

Key Points
Platelet-like particles are able to leave circulation and infiltrate the bone marrow in vivo.
Platelet-derived extracellular vesicles bind to and functionally alter hematopoietic cells in vitro to restore megakaryopoiesis.

Abstract
During inflammation, steady-state hematopoiesis switches to emergency hematopoiesis to repopulate myeloid cells, with a bias toward the megakaryocytic lineage. Soluble inflammatory cues are thought to be largely responsible for these alterations. However, how these plasma factors rapidly alter the bone marrow (BM) is not understood. Inflammation also drives platelet activation, causing the release of platelet-derived extracellular vesicles (PEVs), which package diverse cargo and reprogram target cells. We hypothesized that PEVs infiltrate the BM, providing a direct mode of communication between the plasma and BM environments. We transfused fluorescent, wild-type (MPL+) platelets into recipient cMpl−/−mice before triggering systemic inflammation. Twenty hours postinfusion, we observed significant infiltration of donor platelet-derived particles in the BM, which we tracked immunophenotypically (MPL+ immunohistochemistry staining) and quantified by flow cytometry. To determine if this phenomenon relates to humans, we extensively characterized both megakaryocyte-derived and PEVs generated in vitro and in vivo, and found enrichment of extracellular vesicles in bone marrow compared with autologous peripheral blood. Last, BM from cMpl−/− mice was cultured in the presence or absence of wild-type (MPL+) PEVs. After 72 hours, flow cytometry revealed increased megakaryocytes only in cultures with added PEVs. The majority of CD41+ cells were bound to PEVs, suggesting a PEV-mediated rescue of megakaryopoiesis. In conclusion, we report for the first time that plasma-residing PEVs infiltrate the BM. Further, PEVs interact with BM cells in vivo and in vitro, causing functional reprogramming that may represent a novel model of inflammation-induced hematopoiesis.
Introduction
During inflammation, steady-state hematopoiesis switches to emergency hematopoiesis to repopulate myeloid cells, with a bias toward the megakaryocyte (MK) lineage.1-4 MKs are derived from hematopoietic stem cells (HSCs) that reside mainly in the bone marrow (BM) and are responsible for platelet production.5 During many inflammatory states, platelet production becomes dysregulated and can present as thrombocytopenia or thrombocytosis.2 These changes can occur rapidly (within hours), and are therefore independent of thrombopoietin (TPO), the cytokine that directs HSCs to differentiate into MKs over ∼5 to 7 days. In conditions that present with thrombocytosis, inflammatory cues, such as circulating cytokines and chemokines (ie, interleukin-6 and CCL5), are reported to be largely responsible for regulating platelet production during hematopoietic stress.6,7 However, how these inflammatory signals rapidly infiltrate the BM in concentrations potent enough to stimulate hematopoietic cells to divide and differentiate remains poorly understood.
In addition to enhanced circulating cytokine levels, inflammation also results in constitutive platelet activation that exacerbates disease pathology. Platelet activation results in the secretion of pro-inflammatory mediators, including platelet-derived extracellular vesicles (PEVs, reviewed in Mause and Weber8 ). PEVs are small (0.1-1 μm), heterogenous vesicles released from the surface of platelets. Although the majority of circulating extracellular vesicles (EVs; ∼80%) in plasma from healthy people contain CD41 on their surface (CD41+),9,10 there is debate over what proportion arises from platelets vs MKs.11 Generally speaking, CD41+ EVs that also express activation markers, such as P-selectin (CD62P) and phosphatidylserine (PS), are considered to have been derived from activated platelets and are therefore a biomarker of pathological platelet activation. In support of this, the number of CD41+/CD62P+ EVs dramatically increases in conditions with chronic inflammation and ongoing platelet activation, including sepsis,12,13 cardiovascular disease,14,15 diabetes,16 cancer,17 and autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis.18-20 In addition to increased abundance, PEVs play a direct pathologic and deleterious role in numerous disease states such as SLE,21,22 arthritis,23 atherosclerosis,24,25 and cancer.26,27
PEVs are well-established regulators of intracellular communication (reviewed in Mause and Weber8 ). Like their MK counterparts, PEVs contain diverse cargo such as microRNAs, cytokines, and organelles like mitochondria.24,28-30 Both PEVs and MK-derived EVs (MkEVs) can transfer their cargo to alter the function of recipient cells. They exert their influence on these cells through specific modes of cell–EV interactions, including surface receptor signaling, plasma membrane fusion, and internalization.31,32 PEVs can functionally reprogram macrophages and endothelial cells.33-36 In 1 study, the fusion of PEVs to target cell membranes transferred the surface protein CXCR4 to null recipient cells, causing these previously inaccessible cells to be susceptible to HIV infection.37 Appropriation of GPIbα from PEVs to monocytes was also recently described as a major player in the formation of leukocyte aggregates that propagate inflammatory thrombosis.38 MkEVs are far less studied than PEVs; however, there is a growing body of evidence showing their ability to communicate within the BM microenvironment by reprogramming HSCs to stimulate megakaryopoiesis in vitro and in vivo.39-41 In culture, MKs constitutively shed EVs, distinct from proplatelets.11 Currently, there is no evidence suggesting that MkEVs are pathogenic.
Previous studies have examined how PEVs affect cells within or in contact with circulation8 and how MkEVs communicate within the BM niche.42 In this manuscript, we examine the novel hypothesis that PEVs generated via activated platelets can travel between the circulation and BM environment, carrying and delivering cargo directly from the plasma to the BM. Our data reveal that PEVs can penetrate the BM space and directly interact with cells residing in the BM, including MKs and MK-precursor cells. We propose that PEVs are a previously unidentified contributor to alterations in the BM microenvironment during conditions with ongoing platelet activation, such as thrombo-inflammation. This may represent a novel mode of communication for delivering alterations in the plasma milieu directly into the BM, allowing it to rapidly sense and respond to pathologic changes.
Materials and methods
Nomenclature
Throughout this manuscript, the term “extracellular vesicle” is used to describe heterogeneous particles that are derived from the plasma membrane (often referred to as microvesicles or microparticles). There are some instances where we were experimentally unable to distinguish between extracellular vesicles (50-1500 nm in diameter) and platelets (1000-2000 nm) by size in vivo because of sensitivity limitations of currently availability technology. In those instances, we used the term “platelet-like particles.”
Sample collection
Blood collection.
Human whole blood was collected via venipuncture from healthy donors after obtaining written informed consent. Blood was drawn into citrate (3.2% w/v) vacutainers using a 21-gauge needle. For mouse blood, mice were anesthetized with ketamine/xylazine (87.5 mg/kg ketamine, 12.5 mg/kg xylazine, administered by intraperitoneal injection) and whole blood was collected from the inferior vena cava into syringes containing heparin (20 U/mL).
Patient blood and autologous bone marrow.
Blood was collected from 10 patients in remission from acute lymphoblastic leukemia after obtaining informed consent according to the guidelines of the Dmitry Rogachev National Medical Research Center of Pediatric. Patient characteristics are shown in Table 1. Peripheral blood was drawn from an antecubital vein using a 22-gauge needle and collected in 4.9-mL K3EDTA S-Monovette tubes (Sarstedt AG & Co. KG, Nümbrecht, Germany), and BM aspirates were collected according to the hospital protocol under general anesthesia. BM was collected in 2.6 mL K3EDTA S-Monovette tubes. The preparation of platelet free plasma samples was started less than 1 hour after blood collection. From each sample, 1 mL of blood or BM was centrifuged twice at 2500g for 15 minutes at 20°C (Rotina 420 R; Hettich, Tuttlingen, Germany) to pellet cells.
Characteristics of patients for experiments performed at the Dmitry Rogachev National Medical Research Center of Pediatric Hematology (Figure 3)
| Patient no. | Age, y | Sex | PLT count | Diagnosis | Therapy regimen |
|---|---|---|---|---|---|
| 1 | 13 | Female | 201 | ALL | Cytarabine, dexamethasone, methotrexate |
| 2 | 7 | Male | 105 | ALL | Cytarabine, dexamethasone, methotrexate |
| 3 | 6 | Female | 356 | ALL | Cytarabine, dexamethasone, methotrexate |
| 4 | 9 | Male | 415 | ALL | Cytarabine, dexamethasone, methotrexate |
| 5 | 3 | Male | 421 | ALL | Cytarabine, dexamethasone, methotrexate |
| 6 | 4 | Female | 103 | ALL | Cytarabine, dexamethasone, methotrexate |
| 7 | 3 | Male | 293 | ALL | Cytarabine, dexamethasone, methotrexate |
| 8 | 3 | Female | 489 | ALL | Cytarabine, dexamethasone, methotrexate |
| 9 | 1 | Male | 280 | ALL | Cytarabine, dexamethasone, methotrexate |
| 10 | 13 | Female | 198 | ALL | Cytarabine, dexamethasone, methotrexate |
| Patient no. | Age, y | Sex | PLT count | Diagnosis | Therapy regimen |
|---|---|---|---|---|---|
| 1 | 13 | Female | 201 | ALL | Cytarabine, dexamethasone, methotrexate |
| 2 | 7 | Male | 105 | ALL | Cytarabine, dexamethasone, methotrexate |
| 3 | 6 | Female | 356 | ALL | Cytarabine, dexamethasone, methotrexate |
| 4 | 9 | Male | 415 | ALL | Cytarabine, dexamethasone, methotrexate |
| 5 | 3 | Male | 421 | ALL | Cytarabine, dexamethasone, methotrexate |
| 6 | 4 | Female | 103 | ALL | Cytarabine, dexamethasone, methotrexate |
| 7 | 3 | Male | 293 | ALL | Cytarabine, dexamethasone, methotrexate |
| 8 | 3 | Female | 489 | ALL | Cytarabine, dexamethasone, methotrexate |
| 9 | 1 | Male | 280 | ALL | Cytarabine, dexamethasone, methotrexate |
| 10 | 13 | Female | 198 | ALL | Cytarabine, dexamethasone, methotrexate |
Samples from bone marrow aspirates and autologous blood were taken from each patient, who were undergoing bone marrow biopsy to confirm remission from ALL.
ALL, acute lymphoblastic leukemia; PLT, platelet.
Generation and characterization of platelet and MkEVs
EV preparations were isolated and characterized according to the guidelines of the International Society for Extracellular Vesicles,43 International Society of Thrombosis and Hemostasis,44 and recently outlined by Coumans and colleagues.45 MkEVs were collected from media containing mature, cultured MKs 24 hours after MK isolation and purification. PEVs were generated as previously described.46 Isolated platelets were stimulated with 1 or a combination of the following: thrombin (0.1 U/mL), collagen (1 μg/mL), calcium ionophore A23187 (25 μM), and lipopolysaccharide (LPS, 5 μg/mL) (Sigma-Aldrich, St. Louis, MO) for 2 hours at 37°C.
Detection and quantification of platelet-like particles in murine BM
c-Mpl−/− mice were transfused with CMPTX-labeled wild-type (MPL+) platelets (2 donors per recipient, 4 × 108 total platelets) and injected with LPS (5 μg/g) or saline vehicle control. Mice were euthanized 20 hours postinflammatory trigger and perfused with phosphate-buffered saline to replace blood volume. Legs were dissected and BM flushed using a 21-gauge needle into CATCH buffer (1× phosphate-buffered saline, 1.5% fetal calf serum, 1 mM adenosine, 2 mM theophylline, and 0.38% sodium citrate). Isolated BM was homogenized and filtered through a 100-μm filter. Cells were pelleted via centrifugation (1000g, 5 minutes), and supernatant containing unbound vesicles was removed. Cell pellets were resuspended in CATCH buffer and stained with anti-CD41 for 1 hour on ice. Cells from whole BM were analyzed by flow cytometry to determine the presence of donor-derived, CMPTX-positive events. One femur per mouse was kept intact and fixed with 4% paraformaldehyde, embedded, sectioned, and stained for MPL by immunohistochemistry (IHC) to visualize platelet-like particle infiltration. In some experiments, BM was sorted for CMPTX-positive cells, which were then fixed and placed on coverslips for immunofluorescence analysis.
To mimic an autoimmune-mediated inflammatory response, FcγRIIATGNCD41-YFP mice were transfused with CMFDA-labeled FcγRIIATGN platelets (2 donors per recipient; 4 × 108 total platelets) and injected with heat aggregated-IgG (600 μg per mouse, Sigma-Aldrich) to trigger systemic inflammation and platelet activation, as previously described.47 Mice were euthanized after 20 hours. One femur per mouse was fixed in 4% PFA, embedded, sectioned, and stained for P-selectin by IHC to measure BM inflammation. The remaining femurs were flushed as described above and BM cells were incubated with antibodies against CD41 and analyzed by flow cytometry to detect CMFDA (donor platelet)-positive events.
Additional methods can be found in the supplemental Material.
Results
Platelet-like particles leave circulation and infiltrate the bone marrow
To determine whether platelet-derived vesicles generated by inflammatory stimuli can infiltrate the BM, we used a mouse model mimicking a systemic response to infection (LPS-induced sepsis). To definitively track the proportion of exogenous platelet-like particles able to infiltrate the BM, we used mice deficient in the TPO receptor, MPL (c-Mpl−/−) (Figure 1A). As these mice lack MPL, they are unresponsive to TPO, the key signaling molecule that drives MK differentiation from HSCs.48 These mice have very low numbers of BM MKs and are thrombocytopenic (∼250 × 103/µL), allowing us to infuse and track wildtype MPL+ donor platelets by both fluorescence and MPL expression. c-Mpl−/− mice were transfused with wildtype, fluorescent platelets (Figure 1A). Whole blood counts immediately post platelet infusion indicated that fluorescent donor platelets constituted 68% (mean, n = 3) of platelets in circulation (Figure 1B). Next, LPS (or saline control) was administered to induce a systemic inflammatory response. Four hours post LPS administration, donor platelets still accounted for 39% (mean, n = 3) of platelet volume (Figure 1B). Mice were euthanized after 20 hours and femurs were harvested. Figure 1C-D shows positive and negative staining controls, respectively. Figure 1C inset reveals 2 small platelet-like particles, suggesting that they are present but rare in wildtype murine bone marrow. IHC staining of the BM of c-Mpl−/− mice that received wildtype platelet transfusions (Figure 1E-F) revealed small, punctate MPL-positive staining throughout. Note increased staining after LPS infusion (Figure 1F). Insets reveal MPL+ events that appear to be bound to a BM cell (Figure 1E) and extravasating through a BM sinusoid (Figure 1F). Further, we used flow cytometry to quantify the presence of fluorescent particles in flushed BM. While there appeared to be platelet-like-particle infiltration in both the presence and absence of LPS administration, this was only significant with LPS, where up to 8.3% of all BM events were positive for PEV fluorescence (Figure 1G). As both MPL expression and fluorescence could only be derived from the infused platelets, these data reveal that platelet-like particles migrate from the plasma into the BM and interact with BM cells. Together, these data indicate a significant penetrance of platelet-like particles into the BM.
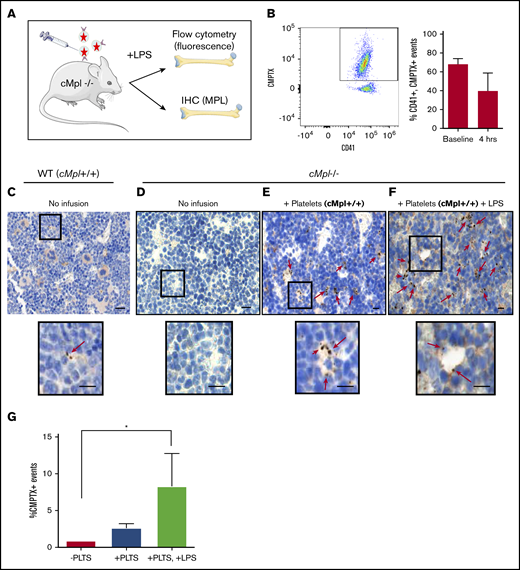
Infused platelet-like particles infiltrate the bone marrow. (A) cMpl−/− mice were infused with wild-type (MPL+), CMPTX-labeled platelets (2 donors/recipient; 4 × 108 total platelets) before an injection of LPS (5 µg/g) or saline control. Mice were euthanized after 20 hours. (B) Transfusion efficiency was measured immediately after platelet infusion (baseline) and 4 hours post–platelet infusion and LPS administration. Shown is a representative scatter plot and gating strategy for selection of CD41+, CMPTX+ events. Flow cytometry of whole blood revealed donor platelets constituted a mean of 68% of circulating platelets at baseline and 39% after 4 hours. Twenty hours postplatelet infusion, MPL expression in bone marrow was measured by immunohistochemistry on whole bone marrow (femurs). (C) Positive control: wild-type C57BL/6 mouse showing positive MPL staining of MKs. Insert shows 2 EVs. (D) Negative control: cMpl−/− mouse showing no MPL staining. (E) cMpl−/− mouse infused with MPL+ platelets: note an observable increase in brown punctate staining corresponding to MPL+ platelet-like particles. Insert shows platelet-like particles that appear to be bound to a BM cell. (F) cMpl−/− mouse infused with MPL+ platelets and LPS: insert shows platelet-like particles that appear to be extravasating through a sinusoid. Scale bars, 50 µm. (G) Flow cytometry of flushed bone marrow was then used to quantify CMPTX+ events in the BM after platelet infusion (±LPS) compared with unstained bone marrow control. Data are mean ± standard error of the mean (SEM) of 3 independent experiments, *P < .05 (Student t test).
Infused platelet-like particles infiltrate the bone marrow. (A) cMpl−/− mice were infused with wild-type (MPL+), CMPTX-labeled platelets (2 donors/recipient; 4 × 108 total platelets) before an injection of LPS (5 µg/g) or saline control. Mice were euthanized after 20 hours. (B) Transfusion efficiency was measured immediately after platelet infusion (baseline) and 4 hours post–platelet infusion and LPS administration. Shown is a representative scatter plot and gating strategy for selection of CD41+, CMPTX+ events. Flow cytometry of whole blood revealed donor platelets constituted a mean of 68% of circulating platelets at baseline and 39% after 4 hours. Twenty hours postplatelet infusion, MPL expression in bone marrow was measured by immunohistochemistry on whole bone marrow (femurs). (C) Positive control: wild-type C57BL/6 mouse showing positive MPL staining of MKs. Insert shows 2 EVs. (D) Negative control: cMpl−/− mouse showing no MPL staining. (E) cMpl−/− mouse infused with MPL+ platelets: note an observable increase in brown punctate staining corresponding to MPL+ platelet-like particles. Insert shows platelet-like particles that appear to be bound to a BM cell. (F) cMpl−/− mouse infused with MPL+ platelets and LPS: insert shows platelet-like particles that appear to be extravasating through a sinusoid. Scale bars, 50 µm. (G) Flow cytometry of flushed bone marrow was then used to quantify CMPTX+ events in the BM after platelet infusion (±LPS) compared with unstained bone marrow control. Data are mean ± standard error of the mean (SEM) of 3 independent experiments, *P < .05 (Student t test).
To confirm that our findings were not specific to 1 type of inflammation, we used a model of autoimmune-induced inflammation. We infused either fluorescent PEVs or CMFDA-labeled fluorescent donor platelets into FcγRIIATGN mice prior to injecting heat-aggregated immunoglobulin-G to trigger inflammation (supplemental Figure 1A). After 20 hours, IHC of femurs revealed increased CD62P staining after inflammatory challenge, suggesting a robust inflammatory response (supplemental Figure 1B). In addition, we quantified the amount of donor-derived fluorescent particles in the BM bound to CD41+ cells (supplemental Figure 1C), and found these were significantly enhanced after direct PEV infusion. These data further support the finding that platelet-derived particles and PEVs are able to infiltrate the BM and interact with resident BM cells. Furthermore, confirmation in a second distinct model suggests that this outcome is not specific to 1 inflammatory state.
Characterization of megakaryocyte and platelet-derived extracellular vesicles in vitro and in vivo
Ultimately, we aim to quantify and characterize native PEVs directly from the BM to determine their prevalence and biological relevance in health and disease. However, for direct measurement of PEVs and MkEVs in the BM, distinct markers to differentiate these 2 populations must be established to faithfully distinguish PEVs from MkEVs Therefore, we next characterized PEVs and MkEVs in accordance with current International Society for Extracellular Vesicles guidelines.43 Because culture conditions and cell sources have significant effects on MK phenotype and the EVs derived from them, we characterized MkEVs collected in vitro from primary murine BM MKs, primary murine fetal liver MKs, and human CD34+ cord blood-derived MKs. PEVs were generated in vitro from isolated, washed human platelets by stimulation with either thrombin and collagen, or LPS to mimic our in vivo experimental conditions. EVs were characterized using nanoparticle tracking analysis (supplemental Figure 2A) in conjunction with immunogold labeling and electron microscopy (supplemental Figure 2B) to quantify CD41+ expression (Figure 2). Murine BM-derived MKs produced more EVs/cell in culture than their fetal liver-derived counterparts and human CD34+ cord blood-derived MKs (Figure 2A), with a similar average size of ∼200 nm (Figure 2B). The percentage of CD41+ murine BM-derived MkEVs and human CD34+-derived MkEVs was similar; however, we found more CD41-bound gold particles in human CD34+-derived MkEVs than their murine counterparts (Figure 2C). We next looked at EVs generated from human platelets activated with traditional agonists (thrombin and collagen) and inflammatory stimuli (LPS, to mimic our in vivo model). Platelets generated a similar number of EVs/platelet regardless of agonist (Figure 2D) and were similar in size (Figure 2E). EVs made from thrombin/collagen and LPS stimulation also expressed CD41 to a similar extent (Figure 2F).
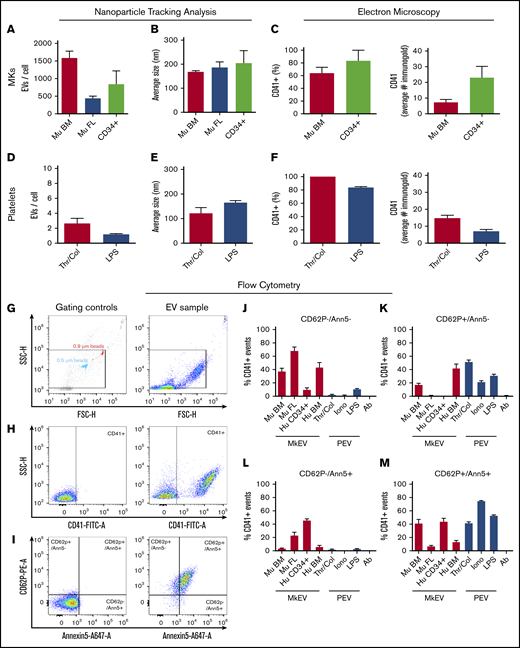
Differential expression of activation markers on megakaryocyte and platelet extracellular vesicles. EVs were harvested from MKs and platelets and characterized via nanoparticle tracking analysis (NTA), electron microscopy, and flow cytometry. (A-B) Murine BM-derived MK (Mu BM), murine fetal liver-derived MK (Mu FL), and human CD34+-derived MKs (CD34+) produced between ∼500 and 1500 EVs per cell, after 24 hours in culture, with an average size of ∼200 nm. (C) Immunogold labeling confirmed the majority of EVs expressed CD41 (60% to 80%), with CD34+-derived EVs expressing more gold particles per EV than their murine counterparts. (D-F) Isolated human platelets were stimulated with either thrombin (0.1 U/mL) and collagen (1 µg/mL) (traditional platelet agonists) or LPS (inflammatory agonist) to generate platelet-derived EVs (PEVs). (D-E) NTA analysis showed thrombin/collagen (Thr/Col) activation generated ∼3 EVs/cell and LPS stimulation generated an average of ∼1 EV/cell, with an average size of 100 to 200 nm. (F) Immunogold labeling showed >80% of EVs generated expressed CD41; at least 50 particles were quantified manually by 2 blinded reviewers per n, per condition. Flow cytometry was used to determine the presence of P-selectin (CD62P) and phosphatidyl serine (Annexin V, Ann5). Calcium ionophore-stimulated platelet EVs (Iono) were used as a positive control for CD62P+, Ann5+ staining and antibody-alone controls (Ab) were used to control for background fluorescence and antibody aggregates. EV preparations were incubated with anti-CD41, anti-CD62P, and Annexin V (10 µg/mL) and analyzed by flow cytometry. Representative examples of (G) flow cytometry gating strategy and for (H-I) sample and negative controls. Megamix beads of known size were used to gauge approximate size of particles. Strict fluorescence minus 1 controls were used to determine negative population spread and set gates. Red bars are MkEVs and blue bars are PEVs percentage of CD41+ EVs, which are (J) negative for both CD62P and Annexin V expression, (K) bind CD62P but not Annexin V, (L) bind Annexin V but not CD62P, and (M) bind both Annexin V and CD62P. Data are mean ± SEM of 3 to 4 independent experiments.
Differential expression of activation markers on megakaryocyte and platelet extracellular vesicles. EVs were harvested from MKs and platelets and characterized via nanoparticle tracking analysis (NTA), electron microscopy, and flow cytometry. (A-B) Murine BM-derived MK (Mu BM), murine fetal liver-derived MK (Mu FL), and human CD34+-derived MKs (CD34+) produced between ∼500 and 1500 EVs per cell, after 24 hours in culture, with an average size of ∼200 nm. (C) Immunogold labeling confirmed the majority of EVs expressed CD41 (60% to 80%), with CD34+-derived EVs expressing more gold particles per EV than their murine counterparts. (D-F) Isolated human platelets were stimulated with either thrombin (0.1 U/mL) and collagen (1 µg/mL) (traditional platelet agonists) or LPS (inflammatory agonist) to generate platelet-derived EVs (PEVs). (D-E) NTA analysis showed thrombin/collagen (Thr/Col) activation generated ∼3 EVs/cell and LPS stimulation generated an average of ∼1 EV/cell, with an average size of 100 to 200 nm. (F) Immunogold labeling showed >80% of EVs generated expressed CD41; at least 50 particles were quantified manually by 2 blinded reviewers per n, per condition. Flow cytometry was used to determine the presence of P-selectin (CD62P) and phosphatidyl serine (Annexin V, Ann5). Calcium ionophore-stimulated platelet EVs (Iono) were used as a positive control for CD62P+, Ann5+ staining and antibody-alone controls (Ab) were used to control for background fluorescence and antibody aggregates. EV preparations were incubated with anti-CD41, anti-CD62P, and Annexin V (10 µg/mL) and analyzed by flow cytometry. Representative examples of (G) flow cytometry gating strategy and for (H-I) sample and negative controls. Megamix beads of known size were used to gauge approximate size of particles. Strict fluorescence minus 1 controls were used to determine negative population spread and set gates. Red bars are MkEVs and blue bars are PEVs percentage of CD41+ EVs, which are (J) negative for both CD62P and Annexin V expression, (K) bind CD62P but not Annexin V, (L) bind Annexin V but not CD62P, and (M) bind both Annexin V and CD62P. Data are mean ± SEM of 3 to 4 independent experiments.
We next used flow cytometry to perform quantitative studies of activation marker surface expression in MkEV and PEV populations. Figure 2G shows the EV gating scheme and a corresponding EV sample. Figure 2H-I shows gating controls (left) and a corresponding representative sample (right) for identification of the CD41 population, and then identification of CD62P and Annexin V+ events, respectively. We quantified the percentage of: (1) CD41+ particles that were negative for both activation markers (Figure 2J), (2) CD41+ particles that were positive for CD62P but not Annexin V (Figure 2K), (3) CD41+ particles that were positive for Annexin V but not CD62P (Figure 2L), and (4) CD41+ particles that were positive for both CD62P and Annexin V (Figure 2M). Calcium ionophore-generated PEVs were used as a positive control for activation marker expression where >75% of CD41+ EVs expressed both CD62P and PS (Annexin V+, Figure 2M). Consistent with previous data, the majority (>60%) of EVs generated from fetal liver-derived MKs did not express activation markers.11 Of note, human MkEVs derived from CD34+ cells vs bone marrow had very different profiles; whereas the majority of the CD41+ events from human BM-derived MKs were PS−, the CD34+-derived MkEVs were largely PS+. These data reveal that MkEVs generated in culture are a mixed population, the characteristics of which are highly influenced by their cell of origin. Further, PEVs generated via inflammatory agonists (LPS) vs traditional agonists (thrombin and collagen) expressed a similar repertoire of platelet activation markers. Together, these data suggest that CD41+ MkEVs generally express fewer activation markers (CD62P and PS) than PEVs, which is consistent with a previous study.11 However, these results also reveal that platelet and MkEVs derived from both humans and mice are heterogenous populations. As such, the cell of origin (MK vs platelet) of CD41+ EVs cannot be fully determined by the presence or absence of the traditionally used activation markers CD62P and Annexin V.
Characterization of megakaryocyte and platelet-derived extracellular vesicles in human bone marrow
We next aimed to validate the physiological relevance of our findings by examining if MkEVs and PEVs were present in human BM; this has not been previously evaluated. Consistent with Figure 2, EVs from cultured MKs were largely CD41+ (1452 ± 88 EV/µL) and CD62P+ (1276 ± 176 EV/µL), with only a small population of PS+ events (110 ± 17 EV/µL). We next investigated the presence of CD41+ EVs in human BM aspirates using autologous peripheral blood (PB) as a control for comparison. Figure 3B-C show the raw flow plots and gating strategy for the human BM and PB samples. There were significantly more CD41+, CD62P+, and PS+ EVs in the BM compared with autologous PB samples (P < .05, Figure 3D-E). We then measured whether CD41+ PEV events were also positive for general platelet activation markers, and whether these markers constituted different subsets within the BM vs PB EV populations. Our data revealed that the BM had a significantly higher percentage of CD41+/CD62P−/PS+ (Figure 3F) and CD41+/CD62P+/PS+ (Figure 3G) EVs, and no significant difference in CD41+/CD62P+/PS− EVs (Figure 3I). Of note, the percentage of CD41+/CD62P−/PS− EVs was significantly higher in PB than in BM samples (Figure 3H). These data reveal that the overall amount of EVs is increased in BM compared with PB. Finally, to investigate the relationship between absolute PEV counts in BM and PB, we matched BM CD41+/CD62P−/PS+, CD41+/CD62P+/PS+, CD41+/CD62P−/PS−, and CD41+/CD62P+/PS− particle counts with autologous PB EV counts. We found a significant positive correlation in CD41+/CD62P−/PS+, CD41+/CD62P+/PS+ (Figure 3J-K) population but no correlation in CD41+/CD62P−/PS− and CD41+/CD62P+/PS+ (Figure 3L-M) matched event counts. These results demonstrate that human BM had significantly more CD41+/PS+ events, markers shown in Figure 2J-M to be largely absent from MkEVs and therefore suggest the CD41+/PS+ EV population in human BM are derived from platelets.
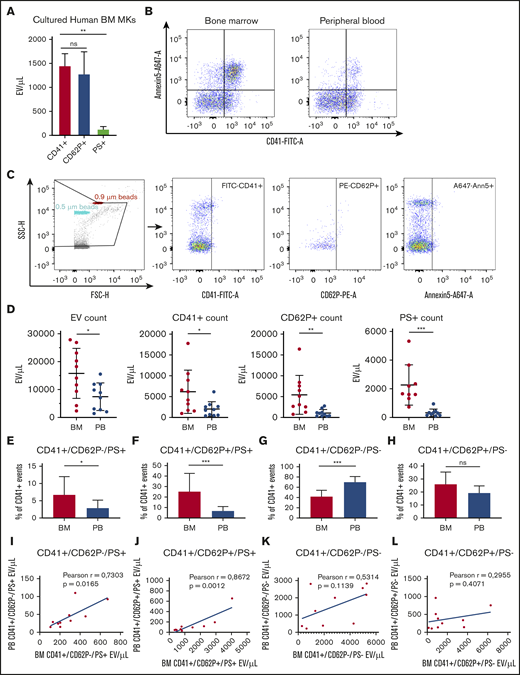
Characterization of extracellular vesicles from human bone marrow and peripheral autologous blood samples. EVs were harvested from (1) BM-derived MKs cultured in EV-depleted media for 24 hours, (2) BM aspirates, and (3) PB patient samples, and characterized by flow cytometry. (A) MKs cultured from human bone marrow produced CD41+ (1452 ± 88 MV/µL) and CD62P+ (1276 ± 176 MV/µL) EVs. (C) To analyze EVs from patient BM and PB samples (n = 10), Megamix beads of known size were used to gauge approximate size of particles. Strict fluorescence minus 1 controls were used to determine negative population spread and set gates. (B) An example of flow cytometry gates showing CD41+ and Annexin 5+ populations from both BM and PB from the same patient. (D) Flow cytometry analyses of EV absolute counts showed significantly more EVs of all types in BM in comparison with autologous PB. (E-H) P < .05 by Mann-Whitney U test. Data were further analyzed to assess platelet activation markers within the CD41+ population. (I-L) *P < .05, **P < .01, ***P < .001, or nonsignificant (ns) by Mann-Whitney U test. Correlation plots of absolute counts matched from BM and PB from the same patients (see Table 1 for patient characteristics).
Characterization of extracellular vesicles from human bone marrow and peripheral autologous blood samples. EVs were harvested from (1) BM-derived MKs cultured in EV-depleted media for 24 hours, (2) BM aspirates, and (3) PB patient samples, and characterized by flow cytometry. (A) MKs cultured from human bone marrow produced CD41+ (1452 ± 88 MV/µL) and CD62P+ (1276 ± 176 MV/µL) EVs. (C) To analyze EVs from patient BM and PB samples (n = 10), Megamix beads of known size were used to gauge approximate size of particles. Strict fluorescence minus 1 controls were used to determine negative population spread and set gates. (B) An example of flow cytometry gates showing CD41+ and Annexin 5+ populations from both BM and PB from the same patient. (D) Flow cytometry analyses of EV absolute counts showed significantly more EVs of all types in BM in comparison with autologous PB. (E-H) P < .05 by Mann-Whitney U test. Data were further analyzed to assess platelet activation markers within the CD41+ population. (I-L) *P < .05, **P < .01, ***P < .001, or nonsignificant (ns) by Mann-Whitney U test. Correlation plots of absolute counts matched from BM and PB from the same patients (see Table 1 for patient characteristics).
PEVs interact with BM MKs and alter hematopoiesis in vitro
We have established that PEVs are present in humans and mice where they can leave circulation and enter the BM. In our murine models, they bind to BM-resident CD41+ cells in vivo (supplemental Figure 1C). We therefore sought to confirm and extend these findings by examining and characterizing the interaction between PEVs and CD41+ MKs in vitro and ex vivo. We first incubated fluorescent (PKH67-labeled, membrane dye), purified PEVs (1 × 108 total) with murine BM MKs over 24 hours. Confocal microscopy revealed that MKs rapidly interacted with and took up PEVs within 30 minutes of incubation (Figure 4A). Next, PEVs were labeled with CMPTX (cytosolic dye) or MitoTracker (mitochondrial dye). Again, confocal microscopy revealed fluorescent particles bound to MKs (Figure 4B-C; supplemental Figure 3A-B), suggesting that the labeled components (membrane, cytosol, and mitochondria) of EVs are able to integrate into target MKs.
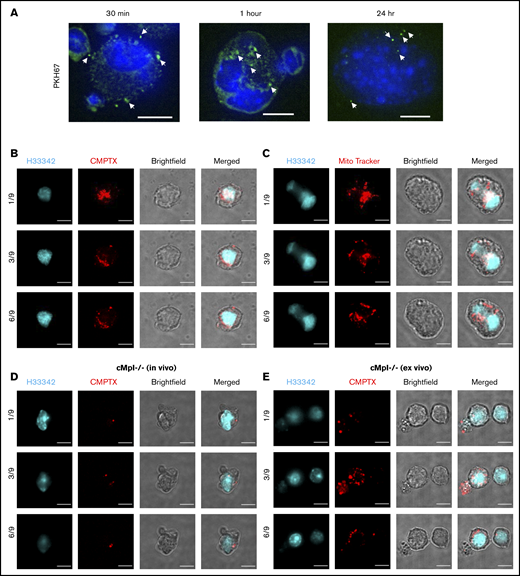
PEVs interact with BM MKs in vitro and in vivo. Human platelets were labeled with either PKH67 (membrane marker), CMPTX (cytosol stain), or MitoTracker Red (mitochondrial marker) and activated with thrombin (0.1U/mL) and collagen (1 μg/mL) to generate PEVs. (A) PKH67-labeled PEVs were incubated with murine BM-derived MKs and fixed over a time course of 30 minutes, 1 hour, and 24 hours. Cells were then harvested and stained with Hoechst (H33342) before confocal microscopy analysis. Representative images are z-stack maximum projections. (B) CMPTX-labeled and (C) MitoTracker–labeled PEVs were incubated with isolated, cultured MKs for 24 hours. Cells were then fixed, permeabilized, stained with Hoechst, and examined by confocal microscopy. The images were taken at different confocal planes with 5 µm z-stack steps. The numbers represent a specific slice (z stack) number/total number of slices. (D) Wild-type (MPL+), CMPTX-labeled platelets were infused into cMpl−/− mice before an LPS injection, as in Figure 1. Mice were euthanized after 20 hours, BM was flushed, incubated with anti-CD41 Ab, and sorted for CMPTX+/CD41+ BM cells by flow cytometry. Sorted populations were fixed, stained with Hoechst, and imaged by confocal microscopy. (E) Wild-type (MPL+), CMPTX-labeled PEVs were cultured with freshly isolated cMpl−/− BM over 72 hours. Cultures were then stained with anti-CD41 and sorted for CMPTX+/CD41+ cells. Sorted populations were fixed, stained with Hoechst, and imaged by confocal microscopy. Scale bars, 10 µm.
PEVs interact with BM MKs in vitro and in vivo. Human platelets were labeled with either PKH67 (membrane marker), CMPTX (cytosol stain), or MitoTracker Red (mitochondrial marker) and activated with thrombin (0.1U/mL) and collagen (1 μg/mL) to generate PEVs. (A) PKH67-labeled PEVs were incubated with murine BM-derived MKs and fixed over a time course of 30 minutes, 1 hour, and 24 hours. Cells were then harvested and stained with Hoechst (H33342) before confocal microscopy analysis. Representative images are z-stack maximum projections. (B) CMPTX-labeled and (C) MitoTracker–labeled PEVs were incubated with isolated, cultured MKs for 24 hours. Cells were then fixed, permeabilized, stained with Hoechst, and examined by confocal microscopy. The images were taken at different confocal planes with 5 µm z-stack steps. The numbers represent a specific slice (z stack) number/total number of slices. (D) Wild-type (MPL+), CMPTX-labeled platelets were infused into cMpl−/− mice before an LPS injection, as in Figure 1. Mice were euthanized after 20 hours, BM was flushed, incubated with anti-CD41 Ab, and sorted for CMPTX+/CD41+ BM cells by flow cytometry. Sorted populations were fixed, stained with Hoechst, and imaged by confocal microscopy. (E) Wild-type (MPL+), CMPTX-labeled PEVs were cultured with freshly isolated cMpl−/− BM over 72 hours. Cultures were then stained with anti-CD41 and sorted for CMPTX+/CD41+ cells. Sorted populations were fixed, stained with Hoechst, and imaged by confocal microscopy. Scale bars, 10 µm.
To examine platelet-like particle-BM cell interactions in vivo, CMPTX+ events from the BM of c-Mpl−/− mice transfused with CMPTX-labeled, MPL+ donor platelets (Figure 1D) were visualized. Cells were sorted by CD41+ and CMPTX+ dual positivity by flow cytometry and then examined by confocal microscopy. We observed fluorescent platelet-like particles bound to polyploid MKs (Figure 4D; supplemental Figure 3C). Brightfield images are included to show cell outlines; addition of a second fluorescent dye to image cells was excluded to limit background and antibody aggregation/artifacts that could interfere with PEV visualization. We also recapitulated this experiment ex vivo by incubating murine CMPTX+, MPL+ PEVs with flushed BM from c-Mpl−/− mice, and then isolating CMPTX+/CD41+ events. Consistent with our in vivo findings, we observed MKs (cells with large, polyploid nuclei) with bound CMPTX+ PEVs (Figure 4E; supplemental Figure 3D).
We have shown that platelet-like particles/PEVs can infiltrate the BM, bind to resident cells, and be internalized. We next wanted to establish a potential functional consequence of these interactions. To examine the ability of PEVs to reprogram hematopoietic cells, BM was extracted from c-Mpl−/− mice and cultured in the presence of wild-type, fluorescent PEVs (MPL+), in the absence and presence of additional TPO for 3 days (Figure 5A). To recapitulate our in vivo data, PEVs were generated from the platelets of 2 donors and incubated with whole BM from 1 recipient mouse (12 × 107 PEVs: 9 × 106 BM cells). Representative images of cultures with and without added PEVs are shown in Figure 5B. After 72 hours, an increase in large cells was observed in only the PEV-treated cultures, regardless of whether additional TPO was present. Of note, these large cells were fluorescent, indicating that they had interacted with and bound/internalized PEVs (Figure 5B). To validate that these large cells were MKs/MK precursors and quantify their prevalence in culture after PEV addition, flow cytometry was performed, and revealed a significant increase in CD41+ cells in PEV-treated cultures (Figure 5C). When examined on forward and side-scatter plots, it was evident that the CD41+/PEV+ population represented the largest cells in the population, suggesting that these CD41+ cells were bona fide MKs and not PEV aggregates or PEVs bound to smaller BM cells (Figure 5D). Further supporting this, flow cytometry revealed that 58% to 65% of CD41+ events were dual positive for CMPTX, suggesting these cells had incorporated PEV membrane at the 72-hour timepoint (Figure 5E). These data show that PEVs are able to functionally reprogram BM cells by restoring megakaryopoiesis in MPL-deficient cells.
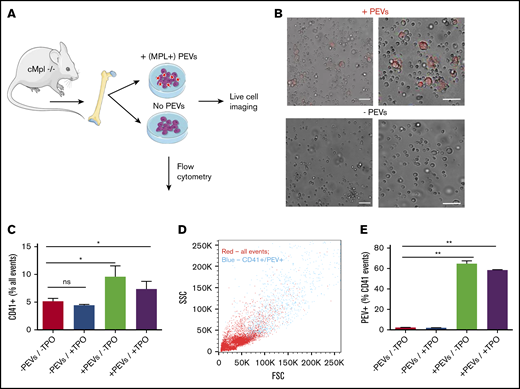
Platelet-derived extracellular vesicles alter hematopoiesis in vitro. (A) BM from cMpl−/− mice was flushed and cultured in the presence or absence of wild-type (MPL+), CMPTX-labeled PEVs (PEVs from 2 donors: 1 recipient; 12 × 107 particles: 9 × 106 BM cells) for 72 hours. Indicated cultures were also supplemented with TPO (70 ng/mL). (B) Cultures were monitored by live-cell microscopy that revealed an increase in large MKs in the cultures treated with PEVs, which were also fluorescent. After 72 hours in culture, the percentage of CD41+ cells was quantified by flow cytometry. (C) Cultures treated with PEVs had significantly increased numbers of CD41+ cells. (D) Representative forward and side scatter plot showing that CD41+/ CMPTX (PEV)+ events (blue) are the largest in the sample. (E) Percentage of CD41+ cells that are also positive for CMPTX (PEV-derived) fluorescence. Data are mean ± SEM of 3 independent experiments; scale bars, 50 µm, *P < .05 **P < .01 (1-way analysis of variance with multiple comparison post hoc test).
Platelet-derived extracellular vesicles alter hematopoiesis in vitro. (A) BM from cMpl−/− mice was flushed and cultured in the presence or absence of wild-type (MPL+), CMPTX-labeled PEVs (PEVs from 2 donors: 1 recipient; 12 × 107 particles: 9 × 106 BM cells) for 72 hours. Indicated cultures were also supplemented with TPO (70 ng/mL). (B) Cultures were monitored by live-cell microscopy that revealed an increase in large MKs in the cultures treated with PEVs, which were also fluorescent. After 72 hours in culture, the percentage of CD41+ cells was quantified by flow cytometry. (C) Cultures treated with PEVs had significantly increased numbers of CD41+ cells. (D) Representative forward and side scatter plot showing that CD41+/ CMPTX (PEV)+ events (blue) are the largest in the sample. (E) Percentage of CD41+ cells that are also positive for CMPTX (PEV-derived) fluorescence. Data are mean ± SEM of 3 independent experiments; scale bars, 50 µm, *P < .05 **P < .01 (1-way analysis of variance with multiple comparison post hoc test).
Discussion
Platelet activation and aberrant hematopoiesis are hallmarks of thrombo-inflammatory disorders. The effects of this are compounding; persistent platelet activation can lead to pathological thrombosis and platelet consumption, leading to thrombocytopenia.49 Conversely, inflammatory signaling can trigger thrombocytosis via increased differentiation of MKs from HSCs in the BM.1,2 PEVs are increased in inflammatory conditions and are able to reprogram various cell types via different pathways. However, to date, the role of PEVs in regulating the BM environment has not been examined. In this study, we show that platelet-derived particles are able to infiltrate the BM and interact with BM-residing cells in vivo and in vitro. The influence of EVs on recipient cell function is based on the EV composition. Because platelets are known to directly endocytose plasma components,50-52 and PEVs contain a subset of platelet content, PEVs also likely reflect the plasma environment. Indeed, changes in EVs have been detected in many pathological disorders such as cardiovascular disease53, infections,54 autoimmune diseases (multiple sclerosis,55 rheumatoid arthritis,19 SLE21,22 ), and cancer.56 Therefore, PEVs may provide a mechanism by which to directly deliver pathological plasma changes to cells in the BM. This would provide a direct and fast mode of communication between the plasma and BM cells, allowing the BM to quickly respond and adapt to inflammation. This suggests that targeting (1) PEV formation or (2) PEV interactions with BM cells may hold promise for future therapeutics to downregulate inflammatory responses.
One major challenge in characterizing the role and presence of PEVs in the BM is the difficultly of distinguishing PEVs from their MK-derived counterparts. In this study, we aimed to clearly discriminate between platelet-like particles that originated in circulation and particles from resident MKs in the BM by fluorescently labeling platelets or PEVs before infusion and exclusively quantifying fluorescent platelet-like particles in the BM. This distinction underestimates the contribution of PEVs by discounting any PEV contribution from endogenous platelets, but also excludes the possibility of contamination from BM MKs. To fully understand the prevalence and biological relevance of PEVs in the BM, the ability to quantify and characterize native, CD41-expressing EVs directly from the BM is critical. We therefore performed a head-to-head characterization of platelet and MkEVs derived from various commonly used preparation techniques and cells of origin. Ultimately, we determined that CD41+ EVs from both platelets and MKs, residing in vivo or generated in vitro, consist of subpopulations that express 1 or both CD62P and PS (Figures 2 and 3). In general, MkEVs expressed less activation markers (CD62P and PS) than PEVs, whereas PEV were largely CD62P+ and PS+. This is consistent with the idea that MkEVs are constitutively released, whereas PEVs are a by-product of platelet activation and therefore resemble an activated platelet.
To validate the physiological relevance of our findings in humans, we analyzed human BM and autologous PB. Analysis of BM from biopsies revealed that human BM contained CD41+ EVs that were significantly enriched compared with autologous PB (Figure 3D). In addition, CD41+ EVs found in human BM expressed more PS and CD62P than their PB counterparts (Figure 3D-F); this is consistent with the surface markers found on human PEVs (and not human BM-derived MkEVs, Figure 2J-M) and supports our hypothesis of PEV infiltration from blood to BM.
Finally, we show that PEVs can restore megakaryopoiesis in cultured Mpl−/− BM cells, uncovering a functional outcome of the interaction between PEVs and target BM cells. Although we do not know the specific mechanism by which PEVs restore megakaryopoiesis, it may be through transfer of the MPL receptor. Indeed, Figure 4 showed that all components of PEVs can be incorporated into target MKs. However, the increase in CD41+ cells after PEV addition was not dependent on addition of TPO to the media, suggesting that the mechanism by which PEVs restore MK production could be independent of TPO. However, the small contribution of TPO from the added PEVs cannot be ruled out and therefore experiments done with PEVs but without added TPO in the media may not be entirely TPO independent. Future studies will determine the specific mechanisms by which PEVs restore hematopoiesis in vitro and examine their impact in vivo.
The modes of PEV infiltration into the BM remain to be determined. Of interest, a recent study by Escobar et al examines the biodistribution of in vitro-produced MkEVs that were infused into healthy mice and found that after 24 hours, the organ that contains the most EVs is the BM.41 These data suggest that CD41+ EVs may constitutively home to the BM, and that the limiting factor determining the amount of PEV infiltration into the BM may be their creation through platelet activation. In the future, we will determine whether this is a phenomenon that occurs strictly during inflammatory and other pathological conditions, or whether it is a more constitutive process. The complexity of inflammatory conditions is important to consider and in pathological states, the source of CD41+ EVs may also be from platelet-destructive processes mediated by cells such as macrophages.57 These factors will have to be considered moving forward. In addition, future studies will examine additional ways by which PEVs may modify BM cells. For example, we have previously shown that MKs can uptake protein antigens and act as antigen-presenting cells.58 This presents the intriguing possibility that PEVs may further educate or activate MKs while they are still in the BM.
In summary, we report for the first time that plasma-residing PEVs can penetrate the BM. Further, PEVs interact with BM cells in vivo and in vitro, causing functional reprogramming. This finding suggests a novel regulator of inflammation-induced hematopoiesis, perhaps representing a model in which PEVs traffic proliferative stimuli to the BM, which helps to instruct progenitor cells to manufacture enforcements and alert the BM environment of an inflammatory trigger.
All renewable materials, datasets, and protocols will be available to other investigators by contacting the corresponding author, Kellie R. Machlus (kmachlus@bwh.harvard.edu).
Acknowledgments
The authors thank Maria Ericsson and the Electron Microscopy Facility at Harvard Medical School for processing of electron microscopy samples, Ron Mathieu and the Boston Children’s Hospital Flow Cytometry Core for assistance in flow cytometry, and Teri Bowman at the Brigham and Women's Hospital (BWH) Specialized Histopathology Core for preparation and staining of IHC samples, Zhi-Jian Lui for culture of CD34+ human cells, and Laura Goldberg for her assistance and insight in editing the manuscript. The Sysmex XN veterinary analyzer used to take murine blood cell counts was a generous loan from Sysmex America, Inc. The authors also thank Alexey Kibardin and Alexandra Posvyatenko from the Larin Laboratory, and Nadezhda Podoplelova at Dmitry Rogachev Pediatric Hematology Hospital and Polina Vishnyakova at Kulakov’s Research Center for Obstetrics, Gynecology and Perinatology for their generous help and resourceful comments.
This study was supported by grants from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases K01DK111515 (K.R.M.) and National Heart, Lung, and Blood Institute 5T32HL007917-20 (P.D.), P01HL046925 (M.S.V.), and R01HL068130 (J.E.I.). M.A.P. is supported by a grant from the endowment foundation “Doctors, Innovations, Science for Children.” M.A.M. acknowledges the Breast Cancer Research Foundation and the Advanced Medical Research Foundation for their support of this work. E.B. is supported by the Canadian Institutes of Health Research and is a Fonds de Recherche en Santé du Québec scholar. A.L. is a recipient of a fellowship from the Fondation de Recherche sur l’Arthrite Pierre Borgeat. K.R.M. is an American Society of Hematology Scholar.
Authorship
Contribution: S.L.F. and K.R.B. planned and performed experiments, analyzed data, and wrote the manuscript; I.A., A.L., P.D., J.C., G.M., and N.M.T. performed experiments and collected and analyzed data; J.E.I., M.A.M., M.S.-V., M.A.P., and E.B. planned and supervised experiments and helped edit the manuscript; and K.R.M. designed and supervised the study and wrote the manuscript.
Conflict-of-interest disclosure: J.E.I. has financial interest in and is a founder of Platelet BioGenesis, a company that aims to produce donor-independent human platelets from human-induced pluripotent stem cells at scale. The interests of J.E.I. were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. The remaining authors declare no competing financial interests.
Correspondence: Kellie R. Machlus, Harvard Institutes of Medicine, BWH Hematology Division, 77 Ave Louis Pasteur, HIM 733, Boston, MA 02115; e-mail: kmachlus@bwh.harvard.edu.

