recombinant homozygous in-frame deletions. HEK293 cells were transfected with WT rVWF only (A-D) and either rVWF3del (E-H), rVWF32-34del (I-L), or rVWF33-34del (M-P). Cells were fixed and stained for VWF (green), ER (red), and Golgi (magenta). Scale bars, 5 μm.
Under SIM analysis, WT pseudo-WPB were ∼1 to 2 µm in length with a distinctive hollow appearance and bright VWF staining along the outer membrane (Figure 4A-B). Significant variation in shape and size of these structures was observable, with some appearing as short round structures and others formed in the classic elongated cigar shape. For rVWF3del expression, SIM analysis revealed a network-like structure of VWF, presumably reflecting its location, interspersed within the ER (Figure 4C-D). In contrast, SIM analysis revealed that rVWF32-34del expression formed VWF-positive puncta, more accurately described as VWF aggregates (Figure 4E-F). These were clearly distinct from pseudo-WPB in that they were disordered, random accumulations of VWF, without the characteristic hollow appearance observed in the WT pseudo-WPB (Figure 4B). Instead, the VWF-positive signal exhibited a bright uniform intensity across the entire structure (Figure 4F), likely reflecting the disordered nature of the contained VWF.
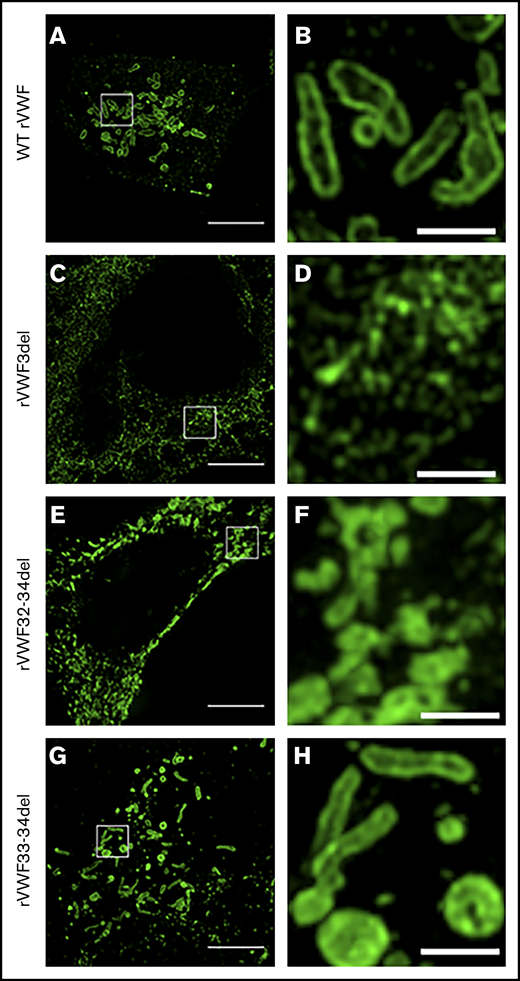
SIM imaging of pseudo-WPB formation in recombinant homozygous in-frame deletions. Expression of WT rVWF produced numerous pseudo-WPB (A), which, on close inspection, had bright outer membrane staining (B). (C) rVWF3del expression produced a diffuse, speckled network-like staining pattern. (D) No large VWF-positive structures were visible. (E) rVWF32-34del expression also produced a more diffuse staining pattern, although with apparent VWF-positive structures. (F) However, the VWF-positive structures displayed uniform staining intensity and nonuniform structure, possibly representing aggregates of VWF oligomers. rVWF33-34del expression was able to form pseudo-WPB like structures (G); however, numerous rounded structures were also visible (H). Scale bars, 5 μm (A,C,E,G) and 1 μm (B,D,F,H). Panels B, D, F, and H are higher magnifications of the region of interest in panels A, C, E, and G (denoted by the white box).
SIM imaging of pseudo-WPB formation in recombinant homozygous in-frame deletions. Expression of WT rVWF produced numerous pseudo-WPB (A), which, on close inspection, had bright outer membrane staining (B). (C) rVWF3del expression produced a diffuse, speckled network-like staining pattern. (D) No large VWF-positive structures were visible. (E) rVWF32-34del expression also produced a more diffuse staining pattern, although with apparent VWF-positive structures. (F) However, the VWF-positive structures displayed uniform staining intensity and nonuniform structure, possibly representing aggregates of VWF oligomers. rVWF33-34del expression was able to form pseudo-WPB like structures (G); however, numerous rounded structures were also visible (H). Scale bars, 5 μm (A,C,E,G) and 1 μm (B,D,F,H). Panels B, D, F, and H are higher magnifications of the region of interest in panels A, C, E, and G (denoted by the white box).
Similar to heterozygous expression, homozygous rVWF33-34del demonstrated a mild reduction in total VWF:Ag (∼53% [P < .0001]; Figure 1A), but very little impact on secretion (ratio, 0.73; Figure 1C). Microscopy revealed that rVWF33-34del produced pseudo-WPB that appeared very similar to WT (Figures 3M-P and 4G-H). Although a number of smaller and rounder pseudo-WPB structures were present, similar structures were present in the WT. Therefore, it is unclear if rVWF33-34del pseudo-WPB displayed any significant morphological differences or aberrations compared with WT. The level of ER-localized VWF was also comparable to that observed in WT.
Stimulated release of VWF
The ability of the mutants to release VWF after external stimulation of transfected HEK293 cells with PMA was investigated. There was a clear reduction in the ability to secrete VWF after stimulation in cells transfected solely with mutant VWF (Figure 5); however, this was diminished when cells were cotransfected with both WT and mutant VWF to mimic the heterozygote state (Figure 5).
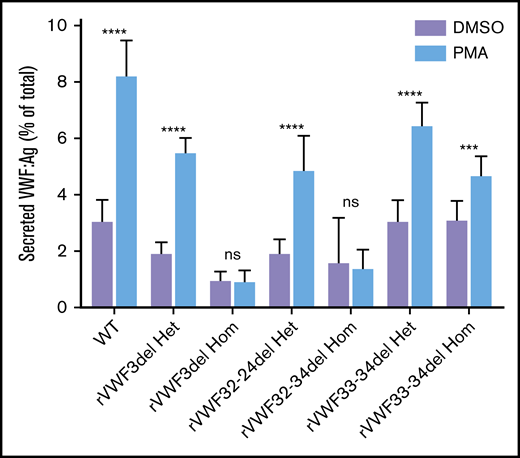
Stimulated secretion of in vitro-expressed rVWF. Data shows VWF:Ag measured in supernatant divided by total VWF (lysate plus supernatant) following 1 hour of stimulation with PMA or vehicle (DMSO). Data were analyzed using Student t test; DMSO versus PMA (***P < .001; ****P < .0001). Mean values for n = 3 triplicate measurements are shown (bars indicate standard deviation).
Stimulated secretion of in vitro-expressed rVWF. Data shows VWF:Ag measured in supernatant divided by total VWF (lysate plus supernatant) following 1 hour of stimulation with PMA or vehicle (DMSO). Data were analyzed using Student t test; DMSO versus PMA (***P < .001; ****P < .0001). Mean values for n = 3 triplicate measurements are shown (bars indicate standard deviation).
Ex3del disrupts VWF SP cleavage
The loss of 55 amino acids in ex3del includes 4 residues from the C terminus of the VWF SP, which encompasses the SP cleavage site. This region exhibits significant interspecies conservation around the cleavage site (Figure 6A); suggesting that changes to this specific amino acid motif may disrupt signal peptidase (SPase)–dependent cleavage. In order to determine what impact this may have on SP cleavage, further in silico analysis was performed. The first 40 residues of WT and ex3del VWF were analyzed. In WT sequence, predicted cleavage after p.Cys22 agreed with the published VWF SP cleavage site.29 This was indicated by a steep drop in the SP score slope (S-score) after p.Cys22, a high cleavage score (C-score), and a peak in the combined cleavage score (Y-score) at p.Ala23 (Figure 6B). Analysis of ex3del also showed a similarly high S-score, indicative of a true SP, which dropped off sharply but was staggered across residues p.Gly19_Phe21 (Figure 6C). However, the C-score and Y-score were both low, with no clear indication of a functional cleavage site (Figure 6C).
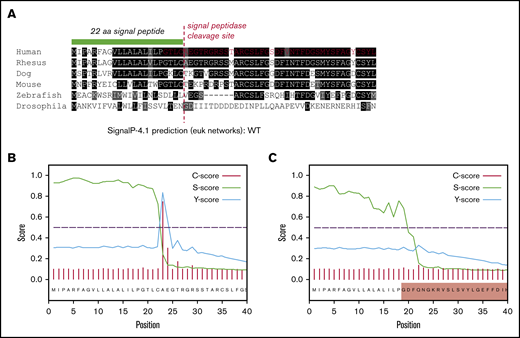
In silico analysis of VWF SP cleavage. (A) Multiple sequence alignment showing the first 40 amino acids of VWF including the 22 residue SP (green bar). Conserved residues are in shaded boxes. The red dashed line marks the SPase cleavage site and the red residues in the human alignment mark the start of ex3del. Note the conservation of the ValLeuLeuAlaLeuAlaLeu hydrophobic motif, Cys prior to the cleavage site, Thr 3 residues prior to the cleavage site, and small residues such as Ala, Thr, and Gly after the cleavage site. (B) SP and cleavage site prediction output for WT VWF. (C) SP and cleavage site prediction output for ex3del VWF. The shaded red box indicates residues in exon 4.
In silico analysis of VWF SP cleavage. (A) Multiple sequence alignment showing the first 40 amino acids of VWF including the 22 residue SP (green bar). Conserved residues are in shaded boxes. The red dashed line marks the SPase cleavage site and the red residues in the human alignment mark the start of ex3del. Note the conservation of the ValLeuLeuAlaLeuAlaLeu hydrophobic motif, Cys prior to the cleavage site, Thr 3 residues prior to the cleavage site, and small residues such as Ala, Thr, and Gly after the cleavage site. (B) SP and cleavage site prediction output for WT VWF. (C) SP and cleavage site prediction output for ex3del VWF. The shaded red box indicates residues in exon 4.
Discussion
This study aimed to investigate the contribution of CNV to disease phenotype within the MCMDM-1VWD cohort. MLPA analysis identified heterozygous deletions in 6 IC, which tracked with disease phenotype in respective families. Therefore, CNV accounts for the cause of VWD in ∼4% of the MCMDM-1VWD IC, similar to the ∼6% contribution previously reported in French VWD1 IC,13 but lower than the ∼12% contribution recently reported in Czech VWD1 IC.14 In the remaining 98 IC in which no CNV was identified, deep intronic variation or alternative disease mechanisms (eg, disruption of microRNA motifs or protein folding) may contribute to clinical phenotype.2
Four of the IC with CNV had no previously identified causal variant.19 Three of the 4 IC were shown to have the previously identified and characterized ex4-5del known to cause both VWD1 and VWD315 ; the remaining IC had ex32-34del. In addition to being previously reported in French VWD1 IC,13 a de novo ex32-34del has been reported and characterized in an Italian IC with unspecified VWD.18 Unlike ex4-5del IC, which had the same deletion breakpoints as previously reported, the ex32-34del IC had a unique breakpoint to the Italian IC; c.5455+279_5842+998delins(7) (∼5.2 kb) and c.5456-66_5842+1316del (∼3.4 kb),18 respectively. This is not surprising because this study indicates that ex32-34del likely results from nonhomologous end joining (NHEJ; as opposed to Alu-mediated homologous recombination [AMHR] for ex4-5del15 ), suggesting that ex32-34del breakpoints are likely to occur randomly.30 However, the phenotype of both characterized ex32-34del IC is very similar, including the slight increase in VWF clearance and abnormal multimer profile.18
The remaining 2 IC both had previously reported variants. In 1, a novel ex3del occurred in cis with p.Pro2722Ala, but given that p.Pro2722Ala had no effect on in vitro VWF expression, it is likely that ex3del is the actual cause of VWD1 in this IC. Like ex4-5del, ex3del results from AMHR, suggesting that deletion breakpoints would be conserved in other VWD patients with the same deletion. The remaining IC had an ex33-34del occurring in trans with p.Arg854Gln. The ex33-34del has been previously reported in both VWD1 and VWD2,6,7,13 but has not been characterized. In this study, NHEJ was the likely mechanism for this deletion, suggesting that ex33-34del breakpoints are also likely to be random,30 similar to ex32-34del. However, as observed with ex32-34del, clinical phenotype is likely consistent for all carriers of this deletion, with the IC reported in this study reclassified as VWD2A on the basis of their abnormal multimer profile.25 Although the clinical phenotype in this IC is explained primarily by ex33-34del, coinheritance of p.Arg854Gln probably accounts for the observed reduction in VWF:FVIIIB. As such, both ex3del and ex33-34del IC highlight the importance of CNV screening, where previously identified genetic variation does not fully explain clinical phenotype.
Heterozygous in vitro expression of the identified deletions all demonstrated significant reductions in total VWF:Ag ranging from ∼40% to ∼60%, suggesting a reduction in VWF biosynthesis. Although not widely reported in VWD, previous rVWF expression of variants p.Arg1205His and p.Tyr1584Cys in HEK293T cells also demonstrated reduced VWF biosynthesis,31 as has rVWF expression of p.Leu1696Arg and p.Pro1824His (although only in COS-7 cells, which do not form pseudo-WPB).32 An effect on VWF biosynthesis could occur at the level of transcription or translation. Historically, a reported reduction in VWF mRNA levels in endothelial cells (EC) from VWD1 patients,33 and more recently in EC from VWD1 patients with known variants (including ex4-5del and p.Tyr1584Cys),16 would suggest that it occurs at the level of transcription.
Despite the reduction in total VWF:Ag, there was no significant difference in VWF lysate levels, similar to observations made from rVWF expression of p.Arg1205His and p.Tyr1584Cys.31 A more accurate assessment of intracellular retention and reduced secretion is to calculate the secretion ratio (VWF in supernatant/VWF in lysate) for each mutant and WT. In this study, both heterozygous expression of rVWF3del and rVWF32-34del demonstrated significant reductions in secretion (titration experiments confirming that both have a dominant-negative affect on VWF), whereas heterozygous rVWF33-34del expression resulted in only a mild secretion defect. In support of this observation, widefield microscopy revealed fewer pseudo-WPB/ER localization and increased Golgi/TGN localization for rVWF3del and rVWF32-34del, respectively, compared with normal pseudo-WPB formation/localization for rVWF33-34del. This suggests that the efficient processing of rVWF3del and rVWF32-34del is impaired; further supported by the reduced HMW multimers observed for rVWF32-34del compared with WT rVWF. Although this reduction in HMW multimers was more pronounced in the IC-derived plasma sample, this is likely because of in vitro-expressed VWF in HEK293 cells not fully undergoing the same complex or regulated storage and secretion processes and flow conditions that VWF experiences in vivo.34
In vitro expression therefore suggests that these deletions influence VWF production via a combined influence on biosynthesis and secretion. On the basis of the effective secretion (secretion ratio × percentage of WT), both rVWF3del and rVWF32-34del reduced VWF levels by ∼80%, which is not dissimilar to the reduction in plasma VWF observed in the respective IC when compared against plasma levels in unaffected family members (P9F11 [ex3del], ∼75% reduction; P9F3 [ex32-34del], ∼90% reduction). However, effective secretion for rVWF33-34del, indicating a ∼50% reduction, is still not as pronounced as that observed in the IC (∼70%). Although effective secretion in vitro more accurately reflects patient phenotype, it also further highlights that other factors influencing VWF levels in vivo (eg, flow) cannot be fully captured using a HEK293 cell model.
This study also performed microscopic characterization of homozygous VWF storage and pseudo-WPB formation to gain further insight regarding the impact of in-frame deletions. To date, few studies have explored storage impairment caused by pathogenic CNV; those that have, primarily focused on VWD2.12,16,17 In addition to standard widefield microscopy, use of SIM enabled high-resolution morphological analysis of pseudo-WPB. SIM uses spatially structured or patterned illumination and computational reconstruction to overcome the diffraction limitations of conventional light microscopy, resulting in a twofold resolution enhancement in both the lateral and axial dimensions.35 Although this technology has been used to image WPB in EC,36 it has not been used to characterize WPB morphology in VWD. In this study, ex3del, ex32-34del, and ex33-34del all displayed distinct microscopic phenotypes that highlighted differences in their pathological mechanisms.
Both widefield and SIM imaging highlighted a lack of pseudo-WPB formation following rVWF3del expression, with VWF staining targeted to the ER. Previous studies have attributed ER retention to disruption of key regions in the VWF D1 and D2 domains essential for the trafficking of VWF dimers to the Golgi37 or for forming a transient covalent bond with the D3 domain in the ER prior to Golgi multimerization.38 Although these regions are disrupted by ex4-5del, which also results in ER retention,16 they are not disrupted by ex3del. Instead, in silico analysis indicated that ex3del disrupts the C-terminus of the VWF SP, which targets nascent VWF monomers to the ER membrane before its cleavage by SPase. A SP consists of 3 core regions; the nonvariable region adjacent to the SP cleavage site (−1 to −5 amino acids upstream) contains specific small uncharged residues (eg, alanine or glycine) at −1 and −3 that define the cleavage site.39 ex3del results in the loss of 4 of 5 amino acids in this region, which suggests that, although the VWF SP could still target the ER, SP cleavage would be unlikely. In the heterozygous state, the ex3del allele is likely to remain anchored to the ER (effectively acting as a null allele), although it may also disrupt the processing and targeting of the WT allele. The ex3del, therefore, may represent a novel pathogenic mechanism in VWD1.
Despite similarities between rVWF3del and rVWF32-34del expression, microscopy indicated that ex32-34del formed VWF aggregates in the ER. The ex32-34del extends into the VWF A3 domain; characterization of VWF variants in the A domains (eg, p.Val1822Gly within the A3 domain) suggests they impair VWF storage and result in significant ER retention.40 In addition, reduced or lack of desmopressin response associated with VWD1 variants in the A3 domain suggests that this region is essential for the biogenesis of functional WPB.41 The A1 domain appears to play a role in stabilizing VWF tubules42 and the A3 domain could potentially perform a similar structural role. It is also possible that loss of the A3 domain leads to severe misfolding and accumulation of mutant VWF within the ER, leading to the VWF aggregations observed. In some instances, the observation of large VWF-positive compartments or vacuoles possibly reflects the dilated nature of the ER in these mutants. ER dilation is a common observation in mutant proteins retained in the ER, including a number of VWF mutants that disrupt cysteine residues involved in the formation of intrachain disulfide bonds.43
The fact that rVWF33-34del expression formed similar if not identical pseudo-WPB to WT reflects the relatively mild affect this deletion had on VWF multimers and secretion in vitro. This also supports the suggestion that the A3 domain is important for WPB formation, as this domain remains following ex33-34del. Therefore, in vitro expression data for ex33-34del suggested that decreased VWF biosynthesis is the most significant contributor to reduced VWF levels. Although it is not clear how this variant causes a reduction in VWF levels, despite forming WPB, this process may be less efficient. Removal of exons 33-34 would likely result in disruption to intrachain disulfide bonds and VWF misfolding, potentially compromising packaging and tubulation within WPB. The relative intensity of VWF antibody staining can potentially act as a measure of VWF tubulation and packaging within WPB44 ; disruption of VWF folding and the intraorganelle structure leads to a less compact state and provides an increased area of VWF to which the antibodies can bind. Despite the morphological similarities between pseudo-WPB formed by rVWF33-34del and WT, numerous small round VWF-positive structures were also found in rVWF33-34del. Interestingly, these rounded structures appeared to have a more uniform fluorescence signal, perhaps suggesting disordered VWF tubule formation and, therefore, a reduction in density. The reduced HMW multimers observed for rVWF33-34del and the corresponding IC supports this theory.
In summary, this study has provided further insight and understanding of the genetic mechanisms of VWD through identification and characterization of in-frame VWF deletions. Furthermore, in vitro expression and high-resolution microscopy indicates that in-frame deletions lead to moderate and severe defects in VWF biosynthesis and/or secretion that mimics the observed clinical phenotype.
Send data sharing requests via e-mail to the corresponding author, Daniel J. Hampshire (d.hampshire@hull.ac.uk).
Acknowledgments
The authors thank C. G. Walther for technical assistance with the 3D-SIM imaging, performed in the Wolfson Light Microscopy Facility, University of Sheffield (supported by UK Medical Research Council grant MR/K015753/1). The authors thank R. N. C. P. Vijzelaar (MRC-Holland b.v., Amsterdam, The Netherlands) for kindly providing the VWF-specific MLPA kits, M. L. Bowman (Queen’s University, Kingston, ON, Canada) for kindly providing the pCI-neo-VWF plasmid, and K. J. De Vos (University of Sheffield, Sheffield, United Kingdom) for kindly providing the HEK293 cells.
This study was supported by the UK Medical Research Council (A.C.) and the National Institutes of Health, National Heart, Lung, and Blood Institute program project grant (HL081588: Zimmerman Program for the Molecular and Clinical Biology of von Willebrand Disease) (S.J.W., I.R.P., A.C.G., and D.J.H.).
Authorship
Contribution: I.R.P., A.C.G., and D.J.H. initiated the study; A.C.G. and D.J.H. coordinated the study; A.C., S.J.W., and D.J.H. designed the study and wrote the manuscript; A.C., S.J.W., A.d.J., and D.J.H. analyzed and interpreted the data; A.C., S.J.W., A.d.J., R.J.D., L.D.S.B., A.M.A.-B., U.B., C.H., J.G., J.C.J.E., and D.J.H. generated the data; S.J.W. performed the microscopy; D.H. and J.C.J.E. recruited families; S.J.W. and D.J.H. revised the paper; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the European Group on von Willebrand Disease and Zimmerman Program for the Molecular and Clinical Biology of von Willebrand Disease Study Groups appears in “Appendix.”
Correspondence: Daniel J. Hampshire, Department of Biomedical Sciences, Faculty of Health Sciences, University of Hull, Cottingham Rd, Hull HU6 7RX, United Kingdom; e-mail: d.hampshire@hull.ac.uk.
Appendix: study group members
The members of the European Group on von Willebrand Disease Study Group are: J. Batlle (A Coruña, Spain), E. Berntorp (Malmö, Sweden), I. Bodó (Budapest, Hungary), U.B. (Hamburg, Germany), G. Castaman (Florence, Italy), J.C.J.E. (Leiden, The Netherlands), A. B. Federici (Milan, Italy), A. Gadisseur (Antwerp, Belgium), A.C.G. (Sheffield, United Kingdom), J.G. (Lille, France), M. Laffan (London, United Kingdom), F. Leebeek (Rotterdam, The Netherlands), P. M. Mannucci (Milan, Italy), I.R.P. (Sheffield, United Kingdom), F. Peyvandi (Milan, Italy), F. Rodeghiero (Vicenza, Italy), R. Schneppenheim (Hamburg, Germany), A. Tosetto (Vicenza, Italy), and A. Veyradier (Paris, France). The members of the Zimmerman Program for the Molecular and Clinical Biology of von Willebrand Disease Study Group are: R. Montgomery and S. Haberichter (Milwaukee, WI), D. Lillicrap and P. James (Kingston, ON, Canada), and A.C.G., I.R.P., and D.J.H. (Sheffield, United Kingdom).

