

Key Points
Platelets and influenza virus interact in a sialic acid–dependent manner, which may designate platelets for hepatic clearance.
Degree of thrombocytopenia and platelet activation is dependent on the influenza virus subtype and may contribute to virus pathogenicity.

Abstract
Thrombocytopenia is a common complication of influenza virus infection, and its severity predicts the clinical outcome of critically ill patients. The underlying cause(s) remain incompletely understood. In this study, in patients with an influenza A/H1N1 virus infection, viral load and platelet count correlated inversely during the acute infection phase. We confirmed this finding in a ferret model of influenza virus infection. In these animals, platelet count decreased with the degree of virus pathogenicity varying from 0% in animals infected with the influenza A/H3N2 virus, to 22% in those with the pandemic influenza A/H1N1 virus, up to 62% in animals with a highly pathogenic A/H5N1 virus infection. This thrombocytopenia is associated with virus-containing platelets that circulate in the blood. Uptake of influenza virus particles by platelets requires binding to sialoglycans and results in the removal of sialic acids by the virus neuraminidase, a trigger for hepatic clearance of platelets. We propose the clearance of influenza virus by platelets as a paradigm. These insights clarify the pathophysiology of influenza virus infection and show how severe respiratory infections, including COVID-19, may propagate thrombocytopenia and/or thromboembolic complications.
Introduction
Platelets are small, anuclear cells with their primary physiological role in hemostasis and thrombosis.1 Therefore, an astonishing 100 billion platelets are produced and cleared from the blood each day, to maintain 150 to 450 billion functional platelets per liter.2,3 Because spontaneous bleeding events usually do not occur when counts are above 10 billion platelets per liter,4 their relative abundance suggests that platelets have additional roles. The emerging view of platelets as immune cells may explain their excess, as platelets fulfill a variety of immune-regulatory functions that go far beyond hemostasis.5-13
Thrombocytopenia (low platelet count) is a commonly observed and sometimes life-threatening symptom during sepsis and severe influenza.14-17 For instance, it was reported in 14% of the hospitalized cases globally during the 2009 influenza pandemic.18 Thrombocytopenia was not only found to be a biomarker of poor outcome of severe influenza,19 but was associated with severe respiratory infections in general.20-23 Other clinical observations during acute influenza, such as venous and arterial thrombotic and cardiovascular events24,25 and alveolar hemorrhages,26 highlight the role of platelets described herein.
Zoonotic viruses, including influenza viruses and coronaviruses, emerge from animal reservoirs and remain a continuous threat to humans.27,28 Therefore, better insight in the determinants governing the ability of these viruses to switch host species or to cause severe disease is warranted.29 Influenza A viruses are subtyped on the basis of their hemagglutinin (HA) and neuraminidase (NA) surface glycoproteins, which determine the specificity of a virus for a particular host species and host cell. The influenza virus HA is responsible for binding to the sialic acid (SA)-terminated glycans present at the cell membrane.30 The virus NA has an opposing function in facilitating the release of virus progeny by cleaving the SA residues from the cell surface.31
Currently, the influenza A/H3N2 and A/H1N1 viruses circulate in humans. They were introduced by zoonotic events causing the influenza pandemics of, respectively, 1968 and 2009. Similar zoonotic events are infrequently observed in humans, such as the highly pathogenic avian influenza (HPAI) A/H5N1 virus.32 The overall binding affinity of these viruses depends on the strain, expressed in the occurrence and functional balance of different HA and NA subtypes,33 in combination with the specific form and glycan density presented at a cell membrane.34 For instance, avian viruses show binding preference to 2,3-sialyl-(N-acetyl-lactosamine) glycans (2,3-SLN), which are found on cells of the human lower respiratory tract (LRT).35,36 Human influenza viruses preferentially bind 2,6-SLN glycans,37,38 which are present on human epithelial cells of the upper respiratory tract (URT; Figure 1). The different receptor preferences also explain key differences observed in virus pathogenicity.36,39 Thrombocytopenia is mostly associated with severe A/H5N1 or 2009 A/H1N1 virus infections.15,40,41
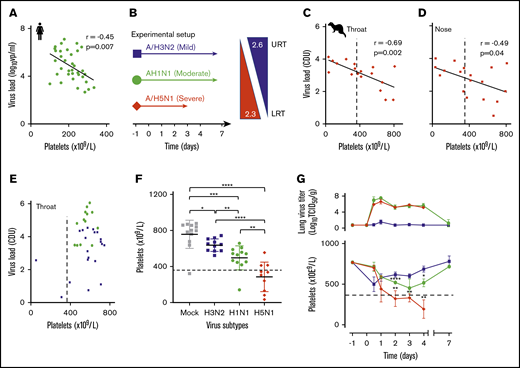
Influenza-induced and subtype-dependent thrombocytopenia in humans and ferrets. (A) The blood platelet count and viral RNA load were inversely correlated in 2009 influenza A/H1N1 virus–infected patients (n = 34). Pearson’s r = −0.45; 95% CI, −0.68 to −0.14. (B) Experimental setup: ferrets inoculated with seasonal A/H3N2 (n = 24), pandemic A/H1N1 (n = 24), or A/H5N1 (n = 20) influenza virus with increasing disease severity in humans and ferrets.39 Arrows: the virus replication sites in the URT and LRT of both humans and ferrets with similar α2,3- and α2,6-sialoglycan receptor distributions. (C) An inverse correlation is shown between platelet count and viral loads (PCR) in throat swabs of A/H5N1 virus–infected ferrets (n = 20). Pearson’s r = −0.69; 95% CI, −0.88 to −0.33. (D) Platelet counts and viral loads (PCR) were inversely correlated in nasal swabs of A/H5N1 virus-infected ferrets (n = 20). Pearson’s r = −0.49; 95% CI, −0.78 to −.03. (E) There was no significant correlation in A/H3N2 (n = 24) and A/H1N1 (n = 24) virus-infected animals. (F) Platelet counts at about the peak of virus infection (days 2-4; n = 12). Error bars, SD; Student t test (unpaired). (G) Top: mean lung virus titer in ferrets (n = 4). Error bars, SD. Bottom: blood platelet count. Mean (n = 4) platelet count. Error bars, SD; Student t test (unpaired). (C-G) Dotted lines indicate the lower limit of normal platelet levels in the ferret (729 × 109/L ± 360 × 109/L). *P < .05; **P < .01; ***P < .001, ****P < .0001.
Influenza-induced and subtype-dependent thrombocytopenia in humans and ferrets. (A) The blood platelet count and viral RNA load were inversely correlated in 2009 influenza A/H1N1 virus–infected patients (n = 34). Pearson’s r = −0.45; 95% CI, −0.68 to −0.14. (B) Experimental setup: ferrets inoculated with seasonal A/H3N2 (n = 24), pandemic A/H1N1 (n = 24), or A/H5N1 (n = 20) influenza virus with increasing disease severity in humans and ferrets.39 Arrows: the virus replication sites in the URT and LRT of both humans and ferrets with similar α2,3- and α2,6-sialoglycan receptor distributions. (C) An inverse correlation is shown between platelet count and viral loads (PCR) in throat swabs of A/H5N1 virus–infected ferrets (n = 20). Pearson’s r = −0.69; 95% CI, −0.88 to −0.33. (D) Platelet counts and viral loads (PCR) were inversely correlated in nasal swabs of A/H5N1 virus-infected ferrets (n = 20). Pearson’s r = −0.49; 95% CI, −0.78 to −.03. (E) There was no significant correlation in A/H3N2 (n = 24) and A/H1N1 (n = 24) virus-infected animals. (F) Platelet counts at about the peak of virus infection (days 2-4; n = 12). Error bars, SD; Student t test (unpaired). (G) Top: mean lung virus titer in ferrets (n = 4). Error bars, SD. Bottom: blood platelet count. Mean (n = 4) platelet count. Error bars, SD; Student t test (unpaired). (C-G) Dotted lines indicate the lower limit of normal platelet levels in the ferret (729 × 109/L ± 360 × 109/L). *P < .05; **P < .01; ***P < .001, ****P < .0001.
In this study, viral load correlated with platelet count in patients with a 2009 A/H1N1 virus infection. Patients with a higher viral load had a higher degree of thrombocytopenia. Influenza viruses were phagocytosed by platelets in vivo, allowing for platelet-mediated transport of virus cargo through the blood circulation. This mechanism, and early platelet response, was dependent on platelet glycan density and virus glycan-binding affinity. Accordingly, observed platelet activation, platelet aggregation, and the degree of thrombocytopenia were subtype dependent, contributing, therefore, to the different degrees of pathogenicity among seasonal, pandemic, and avian influenza viruses. Finally, these observations link respiratory virus infections to acute thrombotic and cardiovascular events.
Methods
Patient inclusion criteria and diagnostics
We identified patients (n = 335) in the Erasmus Medical Centre (Erasmus MC), a large (>40 000 admissions in 2011), tertiary university hospital in The Netherlands. Influenza A virus–positive respiratory specimen were randomly obtained from the patients from August 2009 through July 2012, as previously described.42 We selected adult patients with a healthy immune status (n = 35), defined as no active malignancy, no sepsis with immediate intensive care unit submission, and no other current infections or chemotherapy treatment. Virological and hematological data and patient immune status were obtained by reviewing medical records. One patient (pt. 34) was excluded from the analyses, because the viral load (1.78 log10 virus particles per milliliter) of the respiratory sample was below the limit of quantification of a polymerase chain reaction (PCR) assay (lower limit of quantification, 2.1 log10 viral particles per milliliter).43 The protocol was approved by the hospital medical ethics board (MEC-2012-463).
Human blood donors
The local medical ethics committee of the University Medical Center Utrecht approved the drawing of blood for ex vivo research purposes, including those of this study (MEC-18/774). All donors provided written informed consent and remained anonymous throughout the study. Blood was drawn by venipuncture from the antecubital vein of healthy donors and was collected into Vacuette tubes (Greiner, Kremsmünster, Austria) with 3.2% (w/v) trisodium citrate.
Viruses
The influenza A/H1N1 virus (A/Netherlands/602/2009), isolated from a child during the 2009 pandemic; the highly pathogenic influenza A/H5N1 virus (A/Indonesia/5/2005); and the seasonal influenza A/H3N2 virus (A/Netherlands/177/2008) were used for the ferret studies.39,44 These viruses were propagated on Madin Darby canine kidney (MDCK) cells, as described elsewhere.45 Influenza A virus (A/PR/8/34; Sinai strain) was used to study influenza virus uptake by platelets in vitro. This virus was propagated in 10-day-old embryonated chicken eggs and subsequently purified as has been described.46 Influenza A/Perth/06/2008 wild-type virus and 6+2 recombinant (PR/8/34) viruses with the HA and NA from A/Netherlands/602/2009 and A/Indonesia/5/2005 (without the multibasic cleavage site) were used for the platelet binding and functional assays. These viruses were grown in either MDCK cells or eggs. Viruses propagated on MDCK cells were purified and concentrated as follows. Virus-containing supernatants were cleared from cell debris by centrifugation at 1000g for 10 minutes at 4°C. Subsequently, the supernatant was passed through a 0.2-µm sterile syringe filter (Corning) and concentrated with a centrifugal filter (Ultra-15; Amicon) by centrifugation at 2000g at 4°C until a virus concentration of 1 × 1010 virus particles per milliliter was reached. Viruses propagated in eggs were purified as follows. Virus-containing allantoic fluid was harvested after 48 hours and cleared of debris by centrifugation at 1000g for 10 minutes at 4°C. Viruses were fluorescently labeled first or directly purified on a 20% to 60% discontinuous sucrose gradient in phosphate-buffered saline (pH 7.4; PBS; Lonza) and centrifuged at 25 000g for 45 minutes at 4°C. Fluorescent labeling of influenza viruses was performed by incubating the viruses in PBS with 10 µg/mL N-hydroxysuccinimide ester-fluorescein isothiocyanate (Sigma-Aldrich) for 1.5 hours at RT before density gradient centrifugation. Finally, the viruses were collected by diluting the virus-containing fraction with PBS and pelleting at 27 000g for 90 minutes. The virus pellet was resuspended in PBS, counted using an NS300 Nanoparticle Tracking Analyzer (NTA; Malvern), and stored at −80°C until further use.
Ferrets
Ferrets were housed and experiments were in strict compliance with European Union guidelines (EU Directive on Animal Testing 86/609/EEC), with experiments performed according to Dutch regulations (Experiments on Animals Act, 1997) and reviewed by an independent animal experimentation ethics review committee (National Vaccine Institute, permit: 200900201). All experiments were performed under animal biosafety level 3 conditions. The animals were inoculated under anesthesia with ketamine (4-8 mg/kg; Nimatek, Eurovet Animal Health B.V., Bladel, The Netherlands) and medetomidine hydrochloride (0.1 mg/kg; Domitor; Orion Pharma, Espoo, Finland), with each of the 3 viruses with 106 TCID50 (median tissue culture infectious dose) in a 3-mL volume intratracheally and in a 0.3-mL volume intranasally, evenly divided over the 2 nostrils. After inoculation, the ferrets received atipamezole hydrochloride (0.5 mg/kg Antisedan; Orion Pharma, Espoo, Finland) to antagonize the effect of the medetomidine. Animals were scheduled to be euthanized at 12 hours (0.5 day), and 1, 2, 3, 4, and 7 days after infection for virology, hematological, and histological examination of the extrarespiratory organs. A blood sample was taken before infection (day −1) to obtain normal ferret platelet counts (n = 104; mean ± 2 standard deviation [SD]; 729 × 109/L ± 360 × 109/L). Assessments of viral loads and pathogenicity as well as the data of the histopathological examination of the respiratory tracts were adapted from Van den Brand et al39 with permission.
Isolation of human platelets
Whole blood in citrated vials was centrifuged at 160g for 15 minutes at room temperature without a break. A plastic Pasteur’s pipette was used to transfer platelet-rich plasma (PRP) to a new tube. The plasma was used directly for virus activation assays or transferred into a new tube containing a 1:10 volume of acid-citrate-dextrose (38 mM citric acid, 75 mM sodium citrate, and 135 mM dextrose). PRP was then centrifuged at 400g for 15 minutes at room temperature, with the centrifuge set at brake 1. Afterward, the supernatant was discarded, and the pellet was resuspended in modified Tyrode’s buffer (10 mM HEPES, 137 mM NaCl, 2.8 mM KCl, and 12 mM NaHCO3) with 5.55 mM d-glucose (pH 6.5). Prostaglandin I2 (PGI2; 0.5 µM; Cayman Chemical Co) was added after predilution in modified Tyrode’s buffer (pH 6.5), and the previous centrifugation step was repeated. Finally, the platelets were resuspended in modified Tyrode’s buffer supplemented with 5.55 mM d-glucose (pH 7.4). The platelet count was tested on CELL-DYN Emerald and adjusted to 200 × 109/L.
Platelet-binding assays
Virus-platelet binding measurements were performed by Biolayer Interferometry with an octet RED384 analyzer (Fortebio). Washed platelets (800 × 109/L) were resuspended in PBS supplemented with 0.1 µg/mL prostaglandin E1 (PGE1) and then allowed to bind to Ni-NTA biosensor tips (Fortebio) for 1 hour at room temperature at 100 rpm. Subsequently, the platelets were fixed by incubating the sensors in 0.4% paraformaldehyde (PFA) for 10 minutes. The sensors were stored at 4°C or used immediately afterward to study virus binding kinetics. The binding affinity constant (KD) for PR8 was determined by virus titration from a Langmuir model fit to virus-binding values plotted as a function of virus concentration (range, 0-1.66 × 10−7 M). Virus association was studied in the presence of 100 µM oseltamivir carboxylate (OSC; Hoffmann La-Roche Ltd). Experiments were performed with platelets from a single blood donor. Discontinuous glycan gradients were obtained by mixing 2,3- and 2,6-sialyl-tri-(N-acetyl-lactosamine)-biotin glycans with the nonsialylated di-(N-acetyl-lactosamine)-biotin moieties and functionalization of streptavidin biosensors (n = 8; Fortebio). Initial binding rates were determined with 16.6 pM of virus and plotted as a function of glycan density (range, 0-11.75 pmol/cm2). All virus binding experiments were performed in duplicate.
Transmission electron microscopy
Platelets were fixed for 24 hours in 2.5% glutaraldehyde solution, rinsed with 0.1 M sodium cacodylate buffer (pH 7.2), postfixed in 1% osmium tetroxide, and embedded in EPON 812 (Serva, Heidelberg, Germany), as described.47 For transmission electron microscopy (TEM), platelets infected with influenza viruses were initially fixed with 4% PFA and 0.25% glutaraldehyde in 1 M sodium cacodylate buffer for 24 hours, rinsed with 0.1 M sodium cacodylate, following dehydration in a graded series of alcohol at 4°C. Sections were stained with lead citrate and uranyl acetate and investigated with an EM 10c transmission electron microscope (Carl Zeiss Jena GmbH, Oberkochen, Germany).
Platelet phagocytosis assay
Fluorescein isothiocyanate (FITC)–labeled viruses and washed platelets were mixed at different ratios and incubated at room temperature. To quantify the virus phagocytosis of influenza virus particles by platelets, we adapted previously reported quenching methods, using trypan blue. Platelets were initially labeled with CD42b-APC-Cy7 (Biolegend) to determine the purity of the platelet preparation (>96%) and to set the gate based on the forward and side scatter. Platelets and viruses were incubated for 10 minutes at room temperature (or ice) and then fixed with 0.4% PFA. The platelets were analyzed for FITC fluorescence, with and without quenching, to determine the proportion of virus internalized, which was calculated as the percentage of the median fluorescence intensity (MFI) remaining after quenching. The effect of desialylation on virus uptake was tested on washed platelets pretreated with NA from Clostridium perfringens (Sigma-Aldrich).
Platelet desialylation assay
Viruses and washed platelets were mixed in a 25:1 ratio and incubated in the presence or absence of OSC (100 µM) at room temperature for 20 minutes. The platelets were then fixed with 0.4% PFA, Erythrina cristagalli lectin (ECL)-FITC was added, and the platelets were incubated at room temperature for 1 hour. MFI was determined by flow cytometry on a FACS-Canto cytometer (BD Biosciences).
Platelet activation assay
Serial dilutions of PR8 viruses (range, 0-16.6 pM) and ADP (4 μM) were prepared in HEPES-buffered saline and mixed with anti-P-selectin-Alexa647 (clone B10.6). Subsequently, PRP was added and incubated for 20 minutes, after which the samples were fixed with 0.4% PFA for 10 minutes. MFI was measured by flow cytometry.
Platelet aggregation assay
Platelet aggregometry was performed on a Chrono-Log analyzer (model 700) with PRP (0.5 mL) and viruses at final concentrations ranging from 0 to 100 pM. Experiments were performed at 37°C with stirring at 900 rpm. Addition of ADP (10 µM) was used as a positive control.
Statistical analysis
The relationship between viral load and platelet counts was determined by calculating Pearson’s correlation coefficient. Data are expressed as mean or median ± SD, depending on the distribution. Nonparametric tests were used to analyze the relationship between platelet count and time after infection. Prism software (v.7.04; GraphPad) was used for all data and statistical analysis. We considered P < .05 to indicate statistical significance.
Results
Influenza virus infection leads to thrombocytopenia in humans and ferrets
We quantified viral load and platelet count in adult patients (n = 34) who were admitted to our tertiary hospital from August 2009 through July 2012 with a proven uncomplicated 2009 A/H1N1 virus infection and healthy immune status. Patients were selected as described in the inclusion criteria in “Methods.” We observed that viral load in respiratory swabs correlated inversely with platelet count in the blood of the patients (Figure 1A). We then reproduced this observation experimentally in ferrets, which were infected with a seasonal A/H3N2, pandemic A/H1N1 2009, or highly pathogenic avian influenza (HPAIV) A/H5N1 virus. These viruses induce, respectively, a mild URT or a moderate intermediate or severe LRT infection in humans and ferrets (Figure 1B).39 Groups of animals (n = 4) were infected and euthanized on postinfection day 0.5 and days 1 through 4, and 7, and viral loads in respiratory specimens and blood platelet counts were determined. We observed a strong inverse correlation between platelet count and viral load in swabs of the throat (Figure 1C) and nose (Figure 1D), as we initially observed in humans. Ferrets with a seasonal A/H3N2 or 2009 pandemic A/H1N1 virus infection did not yield such correlations (Figure 1E). Significantly lower platelet counts were observed in the animals infected with either A/H1N1 (P < .0001) or A/H5N1 (P = .003) virus from day 2 onward, in comparison with a mock-infected group (Figure 1F). At the thrombocytopenia trough (days 2-4), platelet counts had decreased 22%, from 639 × 109/L ± 91 × 109/L to 497 × 109/L ± 133 × 109/L in the A/H1N1-infected group (P = .04), which had fully recovered to normal levels at postinfection day 7 (Figure 1G). Platelet loss was most pronounced in the A/H5N1-infected ferrets, which had declined 62% from 778 × 109/L ± 104 × 109/L before infection to 286 × 109/L ± 164 × 109/L (P = .0008) by postinfection day 4 (Figure 1F). In contrast, the A/H3N2 subtype did not cause thrombocytopenia in ferrets.
The ferrets with the A/H3N2 virus presented a URT virus infection similar to that in humans with seasonal influenza (Figures 2A-D). In contrast, the A/H1N1 2009 and HPAIV A/H5N1 viruses caused a severe LRT virus infection in the animals, with severe alveolitis, alveolar edema, and hemorrhage (Figures 2E-F). These observations are very similar to those from experimental lung infections in mice and guinea pigs48-50 and from clinical autopsies of patients who succumbed during the 2009 A/H1N1 virus pandemic.13,24 In the A/H1N1 2009– and HPAIV A/H5N1–infected animals, virus RNA was detected outside the lungs within 12 hours after infection (in the heart, liver, and spleen; Figure 2G-I). Finally, we observed internalized influenza viruses in platelets isolated from the infected ferrets, as observed in patients with influenza51 and reproduced this in vitro by using laboratory virus strain A/PR/8/34 and human platelets (Figure 2J-L).
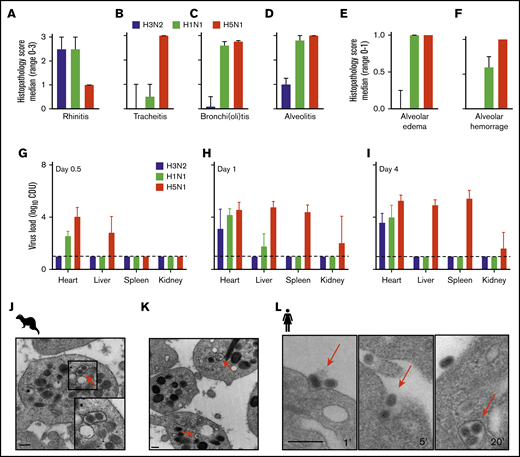
Immunohistopathology and platelet electron microscopy of ferrets infected with different influenza viruses. (A-D) Disease severity scoring of ferrets infected with A/H3N2 (blue), A/H1N1 (green), and A/H5N1 (red) virus at postinfection day 4. Median (n = 4) histopathology score (range 0-3). Error bars, SD. (E-F) Median (n = 4) histopathology score (range, 0-1). (G-I) Virus titer in homogenized organs (heart, liver, and spleen) at days 0.5, 1, and 4. Mean (n = 4). Error bars, SD. Dotted line represents the lower limit of detection. Adapted from Van den Brand et al39 with permission. (J-K) Virus particle internalization (∼120 nm; red arrows) by platelets isolated from the blood drawn from a 2009 A/H1N1 virus-infected ferret. Red arrows: viruses. TEM original magnification ×20 000 (J); ×50 000 (K). The bars represent 250 nm. (L) Internalization of A/PR/8/34 virus by human platelets at 1, 5, and 20 minutes of incubation. The bars bar represent 250 nm.
Immunohistopathology and platelet electron microscopy of ferrets infected with different influenza viruses. (A-D) Disease severity scoring of ferrets infected with A/H3N2 (blue), A/H1N1 (green), and A/H5N1 (red) virus at postinfection day 4. Median (n = 4) histopathology score (range 0-3). Error bars, SD. (E-F) Median (n = 4) histopathology score (range, 0-1). (G-I) Virus titer in homogenized organs (heart, liver, and spleen) at days 0.5, 1, and 4. Mean (n = 4). Error bars, SD. Dotted line represents the lower limit of detection. Adapted from Van den Brand et al39 with permission. (J-K) Virus particle internalization (∼120 nm; red arrows) by platelets isolated from the blood drawn from a 2009 A/H1N1 virus-infected ferret. Red arrows: viruses. TEM original magnification ×20 000 (J); ×50 000 (K). The bars represent 250 nm. (L) Internalization of A/PR/8/34 virus by human platelets at 1, 5, and 20 minutes of incubation. The bars bar represent 250 nm.
Virus binding through interaction with platelet sialoglycans
We hypothesized that the subtype-dependent degree of thrombocytopenia observed in the ferrets was the result of a different interaction between virus and platelet. Therefore, we explored virus binding to isolated human platelets by using biolayer interferometry (BLI) in detail (Figure 3A). The overall virus-binding affinity of the 3 viruses was the same, with association constants (KD) similar to that of the influenza A/PR/8/34 virus (KD = 9.5 ± 1.0 pM), for which we performed full virus-binding titration and virus-uptake experiments (Figures 3B-D). Interestingly, although all binding affinities turned out to be similar, each virus approached a different saturation level (Figure 3D). The saturation level is represented herein by the maximum wavelength shift, which corresponds to the maximum capacity of the virus to bind to platelets (Figure 3C; dotted horizonal line). This level was highest for the A/H5N1 (12.71 nm; 95% confidence interval [CI], 11.43-14.20 nm), lower for the A/H1N1 (10.60 nm; 95% CI, 9.33-12.13 nm), and lowest for the A/H3N2 (3.91 nm; 95% CI, 3.41-4.53 nm) viruses.
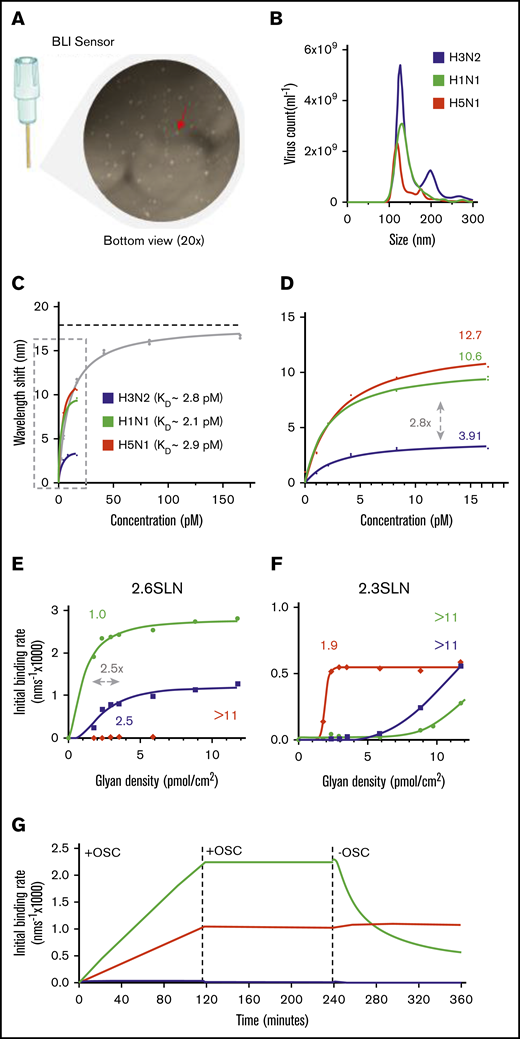
Influenza virus subtypes that bind to platelet sialoglycans. (A) Fixed, washed human platelets attached to a BLI sensor and visualized by light microscopy (original magnification ×20). Red arrow: single platelet. (B) Determination of mean virus particle count by NTA. Total peak values are presented (n = 5). A/H3N2 (5.4 × 1010 ± 3.6 × 109 virus particles per milliliter); A/H1N1 (3.6 × 1010 ± 9.4 × 108 virus particles per milliliter); and A/H5N1 (2.2 × 1010 ± 1.8 × 109 virus particles per milliliter). (C) A highly concentrated A/PR/8/34 (PR8; 1 × 1012 virus particles per milliliter) virus was used for a full titration on fixed platelets in the presence of OSC (100 µM). Mean (n = 2); error bars, SD. The KD value for PR8 (9.5 ± 1.0 pM) was determined from a Langmuir model. The dotted line represents the plateau value (b = 17.89 nm). (D) Enlargement of the dotted box in panel C. Estimated plateau values (b) are 3.91 nm (95% CI, 3.41-4.53 nm) for A/H3N2, 10.6 nm (95% CI, 9.33-12.13 nm) for A/H1N1, and 12.7 nm (95% CI, 11.43-14.20 nm) for A/H5N1. (E) Virus binding to α2,6-sialoglycan density gradients ranging from 0 to 11.75 pmol/cm2. Obtained threshold glycan densities are given above each curve. (F) The same as in panel E, but for the α2,3-sialoglycan density gradient. (G) Virus association and dissociation to BLI-sensors functionalized with α2,3-sialoglycans in the presence and absence of OSC (100 µM).
Influenza virus subtypes that bind to platelet sialoglycans. (A) Fixed, washed human platelets attached to a BLI sensor and visualized by light microscopy (original magnification ×20). Red arrow: single platelet. (B) Determination of mean virus particle count by NTA. Total peak values are presented (n = 5). A/H3N2 (5.4 × 1010 ± 3.6 × 109 virus particles per milliliter); A/H1N1 (3.6 × 1010 ± 9.4 × 108 virus particles per milliliter); and A/H5N1 (2.2 × 1010 ± 1.8 × 109 virus particles per milliliter). (C) A highly concentrated A/PR/8/34 (PR8; 1 × 1012 virus particles per milliliter) virus was used for a full titration on fixed platelets in the presence of OSC (100 µM). Mean (n = 2); error bars, SD. The KD value for PR8 (9.5 ± 1.0 pM) was determined from a Langmuir model. The dotted line represents the plateau value (b = 17.89 nm). (D) Enlargement of the dotted box in panel C. Estimated plateau values (b) are 3.91 nm (95% CI, 3.41-4.53 nm) for A/H3N2, 10.6 nm (95% CI, 9.33-12.13 nm) for A/H1N1, and 12.7 nm (95% CI, 11.43-14.20 nm) for A/H5N1. (E) Virus binding to α2,6-sialoglycan density gradients ranging from 0 to 11.75 pmol/cm2. Obtained threshold glycan densities are given above each curve. (F) The same as in panel E, but for the α2,3-sialoglycan density gradient. (G) Virus association and dissociation to BLI-sensors functionalized with α2,3-sialoglycans in the presence and absence of OSC (100 µM).
Because the KDs for these viruses were the same, we explored the possibility that a difference in receptor preference and/or platelet local glycan density would explain the different levels of saturation.34 To investigate, we immobilized sialoglycans on biosensors with progressively increasing density (Figures 3E-G; range, 0-11.75 pmol/cm2). These sialoglycans are terminated by either α2,3- or α2,6-linked terminal SAs. Both forms are abundantly present at the glycocalyx of human cells. As shown previously, we observed that attachment of the A/H1N1 and A/H3N2 viruses was heavily dependent on α2,6-sialoglycans (Figure 3E). They require a minimal glycan density of 1.0 and 2.5 pmol/cm2, respectively. Notably, this 2.5-fold difference is in the same range as the different platelet saturation levels (2.8-fold) of these 2 viruses (Figure 3D). The A/H5N1 virus binds solely to surfaces with a minimal α2,3-sialoglycan density of 1.9 pmol/cm2 (Figures 3G). Interestingly, although this virus bound irreversibly at high glycan density (11.75 pmol/cm2), the A/H1N1 virus dissociated from the surface when we moved the platelets into an NA inhibitor (OSC)–free buffer (Figure 3G), indicating that virus NA activity also contributes to virus platelet binding and release.
Virus uptake and platelet activation by sialoglycan interaction
We went on to investigate virus uptake by platelets, which we observed in our ferret model and which had been shown recently by others in more detail in humans.51 We used TEM to study the kinetics of uptake of laboratory strain A/PR/8/34 (Figure 4A) by human platelets, which is comparable to the virus (A/WSN/33) used in earlier experiments.51 We observed rapid virus uptake after mixing with platelets in an ∼300:1 ratio, but not by erythrocytes (data not shown). Internalization of virus particles commenced within 1 minute at an estimated initial rate of 1 virus particle every 2 seconds. (Figure 4B; slopes: −0.6 and 0.5 × s−1). Virus uptake of fluorescently labeled A/PR/8/34 viruses was inhibited in experiments on ice (Figure 4C). Uptake was completely abolished by the removal of the SA glycan receptors from the platelet surface, using exogenous NA (Figure 4D). Previous studies had implicated the C-type lectin receptor DC-SIGN (dendritic cell–specific intercellular adhesion molecule-3–grabbing nonintegrin) in dengue virus binding to platelets, by showing the inhibition of primary platelet binding of dengue virus type 2 using an anti-DC-SIGN monoclonal antibody.52 However, blocking of DC-SIGN did not affect influenza virus uptake (Figure 4E), which distinguishes our results from previously published data showing a DC-SIGN–mediated interaction with influenza at the cell surface.53
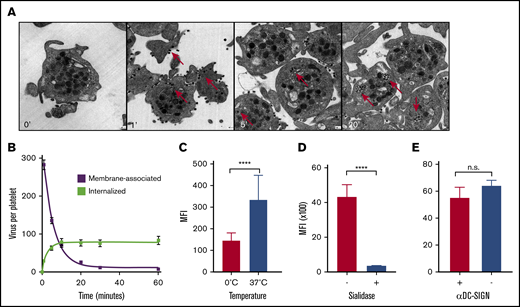
Virus phagocytosis by platelets is dependent on the presence of SAs. (A) Virus uptake over time by human platelets. Original magnification ×25 000. Red arrows: internalized viruses. (B) Quantification of membrane-bound and internalized viruses in ultrathin-sliced platelet sections (n = 100; 50 nm) fixed after 1, 5, 10, 20, 30, and 60 minutes of incubation with influenza A/PR/8/34 virus. Error bars, standard error of the mean. (C) Temperature-dependent virus uptake. Mean (n = 5); error bars, SD. (D) Platelet desialylation with exogenous NA abolishes virus uptake. Control (+) and sialidase-treated (−) platelets. Mean (n = 3); error bars, SD. (E) Virus uptake is independent of DC-SIGN. Uptake in the absence (−) or presence (+) of αDC-SIGN receptor blocking antibody. Mean (n = 4); error bars, SD. ****P < .0001, n.s., nonsignificant, by Student t test.
Virus phagocytosis by platelets is dependent on the presence of SAs. (A) Virus uptake over time by human platelets. Original magnification ×25 000. Red arrows: internalized viruses. (B) Quantification of membrane-bound and internalized viruses in ultrathin-sliced platelet sections (n = 100; 50 nm) fixed after 1, 5, 10, 20, 30, and 60 minutes of incubation with influenza A/PR/8/34 virus. Error bars, standard error of the mean. (C) Temperature-dependent virus uptake. Mean (n = 5); error bars, SD. (D) Platelet desialylation with exogenous NA abolishes virus uptake. Control (+) and sialidase-treated (−) platelets. Mean (n = 3); error bars, SD. (E) Virus uptake is independent of DC-SIGN. Uptake in the absence (−) or presence (+) of αDC-SIGN receptor blocking antibody. Mean (n = 4); error bars, SD. ****P < .0001, n.s., nonsignificant, by Student t test.
Virus subtype-dependent desialylation and platelet activation
A key characteristic of platelet ageing is the gradual loss of SAs from the glycans abundantly present on their cell surface, which can also be accelerated by the intravenous injection of NA in mice.54 Desialylated senescent platelets are designated to the hepatic clearance pathway,10,55-58 which involves recognition of desialylated glycans on platelets by the Ashwell-Morell receptor on liver hepatocytes and macrophages. Therefore, we considered that the influenza virus NA removed SAs from cell surface glycans and proposed the possibility that the interaction between platelets and viruses stimulates accelerated clearance of circulating platelets during influenza and thereby causes thrombocytopenia. Again, we observed that the degree of SA removal was dependent on virus subtype. Sialic acids were removed from the A/H5N1-incubated platelets as shown by increased binding of ECL, which bound solely to nonsialylated glycans (Figure 5A). Virus binding also resulted in the activation of these platelets by the increased expression of P-selectin (CD62P) on the platelet surface (Figure 5B). Platelet activation was dependent on virus concentration (Figure 5C) and was accompanied by platelet aggregation (Figure 5D). The degree of platelet aggregation seemed to differ among virus strains and blood donors. Addition of OSC or the platelet activation inhibitors PGE1 or PGI2 inhibited virus-induced platelet activation (Figure 5B). This observation shows that virus-triggered platelet activation depends on the activity of the virus NA.
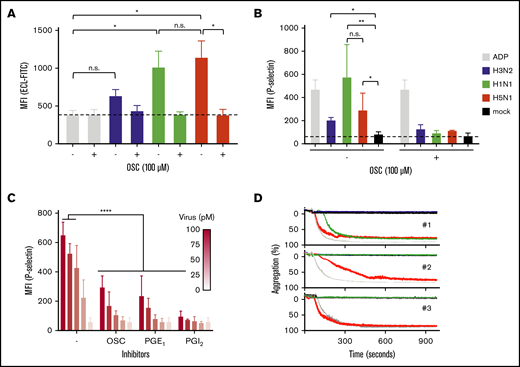
Platelet activation and aggregation upon binding of an influenza virus. (A) Virus NA-dependent desialylation of platelets from human platelets determined by lectin (ECL-FITC) binding. Mean (n = 6); error bars, SD; Tukey’s multiple-comparisons test, *P < .0001; OSC (100 µM). (B) Virus activation of platelets in human plasma. Positive control (ADP; 10 µM). Final virus concentration 50 pM. OSC 100 µM. Mean (6 donors); error bars, SD; Tukey’s multiple-comparisons test, *P < .05; **P < .01. (C) Dose-dependent activation of platelets in plasma by influenza A/PR/8/34 virus. Error bars, SD; Tukey’s multiple-comparisons test, ****P < .0001. (D) Aggregation of platelets by influenza virus subtypes in 3 donors. Positive control (ADP; 10 μM). Final virus concentration (50 pM).
Platelet activation and aggregation upon binding of an influenza virus. (A) Virus NA-dependent desialylation of platelets from human platelets determined by lectin (ECL-FITC) binding. Mean (n = 6); error bars, SD; Tukey’s multiple-comparisons test, *P < .0001; OSC (100 µM). (B) Virus activation of platelets in human plasma. Positive control (ADP; 10 µM). Final virus concentration 50 pM. OSC 100 µM. Mean (6 donors); error bars, SD; Tukey’s multiple-comparisons test, *P < .05; **P < .01. (C) Dose-dependent activation of platelets in plasma by influenza A/PR/8/34 virus. Error bars, SD; Tukey’s multiple-comparisons test, ****P < .0001. (D) Aggregation of platelets by influenza virus subtypes in 3 donors. Positive control (ADP; 10 μM). Final virus concentration (50 pM).
Discussion
Thrombocytopenia is a well-known complication of viral and bacterial infection.17,59 The mechanism of virus-induced thrombocytopenia remains unclear.16,59 Current proposed mechanisms of impaired platelet count include reduced synthesis of the platelet regulator hormone thrombopoietin, enhanced platelet destruction by platelet-associated immunoglobulin G, and platelet-leukocyte aggregation possibly leading to sequestration by macrophages or direct sequestration in the spleen.60,61 Although several factors may influence this acute loss of platelets, such as disseminated intravascular coagulation and bleeding, our study highlighted a previously unrecognized role for platelets in virus clearance during influenza virus infection. We provided evidence to explain the mechanism of influenza virus-induced thrombocytopenia,24,25,62 and showed how the influenza virus is associated with thrombocytopenia by direct interaction with platelets. We confirmed and extended data showing that influenza virus infection leads to significant thrombocytopenia in patients who present with the 2009 A/H1N1 pandemic virus.41 To these data, we added an inverse correlation between patient influenza viral load and platelet count (Figure 1), and we confirmed the data in a preferred influenza ferret model.63 The degree of thrombocytopenia was dependent on influenza virus subtype. Thrombocytopenia was lowest or absent in the seasonal A/H3N2-infected animals and highest in the animals with an A/H5N1 virus infection. We explained this by showing the requirement of a 2.5-fold higher glycan density for the α2.6 sialoglycan for the A/H3N2 virus than for the A/H1N1 virus, and the preference of the A/H5N1 virus for the α2.3 sialoglycan. Clinical observations of thrombocytopenia during severe LRT infections to support this concept are commonly reported during infections with the HPAIV A/H5N1 or A/H7N9 virus.15,40
We showed virus particles in platelets isolated from the blood of infected ferrets (Figure 2J-L) and detected viral RNA in the heart, liver, and spleen of the animals as early as 12 hours after infection (Figure 2G-I). We previously reported that megakaryocytes, the precursors of blood platelets, become activated upon acute influenza A/H5N1 virus infection.64 Therefore, we propose a platelet clearance mechanism by which platelets, upon acute respiratory virus infection, are actively recruited and take up viruses in the lung, after which they reenter the blood circulation. We propose that they are subsequently recognized and cleared from the blood in the liver, spleen, or intestinal tract. These observations together highlight their role in the early innate immune response to respiratory virus infection.
Platelets fulfill key functions in pulmonary immune responses and inflammatory lung diseases.13 Recently, the lung was shown to be a major site for platelet production.3 Also, platelets are increasingly recognized as essential components of the immune system.65 We showed (Figure 5B-C), as have others,16,60 that the platelet activation marker CD62P is expressed on the platelet surface upon incubation with influenza A viruses. Because platelet activation is accompanied by the release of granules, this process may contribute to the cytokine storm associated with acute respiratory distress syndrome (ARDS).66 This fits well with our data showing that platelet activation and aggregation are most pronounced for the more pathogenic A/H1N1 and A/H5N1 viruses and with several clinical observations. For instance, patients with influenza virus infection and thrombocytopenia seem to have an increased risk of (ARDS) and ARDS-related mortality.67 In addition, thrombocytopenia increases mortality from infection,68 and has been shown to be an independent risk factor for infection in patients with primary immune thrombocytopenia.69 Thrombocytopenia and platelet activation are risk factors for mortality in patients with heart failure,70 suggesting that activation of platelets induced by influenza may trigger cardiovascular and hematological diseases.
We previously showed in patients with influenza treated with OSC that platelet counts increase significantly during OSC therapy.71 Although NA inhibitor (NAI) therapy reduces the risk of influenza-induced mortality,72 The efficacy of NAIs remains heavily debated, because of their moderate impact on primary virus end points (eg, reduction of virus titer and days until symptom resolution). We proposed an alternative mode of action by showing that NAI therapy inhibited platelet activation during influenza (Figure 5B). Platelet activation is not currently thought to play a major role during influenza. However, we believe a disturbed platelet function during influenza may have a strong impact on clinical outcome, especially in patients with an underlying cardiovascular medical condition.
In Figure 5, we show the potency of PGI2 to inhibit influenza-induced platelet activation pointing toward the involvement of the G protein (GP)–coupled GPIb-X-IV receptor complex.73 This complex, which is targeted by the von Willebrand factor ligand, is a heavily glycosylated glycoprotein that is abundantly found on platelets. We propose that influenza viruses bind the GP-X-IV complex and, as a side effect, trigger downstream activation signaling, including the thromboxane A2 pathway.
It has been shown that DC-SIGN enhances influenza virus entry into primary human cells53,74 and that entry is blocked by anti-DC-SIGN monoclonal antibodies. We did not find such an inhibitory effect (Figure 5E), indicating that DC-SIGN plays a minor role in the interaction between influenza viruses and platelets.
It has been shown that platelets are among the first cells to arrive at a site of infection from whence they orchestrate innate and adaptive immune responses,75,76 by stimulating recruitment of immune cells,59 including neutrophils,77,78 macrophages,61 and CD8+ T cells.61,79 Also, platelets can capture invading bacterial pathogens,75 directly77 or indirectly, by promoting the formation of neutrophil extracellular traps.80,81 The receptors described as playing a role in the uptake of viruses and bacteria include DC-SIGN, CLEC2, and several Toll-like receptors.82 Our data add to those findings and extend earlier work on uptake of viruses,83-86 showing a critical and direct role for platelet-mediated influenza virus uptake, as mediated by surface SA glycans.
In summary, the evidence supports an early platelet innate immune response in the respiratory tract, further clarifying the process of thrombocytopenia during influenza and other respiratory virus infections that are associated with acute cardiovascular and thrombotic events.
Original data are available in response to an e-mail request to the corresponding author.
Acknowledgments
The authors thank Melle van Schaik for support at the Research Center for Emerging Infections and Zoonoses (RIZ); Richard Wubbolts from the Centre for Cell Imaging (Faculty of Veterinary Medicine, University of Utrecht) for expert help and fruitful discussions; Ron Fouchier for providing the influenza viruses; and Xander de Haan and Frank van Kuppeveld for making the BLI instrument available.
This work was supported by grants from the European Hematology Association (A.J.G.J.) and the German Volkswagen Foundation (E.v.d.V. and J.H.).
Authorship
Contribution: E.v.d.V. and A.J.G.J. conceived the project; E.v.d.V., A.J.G.J., C.M., and M.B. designed the research; A.B., A.O., T.K., C.M., K.R., W.B., E.v.d.V., G.-J.B., and A.J.G.J. provided reagents and material; E.v.d.V. and A.J.G.J. provided clinical data; H.Z.L., E.v.d.V., T.S., A.B., M.T., J.v.d.B., D.D.I., K.R., G.v.A., and K.S. performed the experiments; E.v.d.V., K.R., M.T., D.D.I., and T.S. prepared the figures; E.v.d.V., A.O., and J.H. acquired funding; E.v.d.V. and A.J.G.J. wrote the initial draft of the manuscript, with input from J.H., H.Z.L., W.B., C.M., and M.B.; and all authors reviewed the final draft.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for J.v.d.B. is Department of Pathology, University of Utrecht, Utrecht, The Netherlands.
The current affiliation for E.v.d.V. is Royal GD (Gezondheidsdienst voor Dieren BV), Deventer, The Netherlands.
Correspondence: Erhard van der Vries, Royal GD, Arnsbergstraat 7, 7418 EZ Deventer, The Netherlands; e-mail: e.vd.vries@gdanimalhealth.com.

