

Key Points
GMFG-knockdown murine macrophages exhibit an iron deficiency response and polarization toward the M2 phenotype.
GMFG modulates mtROS generation and respiration chain components, as well as ISCU, protein expression in murine macrophages.

Abstract
In macrophages, cellular iron metabolism status is tightly integrated with macrophage phenotype and associated with mitochondrial function. However, how molecular events regulate mitochondrial activity to integrate regulation of iron metabolism and macrophage phenotype remains unclear. Here, we explored the important role of the actin-regulatory protein glia maturation factor-γ (GMFG) in the regulation of cellular iron metabolism and macrophage phenotype. We found that GMFG was downregulated in murine macrophages by exposure to iron and hydrogen peroxide. GMFG knockdown altered the expression of iron metabolism proteins and increased iron levels in murine macrophages and concomitantly promoted their polarization toward an anti-inflammatory M2 phenotype. GMFG-knockdown macrophages exhibited moderately increased levels of mitochondrial reactive oxygen species (mtROS), which were accompanied by decreased expression of some mitochondrial respiration chain components, including the iron-sulfur cluster assembly scaffold protein ISCU as well as the antioxidant enzymes SOD1 and SOD2. Importantly, treatment of GMFG-knockdown macrophages with the antioxidant N-acetylcysteine reversed the altered expression of iron metabolism proteins and significantly inhibited the enhanced gene expression of M2 macrophage markers, suggesting that mtROS is mechanistically linked to cellular iron metabolism and macrophage phenotype. Finally, GMFG interacted with the mitochondrial membrane ATPase ATAD3A, suggesting that GMFG knockdown–induced mtROS production might be attributed to alteration of mitochondrial function in macrophages. Our findings suggest that GMFG is an important regulator in cellular iron metabolism and macrophage phenotype and could be a novel therapeutic target for modulating macrophage function in immune and metabolic disorders.
Introduction
Macrophages play vital roles in iron homeostasis and immunity and acquire distinct functional activation phenotypes dependent upon environmental cues.1-3 M1 macrophages are activated by lipopolysaccharide (LPS) and interferon-γ (IFN-γ) and characterized by expression of a wide range of proinflammatory genes.4,5 M2 macrophages are induced by interleukin-4 (IL-4)/IL-13, IL-10, transforming growth factor β, or glucocorticoids and characterized by high expression of anti-inflammatory and tissue repair genes.6,7 M2 macrophages play important roles in the resolution of inflammation, allergy, parasite infection, tissue remodeling, and wound repair, as well as in tumor growth and metastasis.8,9 Therefore, manipulation of M2 polarization may represent an attractive pharmacological target to treat chronic inflammation–associated metabolic disease and promote tissue repair.
Activated macrophages exhibit remarkably distinct differences in iron handling between M1 and M2 macrophages.10-12 M1 macrophages adopt an iron sequestration phenotype by upregulating the iron storage protein ferritin (Ft) and downregulating the iron export protein ferroportin 1 (FPN), whereas M2 macrophages acquire an iron deficiency phenotype by upregulating transferrin receptor 1 (TfR1) and FPN as well as downregulating Ft. Recent studies have shown that iron accumulation drives macrophages toward the M1 phenotype under inflammatory conditions,13-15 indicating not only that altered iron metabolism is a characteristic of polarized macrophage phenotypes, but also that intracellular iron status shapes macrophage polarization. However, limited information exists about how scavenging iron affects the polarization of M2 macrophages.
Mitochondria are central regulators for modulating metabolic reprogramming and controlling macrophage phenotype functions.16 Mitochondria also play a key role in iron metabolism in that they synthesize heme, assemble iron-sulfur proteins, and participate in cellular iron regulation.17,18 Disruption of biosynthesis of the mitochondrial iron-sulfur cluster biosynthesis scaffold protein (ISCU) triggers the iron deficiency response.19 Recent studies of the mechanism bridging immune macrophage phenotype and mitochondrial metabolic functions suggest that the mitochondrial electron transport chain (ETC), which regulates mitochondrial reactive oxygen species (mtROS), plays an important role in macrophage activation processes.20-22 However, little is known about how mitochondrial functions, especially mtROS, are integrated into the molecular mechanisms of cellular iron metabolism in the context of macrophage phenotype.
Glia maturation factor-γ (GMFG), a novel regulator of the actin-related protein-2/3 complex, is predominantly expressed in inflammatory cells. GMFG belongs to the actin depolymerization factor/cofilin family and modulates actin cytoskeleton reorganization in microvascular endothelial cells and human airway smooth muscle.23,24 Moreover, GMFG regulates the chemotaxis of neutrophils and T lymphocytes,25,26 migration of ovarian and colorectal cancer cells, and angiogenic sprouting in zebrafish.27-29 In addition, it has been reported that GMFG is downregulated during erythroid maturation30 and in response to LPS.31
The aim of this study was to investigate the role of GMFG in regulating cellular iron homeostasis and macrophage polarization. We found that GMFG knockdown in murine macrophages resulted in an iron deficiency response with increased iron levels and concomitantly primed macrophages toward an anti-inflammatory functional M2 phenotype, which occurred through moderately increased mtROS induced by partial suppression of mitochondrial function. The results of this study provide a new perspective that will help elucidate the mechanism by which iron metabolism regulates macrophage phenotype.
Materials and methods
Macrophage culture and treatment
RAW264.7 macrophages were cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) at 37°C in 5% carbon dioxide. Mouse bone marrow–derived macrophages (BMDMs) were prepared as previously described.32 Briefly, bone marrow cells were isolated from mouse femur and tibia specimens and incubated for 7 days in petri dishes in DMEM containing 10% FBS and 50 ng/mL of murine macrophage colony-stimulating factor (R&D Systems). BMDMs were transiently transfected at day 5 of culture. For M1 or M2 activation of macrophages, day 7 BMDMs or RAW264.7 macrophages were stimulated with murine LPS (100 ng/mL) plus IFN-γ (20 ng/mL) or IL-4 (20 ng/mL) plus IL-13 (20 ng/mL) for 24 hours. For analysis of GMFG or iron metabolism protein expression, RAW264.7 macrophages were treated with ferric ammonium citrate (FAC; 0-150 µM), deferoxamine (DFO; 0-150 µM), or hydrogen peroxide (0-250 µM) for 24 hours. In some experiments, GMFG-knockdown macrophages were treated with the antioxidant N-acetylcysteine (NAC; 8 mM) for 30 minutes before analysis. All animal experiments were approved by the National Institutes of Health Ethics Committee.
RNA interference and transfection
BMDMs (1 × 106 cells) or RAW264.7 cells (American Type Culture Collection; 2 × 106 cells) were transiently transfected for 48 hours with a murine GMFG small interfering RNA (siRNA; s82084) or control siRNA (Thermo Fisher Scientific) using Nucleofector II (Amaxa Biosystems). Human THP-1 cells (American Type Culture Collection; 2 × 106 cells) were transiently transfected for 48 hours with a GMFG siRNA (s18302) or control siRNA using Nucleofector II. All functional experiments with siRNA-transfected cells were performed after 48 hours of transfection, when GMFG expression levels were reduced by 75% in GMFG siRNA-transfected cells compared with control siRNA-transfected cells. For GMFG rescue experiments, cells were first transiently transfected with a control siRNA or GMFG siRNA for 48 hours, followed by cotransfection with green fluorescent protein (GFP) vector or GMFG-GFP expression plasmid (OriGene) and continuous culture in complete growth medium for an additional 24 hours. Cells were then subjected to further analysis.
Measurement of mtROS and contents
To measure mtROS, cells were stained with 2.5 μM of MitoSOX reagent (Thermo Fisher Scientific) in serum-free DMEM for 15 to 30 minutes at 37°C in the dark. Stained cells (1 × 105) were subjected to flow cytometric analysis using FACSCalibur (Becton-Dickinson). For measurement of mitochondrial contents, cells were stained with 200 nM of MitoTracker Green (for total mitochondrial mass) or 200 nM of MitoTracker Red or TMRM (for mitochondrial membrane potential; Thermo Fisher Scientific) in serum-free DMEM for 30 minutes at 37°C in the dark. Stained cells were subjected to flow cytometric analysis using FACSCalibur (Becton-Dickinson).
Flow cytometric assay
For the analysis of expression levels of TfR1 (CD71) on cell surfaces, cells were incubated with Alexa Fluor 488–conjugated CD71 antibody for 30 minutes at room temperature. Cells were then resuspended and subjected to flow cytometric analysis using FACSCalibur. The data were analyzed using CellQuest software (BD Biosciences). Cell surface expression of TfR1 was quantified as the median fluorescence intensity (MFI) determined by flow cytometry.
Immunoblotting
For immunoblotting analysis of the iron metabolism–related protein expression levels, cells were prepared in 10 mM of tris(hydroxymethyl)aminomethane/hydrochloride at a pH of 8.0, 150 mM of sodium chloride, 1% Nonidet P40, 0.1% sodium dodecyl sulfate (SDS), 10 mM of EDTA, and protease inhibitors (Thermo Fisher Scientific) for 30 minutes on ice. For detection of GMFG protein expression levels and all other protein expression levels, cells were lysed in radioimmunoprecipitation assay buffer containing protease inhibitors (Thermo Fisher Scientific). Proteins samples (40 μg per lane) were incubated at 70°C for 10 minutes for GMFG antibody, at 37°C for 10 minutes for FPN antibody, or at 95°C for 8 minutes for the other antibodies. The samples were then separated on 4% to 20% SDS polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes. Membranes were probed with primary antibodies for GMFG (Abgent), CORE2, SOD2, ATPase family AAA domain-containing protein 3A (ATAD3A), or α-tubulin (Santa Cruz Biotechnology); FPN (Alpha Diagnostic International), Ft light chain (FtL), iron regulatory protein 1 (IRP1), IRP2, hypoxia-inducible factor 2α (HIF-2α), SDHD, or COX5A (Abcam); TfR1 or NDUFV2 (Thermo Fisher Scientific); or ISCU or SOD1 (Novus Biologicals). Immunoblots were then incubated with horseradish peroxidase–conjugated secondary antibodies, and the Amersham Biosciences ECL Western Blotting System (GE Healthcare) was used for detection. For densitometric quantification analyses of blot intensities, appropriate film exposures were scanned, and the density of bands was determined with Quantity One software (Life Science Research, Bio-Rad) and normalized to α-tubulin.
Immunoprecipitation analysis
For endogenous coimmunoprecipitation analysis, human THP-1 cells were lysed in IP Lysis Buffer (Thermo Fisher Scientific) and incubated overnight with GMFG monoclonal antibody (LifeSpan BioSciences) or ATAD3A monoclonal antibody (Thermo Fisher Scientific) followed by addition of Dynabeads protein G (Thermo Fisher Scientific) for 3 hours at 4°C. The immunoprecipitates were washed, and the proteins were eluted and then subjected to immunoblotting. For immunoprecipitation analysis using transient transfection, HEK-293T cells were transiently cotransfected with GFP-tagged GMFG plasmid and Myc-DDK–tagged ATAD3A plasmid using Lipofectamine 2000 Reagent (Life Technologies). Forty-eight hours after transfection, cells were lysed and immunoprecipitated overnight with anti-GFP or anti-Myc antibodies (OriGene). Dynabeads protein G (Thermo Fisher Scientific) was added, and the mixture was incubated for 3 hours at 4°C. The immunoprecipitated complexes/beads were washed 3 times in washing buffer, and the proteins were eluted in 30 μL of SDS sample buffer. Proteins were then subjected to immunoblotting.
Measurement of intracellular total and labile iron pools
Total intracellular iron concentrations were determined by inductively coupled plasma mass spectrometry on an Agilent 7900 Triple Quadrupole ICP-MS instrument. Labile iron pool was measured using the calcein AM iron-sensitive probe. Briefly, macrophages were incubated with 0.25 μM of calcein AM in αMEM at 37°C for 15 minutes. After washing, cells were incubated in Hanks balanced salt solution supplemented with 10 mM of glucose (pH, 7.35) with or without 100 μM of salicylaldehyde isonicotinoyl hydrazone (SIH) at 37°C for 30 minutes. Fluorescence was then analyzed using FACSCalibur (Becton-Dickinson). The labile iron pool was calculated by subtracting the MFI of the sample without SIH from the MFI of the sample treated with SIH.
Immunofluorescence staining and confocal microscopy
GMFG-knockdown RAW264.7 macrophages were fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) for 20 minutes, permeabilized with 0.1% Triton X-100/PBS for 10 minutes, and preblocked in 10% FBS/PBS for 1 hour. Cells were then stained with mouse monoclonal anti-TfR1 antibody (Life Technologies; 1:500) for 3 hours, followed by staining with Alexa Fluor 488–conjugated anti-rabbit antibody (Invitrogen; diluted 1:500) for 1 hour at room temperature. Nuclear DNA was stained with DAPI (Sigma-Aldrich) for 5 minutes. For the analysis of internalization of Tf, GMFG-knockdown RAW264.7 macrophages were exposed to 5 μg/mL of Alexa Fluor 568–conjugated Tf (Invitrogen) in 3% FBS/Hanks balanced salt solution for 30 minutes. Internalization was stopped by washing the cells with cold PBS. Bound Tf was removed by washing with PBS at a pH of 5.0. Cells were examined under a Zeiss LSM800 confocal microscope equipped with 405-, 488-, 594-, and 633-nm lasers and Zen 2009 imaging software, using a 63X/1.3 NA oil-immersion objective (Carl Zeiss).
Electrophoretic mobility shift assays
RAW264.7 cells were transiently transfected with GMFG siRNA or control siRNA for 72 hours. Cells were lysed in lysis buffer consisting of 10 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH, 7.6), 3 mM of magnesium chloride, 40 mM of potassium chloride, 5% glycerol, 0.2% Nonidet P40 (Sigma-Aldrich), 1 mM of dithiothreitol, and protease inhibitors (Sigma-Aldrich). Equal amounts of protein from cell lysates were incubated with a molar excess of iron regulatory element (IRE) probe transcribed in vitro from the pSPT-fer plasmid containing the IRE of the human Ft H chain in the presence of [32P]uridine triphosphate using a commercially available kit (Perkin Elmer). After separation on nondenaturing polyacrylamide gels (6%), RNA-protein complexes were visualized by autoradiography.
Statistical analysis
Experiments were carried out in duplicate or triplicate. Parametric data are expressed as the mean ± standard error of the mean. Statistical significance was assessed with the unpaired Student t test. P < .05 was considered statistically significant.
Results
GMFG regulates iron metabolism protein expression in murine macrophages
To explore the effects of GMFG on intracellular iron metabolism, we first investigated whether GMFG expression is regulated by iron in murine macrophages. Treatment of RAW264.7 macrophages with FAC markedly downregulated GMFG protein levels at concentrations from 50 to 150 μM (Figure 1A). Although GMFG mRNA expression was significantly decreased at low concentrations of FAC (25-50 μM), mRNA levels returned to baseline at higher FAC concentrations (100-150 μM; Figure 1C upper panel), suggesting that GMFG downregulation at higher iron concentrations was mainly dependent on posttranscriptional modification. Although no significant changes in GMFG mRNA levels were observed in response to the iron chelator DFO (Figure 1C lower panel), GMFG protein levels were increased at DFO concentrations of ≥100 μM (Figure 1B). Our results suggest that GMFG protein level may be regulated by iron in murine macrophages.

GMFG regulates iron metabolism protein expression in murine macrophages. (A-B) RAW264.7 macrophages were treated with FAC (A; 0-150 µM) or DFO (B; 0-150 µM) in 10% FBS/DMEM for 24 hours. Immunoblot analysis of GMFG in cellular lysates (upper). α-tubulin was used as a loading control. Relative quantification of GMFG protein expression levels from densitometric scans after normalizing to the control α-tubulin (lower graphs). (C) Quantitative polymerase chain reaction (qPCR) analysis of mean relative GMFG messenger RNA (mRNA) expression in FAC- or DFO-treated RAW264.7 macrophages. The data were normalized to 18S mRNA expression. (D) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages or BMDMs transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours. α-tubulin was used as a loading control. Graphs (right) show relative quantification of immunoblot (left). Protein levels from densitometric scans are normalized to the control α-tubulin and presented as fold change relative to control siRNA-transfected cells. (E) qPCR analysis of mean relative mRNA expression levels of iron metabolism proteins in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours. The data were normalized to 18S mRNA expression. (F) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then treated with vehicle, FAC (50-100 µM), or DFO (100 µM) for 24 hours. α-tubulin was used as a loading control. Graphs (right) show relative quantification of immunoblot (left). Protein levels from densitometric scans are normalized to the control α-tubulin and presented as fold change relative to control siRNA-transfected cells before treatments. (G) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, followed by cotransfection of GFP vector or GMFG-GFP plasmid for another 24 hours. α-tubulin was used as a loading control. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control untreated cells or control siRNA-transfected cells. a.u., arbitrary units.
GMFG regulates iron metabolism protein expression in murine macrophages. (A-B) RAW264.7 macrophages were treated with FAC (A; 0-150 µM) or DFO (B; 0-150 µM) in 10% FBS/DMEM for 24 hours. Immunoblot analysis of GMFG in cellular lysates (upper). α-tubulin was used as a loading control. Relative quantification of GMFG protein expression levels from densitometric scans after normalizing to the control α-tubulin (lower graphs). (C) Quantitative polymerase chain reaction (qPCR) analysis of mean relative GMFG messenger RNA (mRNA) expression in FAC- or DFO-treated RAW264.7 macrophages. The data were normalized to 18S mRNA expression. (D) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages or BMDMs transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours. α-tubulin was used as a loading control. Graphs (right) show relative quantification of immunoblot (left). Protein levels from densitometric scans are normalized to the control α-tubulin and presented as fold change relative to control siRNA-transfected cells. (E) qPCR analysis of mean relative mRNA expression levels of iron metabolism proteins in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours. The data were normalized to 18S mRNA expression. (F) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then treated with vehicle, FAC (50-100 µM), or DFO (100 µM) for 24 hours. α-tubulin was used as a loading control. Graphs (right) show relative quantification of immunoblot (left). Protein levels from densitometric scans are normalized to the control α-tubulin and presented as fold change relative to control siRNA-transfected cells before treatments. (G) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, followed by cotransfection of GFP vector or GMFG-GFP plasmid for another 24 hours. α-tubulin was used as a loading control. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control untreated cells or control siRNA-transfected cells. a.u., arbitrary units.
We next examined GMFG-knockdown macrophages to find whether GMFG influenced the expression of the major iron metabolism proteins in macrophages. Knockdown of GMFG in RAW264.7 macrophages and BMDMs remarkably increased the protein levels of TfR1 and FPN, whereas levels of the FtL were decreased, compared with those in control siRNA-transfected cells (Figure 1D). However, the mRNA levels of FPN and FtL in GMFG-knockdown RAW264.7 macrophages were comparable with those of control cells (Figure 1E). Although TfR1 mRNA expression was increased 1.8-fold in GMFG-knockdown RAW264.7 macrophages, TfR1 protein levels were even more upregulated in GMFG-knockdown RAW264.7 macrophages and BMDMs compared with control siRNA-transfected cells. These results suggest that GMFG-knockdown regulation of these iron metabolism–related proteins is coordinated largely at the posttranscriptional level.
To examine the response of GMFG-knockdown macrophages to cellular iron variation, we analyzed the expression of TfR1, FPN, and FtL in GMFG-knockdown RAW264.7 macrophages under conditions of iron repletion and depletion. Knockdown of GMFG induced alteration of iron metabolism protein expression similar to that observed in control siRNA-transfected macrophages treated with the iron chelator DFO, indicating that GMFG knockdown is associated with a cellular iron deficiency response. Treatment with FAC at 100 µM markedly reversed the GMFG knockdown–induced alteration of TfR1, FPN, and FtL protein expression. Conversely, treatment with iron chelator DFO in GMFG-knockdown cells did not affect TfR1 expression, but it markedly decreased FPN expression and further decreased FtL expression (Figure 1F). These results suggest that upregulation of TfR1 may be primarily responsible for the iron deficiency response induced by GMFG knockdown, subsequently leading to a coordinated increase of FPN and decrease of FtL expression. Furthermore, GMFG overexpression, but not empty vector expression, abolished the GMFG knockdown–induced iron deficiency response in RAW264.7 macrophages, confirming that GMFG directly modulates cellular iron homeostasis in macrophages (Figure 1G).
Knockdown of GMFG in macrophages increases intracellular iron content
We further analyzed TfR1 expression in GMFG-knockdown macrophages using immunofluorescence and flow cytometry. Immunofluorescence analyses showed increased TfR1 localization in the cytosol and plasma membranes of GMFG-knockdown RAW264.7 macrophages compared with control siRNA-transfected cells (Figure 2A). Surprisingly, flow cytometric analysis indicated that expression of TfR1 (CD71) on the surface of GMFG-knockdown RAW264.7 cells was not significantly increased compared with that observed for control siRNA-transfected cells (Figure 2B). We next examined the functional Tf uptake in GMFG-knockdown macrophages by incubating the cells with Alexa Fluor–conjugated Tf and then examining the internalized fluorescence by confocal microscopy. We found that Tf internalization at 30 minutes was comparable in GMFG-knockdown and control siRNA–transfected cells (Figure 2C), suggesting that GMFG knockdown did not affect TfR1 endocytosis in RAW264.7 macrophages. Furthermore, we found that both total intracellular iron levels and labile iron levels were significantly increased in GMFG-knockdown RAW264.7 macrophages compared with control cells (Figure 2D-E). These results suggest that GMFG knockdown–induced iron accumulation is associated with increased TfR1 expression to meet the iron deficiency response. Altogether, our results suggest that GMFG is a crucial regulator of macrophage iron homeostasis.

Knockdown of GMFG in macrophages increases intracellular iron content. (A) Representative images of immunofluorescence analysis of TfR1 in RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours. TfR1 was visualized using anti-TfR1 antibody and Alexa Fluor 488–conjugated secondary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Scale bar, 50 μm. Quantification (right) of immunofluorescence intensity (left). Data represent the normalized mean corrected fluorescence intensity (MFI) ± standard deviation of 3 independent experiments. (B) Flow cytometry analysis of cell surface expression levels of TfR1 (CD71) in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours. Representative histogram of unstained control siRNA-transfected cells (gray) or cells stained with the anti-CD71 antibody in control siRNA-transfected cells (red) and GMFG siRNA-transfected cells (blue) in control siRNA (left). Quantification (right) of CD71 cell surface expression flow cytometry results (left). Data represent the normalized MFI ± SD of 3 independent experiments. (C) Representative images of immunofluorescence analysis of Alexa Fluor 568–conjugated Tf internalization in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours. Scale bar, 50 μm. (D) Analysis of total intracellular iron levels in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 72 hours as measured by inductively coupled plasma mass spectrometry. Iron content was normalized to the total iron in control siRNA-transfected cells. (E) Analysis of labile iron pool in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 72 hours as measured by calcein AM. Iron content was normalized to the total iron in control siRNA-transfected cells. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control siRNA-transfected cells.
Knockdown of GMFG in macrophages increases intracellular iron content. (A) Representative images of immunofluorescence analysis of TfR1 in RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours. TfR1 was visualized using anti-TfR1 antibody and Alexa Fluor 488–conjugated secondary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Scale bar, 50 μm. Quantification (right) of immunofluorescence intensity (left). Data represent the normalized mean corrected fluorescence intensity (MFI) ± standard deviation of 3 independent experiments. (B) Flow cytometry analysis of cell surface expression levels of TfR1 (CD71) in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours. Representative histogram of unstained control siRNA-transfected cells (gray) or cells stained with the anti-CD71 antibody in control siRNA-transfected cells (red) and GMFG siRNA-transfected cells (blue) in control siRNA (left). Quantification (right) of CD71 cell surface expression flow cytometry results (left). Data represent the normalized MFI ± SD of 3 independent experiments. (C) Representative images of immunofluorescence analysis of Alexa Fluor 568–conjugated Tf internalization in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours. Scale bar, 50 μm. (D) Analysis of total intracellular iron levels in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 72 hours as measured by inductively coupled plasma mass spectrometry. Iron content was normalized to the total iron in control siRNA-transfected cells. (E) Analysis of labile iron pool in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 72 hours as measured by calcein AM. Iron content was normalized to the total iron in control siRNA-transfected cells. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control siRNA-transfected cells.
Knockdown of GMFG promotes M2 macrophage polarization
Considering that macrophage polarization is closely associated with iron metabolism state, and that M2-polarized macrophages exhibit an iron export phenotype,10,11 we next examined whether GMFG knockdown influenced polarization of macrophages toward the M2 phenotype. GMFG knockdown in BMDMs significantly increased mRNA expression of Arg1, Mrc1, IL-10, Ym2, and Fizz1 in M2 macrophages induced by IL-4/IL-13 compared with control siRNA-transfected M2 BMDMs (Figure 3A-E). However, induction of the M1 gene IL-6 in LPS/IFN-γ–induced M1 macrophages was significantly reduced by GMFG knockdown in BMDMs (Figure 3F). Furthermore, these enhanced responses to GMFG knockdown in IL-4/IL-13–induced M2 macrophages were confirmed in RAW264.7 macrophages (data not shown). Importantly, overexpression of GMFG in GMFG-knockdown BMDMs diminished the increased expression of M2 markers in M2-polarized macrophages, demonstrating that GMFG overexpression directly rescued the effects of GMFG knockdown on the M2 phenotype in macrophages (Figure 3G-I).
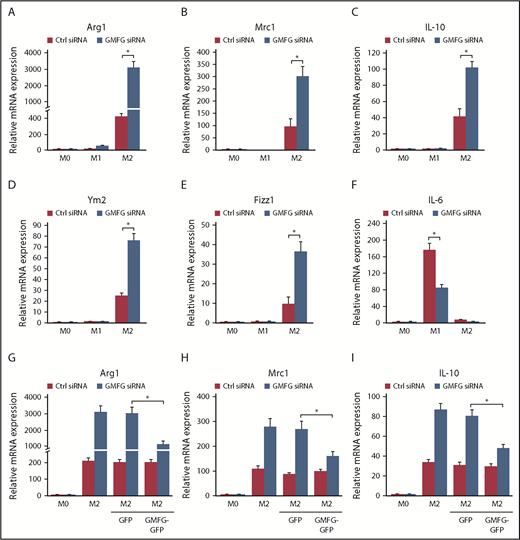
Knockdown of GMFG promotes M2 macrophage polarization. (A-F) BMDMs were transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. Bar graphs represent quantitative polymerase chain reaction (qPCR) analysis of mean relative mRNA expression of M2 (A-E) or M1 (F) macrophage marker genes normalized to 18S mRNA expression. (G-I) qPCR analysis of mean relative mRNA expression of M2 macrophage marker genes in BMDMs transfected with control siRNA or GMFG siRNA for 48 hours, followed by cotransfection of GFP vector or GMFG-GFP plasmid for another 24 hours, then stimulated without (M0) or with M2 macrophage inducers for 24 hours, normalized to 18S mRNA expression. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control siRNA-transfected cells.
Knockdown of GMFG promotes M2 macrophage polarization. (A-F) BMDMs were transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. Bar graphs represent quantitative polymerase chain reaction (qPCR) analysis of mean relative mRNA expression of M2 (A-E) or M1 (F) macrophage marker genes normalized to 18S mRNA expression. (G-I) qPCR analysis of mean relative mRNA expression of M2 macrophage marker genes in BMDMs transfected with control siRNA or GMFG siRNA for 48 hours, followed by cotransfection of GFP vector or GMFG-GFP plasmid for another 24 hours, then stimulated without (M0) or with M2 macrophage inducers for 24 hours, normalized to 18S mRNA expression. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control siRNA-transfected cells.
Knockdown of GMFG in macrophages alters iron metabolism protein expression to mimic that observed in the M2 macrophage phenotype
We further investigated the effect of GMFG knockdown on iron metabolism protein expression in macrophages in response to M1 and M2 macrophage polarization. GMFG-knockdown M0 RAW264.7 macrophages displayed a TfR1high FtLlow FPNhigh pattern, which shared similarities with the pattern displayed in IL-4/IL-13–induced M2 macrophages, but not that observed in LPS/IFN-γ–induced M1 macrophages, in control siRNA-transfected cells (Figure 4A). Among these altered proteins, only TfR1 expression levels were further increased in GMFG-knockdown M2 macrophages compared with control M2 macrophages, suggesting that GMFG knockdown–enhanced TfR1 expression might be mediated by different mechanisms than those in IL-4/IL-13–induced M2 macrophages.
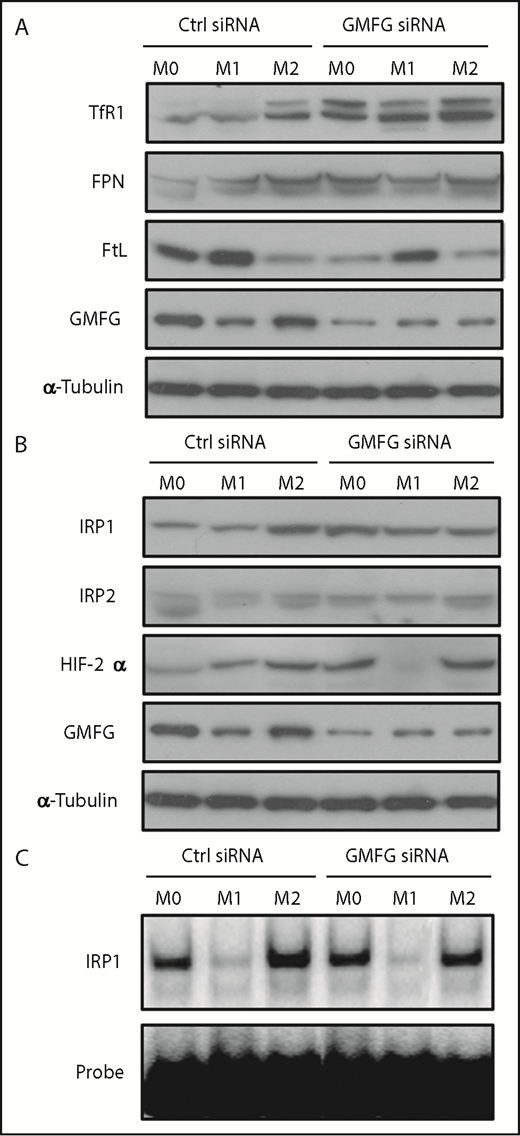
Knockdown of GMFG in macrophages alters iron metabolism protein expression to mimic that observed in the M2 macrophage phenotype. (A) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. α-tubulin was used as a loading control. (B) Immunoblot analysis of IRP1, IRP2, and HIF-2α in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 or M2 macrophage inducers. α-tubulin was used as a loading control. (C) Representative RNA-binding activity of IRP1 analyzed by electrophoretic mobility shift assays. RAW264.7 macrophages were transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 or M2 macrophage inducers for 24 hours. Cytoplasmic lysates were incubated with an excess of a phosphorus-32–labeled iron regulatory element probe. RNA-protein complexes were resolved on nondenaturing polyacrylamide gels and revealed by autoradiography.
Knockdown of GMFG in macrophages alters iron metabolism protein expression to mimic that observed in the M2 macrophage phenotype. (A) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. α-tubulin was used as a loading control. (B) Immunoblot analysis of IRP1, IRP2, and HIF-2α in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 or M2 macrophage inducers. α-tubulin was used as a loading control. (C) Representative RNA-binding activity of IRP1 analyzed by electrophoretic mobility shift assays. RAW264.7 macrophages were transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 or M2 macrophage inducers for 24 hours. Cytoplasmic lysates were incubated with an excess of a phosphorus-32–labeled iron regulatory element probe. RNA-protein complexes were resolved on nondenaturing polyacrylamide gels and revealed by autoradiography.
Because IRP1 and IRP2 are the principal posttranscriptional regulators of cellular iron homeostasis,33 we next examined their expression in GMFG-knockdown M0, M1, and M2 macrophages. IRP1 protein levels were increased in GMFG-knockdown M0 macrophages and in control M2 macrophages compared with control M0 macrophages. However, IRP2 expression was comparable in GMFG-knockdown and control macrophages across all phenotypes examined. Moreover, consistent with previous reports,34,35 HIF-2α expression was increased in GMFG-knockdown M0 and M2 macrophages, as well as in control M2 macrophages, compared with the expression observed in control M0 cells (Figure 4B). To determine whether IRP1 contributed to the induction of TfR1 observed with GMFG downregulation, we investigated the IRE-binding activity of IRP1 in GMFG-knockdown RAW264.7 macrophages. IRE-binding activity of IRP1 in each of the phenotypes (M0, M1, and M2) of GMFG-knockdown cells was similar to that observed in matched control siRNA-transfected cells (Figure 4C), indicating that GMFG knockdown–induced elevation of TfR1 may occur independent of the IRP1 pathway.
GMFG modulates mtROS production and the mitochondrial respiration chain in macrophages
Because iron promotes the generation of ROS via the Fenton reaction, we investigated whether GMFG expression in murine macrophages is regulated by treatment with hydrogen peroxide. GMFG protein expression was remarkably decreased in RAW264.7 macrophages at high concentrations of hydrogen peroxide (200-250 μM); IRP1 and TfR1 protein expression was concomitantly increased at these concentrations of hydrogen peroxide in these cells (Figure 5A). This observation led us to speculate that the alterations in iron metabolism protein expression observed in GMFG-knockdown macrophages were possibly mediated by ROS production. Because iron is a key regulator of mitochondrial biogenesis,36 and mitochondria are the major source of ROS in cells,37 we next determined the effect of GMFG knockdown on mtROS production in M0, M1, and M2 macrophages using the mitochondria-specific ROS indicator MitoSOX. The mtROS levels were moderately increased in GMFG-knockdown M0 and M2 macrophages compared with those observed in control M0 and M2 macrophages, whereas mtROS in GMFG-knockdown M1 macrophages was comparable to that observed in control M1 macrophages. However, M1 macrophages exhibited remarkably high levels of mtROS and M2 macrophages displayed relatively low levels of mtROS in control macrophages (Figure 5B). Moreover, GMFG-knockdown M0 and M2 macrophages had increased mitochondrial mass compared with control M0 and M2 macrophages. In contrast, mitochondrial mass in GMFG-knockdown M1 macrophages was indistinguishable from that observed in control macrophages (Figure 5C). Furthermore, GMFG-knockdown M0 and M2 macrophages displayed moderately increased mitochondrial membrane potential (Δψm) compared with control M0 and M2 macrophages as measured by MitoTracker Red or TMRM (Figure 5D-E). GMFG-knockdown M1 macrophages exhibited slightly increased mitochondrial Δψm compared with control M1 macrophages.
![Figure 5. GMFG modulates mtROS production and the mitochondrial respiration chain in macrophages. (A) Immunoblot analysis of GMFG, TfR1, and IRP1 in cellular lysates of RAW264.7 macrophages treated with hydrogen peroxide (0-250 µM) in 10% FBS/DMEM for 24 hours. α-tubulin was used as a loading control. (B-I) RAW264.7 macrophages were transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. (B-E) mtROS, total mitochondrial mass, and mitochondrial membrane potential (Δψm) were analyzed by labeling cells with MitoSOX (B), MitoTracker Green (C), MitoTracker Red (D), or TMRM (E), respectively. Stained cells were then subjected to flow cytometry. (F-G) Immunoblot analysis of mitochondrial respiratory chain complex subunit proteins (complex I [CI; NDUFV2], complex II [CII; SDHD], complex III [CIII; CORE2], and complex IV [CIV; COX5A]) (F) or ISCU and antioxidant proteins (G). α-tubulin was used as a loading control. (H-I) Oxygen consumption rate (OCR) were measured under basal conditions followed by the sequential addition of oligomycin (1 µM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (0.5 µM), and rotenone (0.5 µM) plus antimycin A (0.5 µM) in control siRNA or GMFG siRNA-transfected Raw264.7 cells. Mean basal (H) and maximal (I) OCRs were measured using a Seahorse XF-24. OCRs were normalized by number of living cells in each condition. Basal OCR was measured over time for a single experiment. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control siRNA-transfected cells of the same phenotype.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/8/10.1182_bloodadvances.2018026070/2/m_advances026070f5.png?Expires=1765922438&Signature=VCl92NANLDHuqG320vCmdjD04p-9XoQbApeVhZbu4V9rx9RcjjI7xcu-QRBQWPK75dMZF8bnW9Fe7EdJg0054Muo1uCLhdftiCYRBZOabp1RvjlLBpZfKzb4cd24OZWlTWH6~vOQt9kVOKGurUMlYTdlawYMfyHFWsu1~njUstcpKgDQc8HCZOdyARsOBVkD2XXlUXK1lv~nAQvNevX-eElgqmqkoOkBCieUZRQlvLDWZd9yAOyRQPM5YHSOho-wG0GFlAhh0ykR4IdEnqUPx7c-A4sks0PfPJ8I4hrZjfhTecRVHkNbPMEAmACw3w1nbx2IT5L0xJ8w9CXvvYu1sQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
GMFG modulates mtROS production and the mitochondrial respiration chain in macrophages. (A) Immunoblot analysis of GMFG, TfR1, and IRP1 in cellular lysates of RAW264.7 macrophages treated with hydrogen peroxide (0-250 µM) in 10% FBS/DMEM for 24 hours. α-tubulin was used as a loading control. (B-I) RAW264.7 macrophages were transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. (B-E) mtROS, total mitochondrial mass, and mitochondrial membrane potential (Δψm) were analyzed by labeling cells with MitoSOX (B), MitoTracker Green (C), MitoTracker Red (D), or TMRM (E), respectively. Stained cells were then subjected to flow cytometry. (F-G) Immunoblot analysis of mitochondrial respiratory chain complex subunit proteins (complex I [CI; NDUFV2], complex II [CII; SDHD], complex III [CIII; CORE2], and complex IV [CIV; COX5A]) (F) or ISCU and antioxidant proteins (G). α-tubulin was used as a loading control. (H-I) Oxygen consumption rate (OCR) were measured under basal conditions followed by the sequential addition of oligomycin (1 µM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (0.5 µM), and rotenone (0.5 µM) plus antimycin A (0.5 µM) in control siRNA or GMFG siRNA-transfected Raw264.7 cells. Mean basal (H) and maximal (I) OCRs were measured using a Seahorse XF-24. OCRs were normalized by number of living cells in each condition. Basal OCR was measured over time for a single experiment. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control siRNA-transfected cells of the same phenotype.
GMFG modulates mtROS production and the mitochondrial respiration chain in macrophages. (A) Immunoblot analysis of GMFG, TfR1, and IRP1 in cellular lysates of RAW264.7 macrophages treated with hydrogen peroxide (0-250 µM) in 10% FBS/DMEM for 24 hours. α-tubulin was used as a loading control. (B-I) RAW264.7 macrophages were transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. (B-E) mtROS, total mitochondrial mass, and mitochondrial membrane potential (Δψm) were analyzed by labeling cells with MitoSOX (B), MitoTracker Green (C), MitoTracker Red (D), or TMRM (E), respectively. Stained cells were then subjected to flow cytometry. (F-G) Immunoblot analysis of mitochondrial respiratory chain complex subunit proteins (complex I [CI; NDUFV2], complex II [CII; SDHD], complex III [CIII; CORE2], and complex IV [CIV; COX5A]) (F) or ISCU and antioxidant proteins (G). α-tubulin was used as a loading control. (H-I) Oxygen consumption rate (OCR) were measured under basal conditions followed by the sequential addition of oligomycin (1 µM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (0.5 µM), and rotenone (0.5 µM) plus antimycin A (0.5 µM) in control siRNA or GMFG siRNA-transfected Raw264.7 cells. Mean basal (H) and maximal (I) OCRs were measured using a Seahorse XF-24. OCRs were normalized by number of living cells in each condition. Basal OCR was measured over time for a single experiment. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with control siRNA-transfected cells of the same phenotype.
Given that generation of mtROS mainly occurs at the mitochondrial ETC,37 we examined the effects of GMFG knockdown on ETC complex subunit proteins expression. Protein expression of complex I (NDUFV2) and complex II (SDHD) subunits was reduced in GMFG-knockdown macrophages compared with control siRNA-transfected macrophages for each of the phenotypes, but expression of complex III (CORE2) and complex IV (COX5A) subunit proteins was similar in GMFG-knockdown and control siRNA-transfected cells (Figure 5F).
Because both ETC complex I and complex II depend on function of ISCU to acquire their iron-sulfur clusters, which are particularly sensitive to oxidation, and the expression levels of ISCU are closely regulated by iron,38-40 we analyzed the protein expression of ISCU in GMFG-knockdown M0, M1, and M2 macrophages. Compared with control M0 macrophages, ISCU protein expression was markedly lower in GMFG-knockdown M0 and M2 macrophages, as well as in control M2 macrophages, but not in M1 macrophages. These results suggest that downregulation of ISCU in GMFG-knockdown macrophages is specifically associated with a suppressed mitochondrial respiration chain. In concordance with this finding, protein expression of the antioxidant enzymes SOD2 and SOD1 was also decreased in GMFG-knockdown M0 and M2 macrophages compared with control M0 macrophages (Figure 5G). On the basis of these data, it seems that GMFG knockdown of M0 macrophages produces a protein expression profile for some iron-sulfur clusters and antioxidant enzymes similar to that observed in M2 macrophages.
To further explore whether GMFG knockdown affects mitochondrial oxygen consumption in macrophages, we used an extracellular flux analyzer to monitor the OCR in GMFG-knockdown macrophages. GMFG-knockdown cells displayed a significant reduction in basal respiration OCRs in both M0 and M2 macrophages when compared with control siRNA-transfected cells of the same phenotype (Figure 5H), whereas there were no significant differences in the maximal OCRs in GMFG-knockdown M0 and M2 macrophages compared with the matched control M0 and M2 macrophages (Figure 5I). Collectively, these findings suggest that the decreased OCRs observed in GMFG-knockdown macrophages may result from reduced complex I and II ETC function.
GMFG-knockdown modulation of iron metabolism protein expression and the M2 macrophage phenotype are associated with increased mtROS
Recent growing evidence has indicated that mtROS functions as an important physiological regulator of redox signaling pathways and that modulation of the redox status alters macrophage M2 polarization.41,42 To determine whether the moderately increased mtROS induced by GMFG knockdown is associated with altered iron metabolism protein expression in macrophages, we treated GMFG-knockdown macrophages with the common antioxidant NAC, which is an ROS scavenger. Treatment with 8 mM of NAC for 30 minutes significantly inhibited the increased mtROS produced by GMFG knockdown in RAW264.7 macrophages (Figure 6A). Moreover, NAC treatment substantially attenuated the effects of GMFG knockdown on TfR1 and FPN, but not FtL, protein expression in M0 and M2 macrophages (Figure 6B). Furthermore, we tested whether modulation of M2 markers by GMFG knockdown in macrophages was mediated by mtROS. NAC treatment significantly inhibited the expression of M2 markers in GMFG-knockdown IL-4/IL-13–induced M2 macrophages, as well as in control siRNA-transfected M2 macrophages (Figure 6C-G). Together, these results suggest that GMFG knockdown–induced moderately increased mtROS may act as the interface between iron metabolism and macrophage M2 polarization by modulating the redox status of the cells.
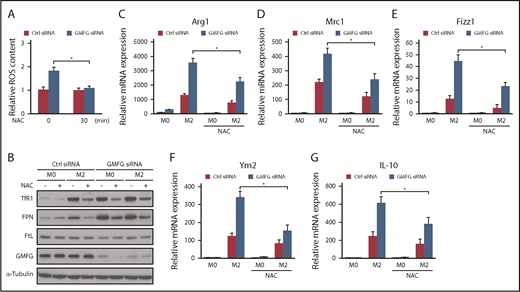
GMFG-knockdown modulation of iron metabolism protein expression and the M2 macrophage phenotype are associated with increased mtROS. (A) mtROS levels in RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then treated with 8 mM of NAC for 30 minutes. Macrophages were then stained with MitoSOX and subjected to flow cytometry, and fluorescence-activated cell sorting MFI values were calculated. Intracellular ROS was expressed as the fold change of MFI normalized to the controls. Data represent the mean ± standard deviation of 3 independent experiments. (B) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M2 (IL-4/IL-13) macrophage inducers for 24 hours. Cells were subsequently treated with or without 8 mM of NAC for 30 minutes. α-tubulin was used as a loading control. (C-G) Quantitative polymerase chain reaction (qPCR) analysis of mean relative mRNA expression of M2 macrophage marker genes in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M2 (IL-4/IL-13) macrophage inducers for 24 hours. Cells were subsequently treated with or without 8 mM of NAC for 30 minutes before RNA isolation. Expression levels were normalized to 18S mRNA expression. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with no NAC treatment GMFG siRNA-transfected M2 cells.
GMFG-knockdown modulation of iron metabolism protein expression and the M2 macrophage phenotype are associated with increased mtROS. (A) mtROS levels in RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then treated with 8 mM of NAC for 30 minutes. Macrophages were then stained with MitoSOX and subjected to flow cytometry, and fluorescence-activated cell sorting MFI values were calculated. Intracellular ROS was expressed as the fold change of MFI normalized to the controls. Data represent the mean ± standard deviation of 3 independent experiments. (B) Immunoblot analysis of iron metabolism proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M2 (IL-4/IL-13) macrophage inducers for 24 hours. Cells were subsequently treated with or without 8 mM of NAC for 30 minutes. α-tubulin was used as a loading control. (C-G) Quantitative polymerase chain reaction (qPCR) analysis of mean relative mRNA expression of M2 macrophage marker genes in RAW264.7 macrophages transfected with control siRNA or GMFG siRNA for 48 hours, then stimulated without (M0) or with M2 (IL-4/IL-13) macrophage inducers for 24 hours. Cells were subsequently treated with or without 8 mM of NAC for 30 minutes before RNA isolation. Expression levels were normalized to 18S mRNA expression. Data represent the mean ± standard deviation of at least 3 independent experiments. *P < .05 compared with no NAC treatment GMFG siRNA-transfected M2 cells.
GMFG is associated with the mitochondrial membrane protein ATAD3A
To gain further insights into the role of GMFG in mitochondrial function, we attempted to identify its potential binding partners using coimmunoprecipitation-coupled mass spectrometry–based proteomics to compare control pull-down (control immunoglobulin G antibody) and experimental pull-down (GMFG antibody) proteins in a quantitative approach.43,44 This analysis in human THP-1 cells identified the mitochondrial membrane ATAD3A as potentially being associated with GMFG immunocomplexes (based on higher experimental/control ratios; Figure 7A). To validate the novel GMFG-ATAD3A interaction, we performed coimmunoprecipitations using GMFG-specific or ATAD3A-specific antibodies. Endogenous ATAD3A was strongly detected in GMFG immunoprecipitates in human THP-1 cells; conversely, endogenous GMFG was precipitated by anti-ATAD3A antibody in human THP-1 cells (Figure 7B-C). To further test this interaction, we exogenously transiently coexpressed GFP-tagged GMFG plasmid together with Myc-DDK–tagged ATAD3A plasmid in human HEK-293T cells, then subjected cell lysates to immunoprecipitation with anti-GFP or anti-Myc antibodies. Anti-GFP antibody precipitated ATAD3A protein; conversely, immunoprecipitation using anti-Myc antibody precipitated GMFG protein (Figure 7D). Although knockdown of GMFG did not affect ATAD3A protein expression in murine macrophages of any phenotype (Figure 7E), our coimmunoprecipitation results suggest that GMFG-knockdown effects on mitochondrial function may be related to its association with ATAD3A.
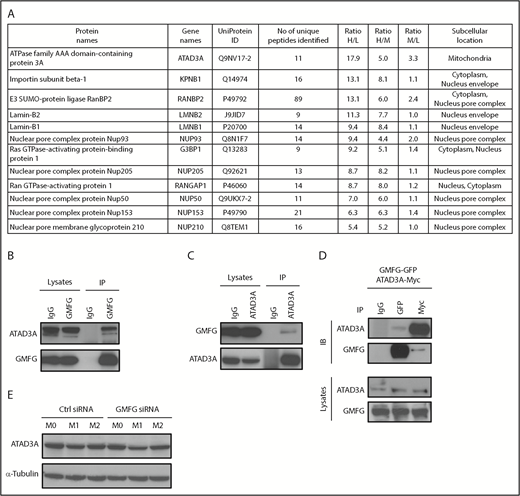
GMFG is associated with the mitochondrial membrane protein ATAD3A. (A) Cellular lysates of human THP-1 cells transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours were immunoprecipitated with control immunoglobulin G (IgG) or monoclonal anti-GMFG antibodies. The immunoprecipitated proteins were isolated using Dynabeads protein G, eluted, and subjected to liquid chromatography-tandem mass spectrometry analysis. Proteome data from Ctrl siRNA- or GMFG siRNA-transfected THP-1 cells were analyzed by MaxQuant, including protein name, intensity L (Ctrl siRNA- or GMFG siRNA-transfected cells immunoprecipitated with IgG antibody), intensity H (Ctrl siRNA-transfected cells immunoprecipitated with GMFG antibody), intensity M (GMFG siRNA-transfected cells immunoprecipitated with GMFG antibody), normalized H/L ratio, H/M ratio, and M/L ratio. (B-C) Immunoprecipitation analysis of GMFG association with the mitochondrial membrane protein ATAD3A in human THP-1 cells. Total cellular lysates were immunoprecipitated with anti-GMFG antibody (B) or anti-ATAD3A antibody (C), then the immunoprecipitants subjected to immunoblot analysis with anti-ATAD3A or anti-GMFG antibody. Samples of the total lysate after immunoprecipitated complexes/beads were isolated (lysates) are shown in the left 2 lanes; immunoprecipitates (IPs) are shown in the right 2 lanes. (D) Cellular lysates of human HEK-293T cells cotransfected with GFP-tagged GMFG plasmid and Myc-DDK–tagged ATAD3A plasmid for 48 hours were immunoprecipitated with control IgG, anti-GFP, or anti-Myc antibody; the IPs were then subjected to immunoblot analysis with anti-ATAD3A or anti-GMFG antibody. Samples of the total lysate after immunoprecipitated complexes/beads were isolated (lysates; bottom). Each sample corresponds to 5% of the cell lysate used in each immunoprecipitation. (E) Immunoblot analysis of ATAD3A proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. α-tubulin was used as a loading control.
GMFG is associated with the mitochondrial membrane protein ATAD3A. (A) Cellular lysates of human THP-1 cells transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours were immunoprecipitated with control immunoglobulin G (IgG) or monoclonal anti-GMFG antibodies. The immunoprecipitated proteins were isolated using Dynabeads protein G, eluted, and subjected to liquid chromatography-tandem mass spectrometry analysis. Proteome data from Ctrl siRNA- or GMFG siRNA-transfected THP-1 cells were analyzed by MaxQuant, including protein name, intensity L (Ctrl siRNA- or GMFG siRNA-transfected cells immunoprecipitated with IgG antibody), intensity H (Ctrl siRNA-transfected cells immunoprecipitated with GMFG antibody), intensity M (GMFG siRNA-transfected cells immunoprecipitated with GMFG antibody), normalized H/L ratio, H/M ratio, and M/L ratio. (B-C) Immunoprecipitation analysis of GMFG association with the mitochondrial membrane protein ATAD3A in human THP-1 cells. Total cellular lysates were immunoprecipitated with anti-GMFG antibody (B) or anti-ATAD3A antibody (C), then the immunoprecipitants subjected to immunoblot analysis with anti-ATAD3A or anti-GMFG antibody. Samples of the total lysate after immunoprecipitated complexes/beads were isolated (lysates) are shown in the left 2 lanes; immunoprecipitates (IPs) are shown in the right 2 lanes. (D) Cellular lysates of human HEK-293T cells cotransfected with GFP-tagged GMFG plasmid and Myc-DDK–tagged ATAD3A plasmid for 48 hours were immunoprecipitated with control IgG, anti-GFP, or anti-Myc antibody; the IPs were then subjected to immunoblot analysis with anti-ATAD3A or anti-GMFG antibody. Samples of the total lysate after immunoprecipitated complexes/beads were isolated (lysates; bottom). Each sample corresponds to 5% of the cell lysate used in each immunoprecipitation. (E) Immunoblot analysis of ATAD3A proteins in cellular lysates of RAW264.7 macrophages transfected with control siRNA (Ctrl) or GMFG siRNA for 48 hours, then stimulated without (M0) or with M1 (LPS/IFN-γ) or M2 (IL-4/IL-13) macrophage inducers for 24 hours. α-tubulin was used as a loading control.
Discussion
In this study, we identify for the first time that the actin-regulatory protein GMFG mediates cellular iron homeostasis and macrophage polarization through modulation of mitochondrial function. We found that knockdown of GMFG in macrophages altered the expression of iron metabolism proteins and enhanced IL-4/IL-13–induced M2 macrophage polarization. The underlying mechanisms for GMFG-knockdown effects on cellular iron homeostasis and M2 macrophage polarization involve production of moderately increased mtROS and are associated with reduced mitochondrial respiration chain, ISCU, and antioxidant protein expression.
The macrophage iron modulatory response was markedly different in M1 and M2 macrophages. Our data showed that knockdown of GMFG produced iron metabolism features mimicking those found in IL-4/IL-13–induced M2 macrophages, which were similar to those seen in the iron chelator DFO-induced iron deficiency response. These findings extend previous work demonstrating an iron deficiency TfR1high FtLlow and FPNhigh phenotype in IL-4–induced M2 macrophages.10,11,45 Although GMFG knockdown increased expression of IRP1 protein in the present study, the unchanged IRP1 RNA-binding activity we observed indicates that IRP1 may not directly mediate the GMFG knockdown–induced iron deficiency response. TfR1 expression is also controlled by other posttranslational modifications, such as palmitoylation46 ; altered palmitoylation of TfR1 could affect its cell surface levels, endocytosis, and turnover/recycling,47 thereby affecting cellular iron homeostasis. Although our results show that GMFG knockdown did not affect TfR1 endocytosis, whether GMFG knockdown influences TfR1 palmitoylation or intracellular trafficking and recycling requires further detailed investigation. Our previous study showed that knockdown of GMFG decreases expression levels of the plasma membrane protein α5β1-integrin in human monocytes.48 These data and those in the current study suggest that GMFG-knockdown alterations of iron metabolism–related proteins TfR1 and FPN are related to the function of GMFG rather than being solely iron-specific effects. It has been reported that iron depletion conditions in macrophages may favor the activation of HIFs,12 because iron is required for constitutive HIF degradation.49 Indeed, our data showed that HIF-2α protein expression was increased in GMFG knockdown M0 and M2 macrophages, as well as in control M2 macrophages, but not in control M0 and M1 macrophages. These findings, together with those of others that have shown that HIF-2α is increased in IL-4–induced M2 macrophages,12,34,35 indicate that the iron deficiency phenotype adopted in M2 macrophages might reduce prolyl hydroxylase activity, leading to stabilized and increased protein level of HIFs, thus mimicking the effect of hypoxia. These findings, combined with the observation that GMFG-knockdown macrophages promoted the IL-4/IL-13–induced M2 phenotype, strengthen the concept that macrophage phenotype and iron metabolism are tightly interconnected and that this connection might be coordinated by common upstream iron- or oxygen-sensing regulatory pathways. Because iron and oxygen are often intimately connected in their metabolism, it is not surprising that their levels could be directly sensed and control various biological processes, as well as influence each other’s metabolism by crosstalk between IRPs and HIFs. Indeed, we observed that GMFG expression could be downregulated by either iron supplementation or hydrogen peroxide exposure. It is important to note that the functional consequences of altered intracellular iron metabolism under conditions of GMFG downregulation promote anti-inflammatory M2 polarization, which further supports the concept that macrophage phenotype and iron metabolism are tightly interconnected.
Iron metabolism is strongly associated with mitochondrial function through ISCU biosynthesis pathways,50-53 because iron-sulfur clusters are incorporated into enzymes that are responsible for mitochondrial respiratory complexes (I, II, and III).54 Alteration of ISCU or other iron-sulfur enzymes can suppress the mitochondrial complex I activity that leads to direct downstream functional consequences for ROS production,55,56 as well as disrupt intracellular iron homeostasis.38,57 In addition, it has been reported that mild hypoxia or mitochondrial stress can modulate iron depletion, which has beneficial protective and prolongevity effects.58 Consistent with these studies, our data showed that GMFG knockdown in macrophages induced the iron deficiency response; reduced expression of mitochondrial complex I (NDUFV2) and complex II (SDHD) subunits, ISCU, and the antioxidants SOD2 and SOD1; and moderately increased mtROS production. We also showed that GMFG knockdown significantly decreased basal oxygen consumption. Together, these results suggest that suppressed mitochondrial function resulting from decreased ISCU and complex I and II component expression is a likely mechanism underlying the moderately increased mtROS and decreased OCR observed upon GMFG knockdown.
Accumulating evidence implicates ROS in macrophage polarization.59 High levels of ROS cause oxidative stress, which increases immunity to defense against bacterial pathogens and is associated with M1 macrophages,60 whereas lower or moderate levels of ROS act as a redox signal to maintain physiological function and promote M2 macrophages and wound healing.42,61,62 Consistent with these studies, our data showed that GMFG-knockdown M0 macrophages exhibited moderately increased mtROS production and that treatment of GMFG-knockdown macrophages with the ROS quencher NAC reversed increased TfR1 and FPN and significantly inhibited IL-4/IL-13–induced M2 macrophage marker gene expression. These findings suggest that moderately increased mtROS induced in GMFG-knockdown macrophages mediates alterations in iron metabolism–related protein expression and M2 macrophage polarization. Therefore, our study provides mechanistic insight into how alteration of mitochondrial function integrates with iron metabolism and macrophage phenotype.
Given that the iron deficiency response can also be modulated by disruption of interorganelle communication between mitochondria and the endoplasmic reticulum,63 the molecules that regulate contact between these organelles could be involved in multiple mitochondrial cellular processes. Indeed, we found that GMFG interacted with the mitochondrial membrane protein ATAD3A, which is crucial for normal mitochondria–endoplasmic reticulum interaction; plays an important role in mitochondrial biogenesis, lipid biosynthesis, and steroid biosynthesis; and is required for iron and heme metabolism.64,65 Therefore, our findings that GMFG knockdown decreased protein expression of ISCU, increased cellular iron levels and iron deficiency phenotype, and enhanced M2 macrophage polarization in macrophages suggest that these effects of GMFG knockdown might be related to its association with ATAD3A. However, additional investigations are needed to confirm the exact role of GMFG in this context.
Understanding how iron availability affects the functional phenotype of immune cells and influences their immunometabolism is profoundly important. Here, we demonstrate that moderately increased mtROS production is the molecular mechanism by which downregulation of the actin-regulatory protein GMFG induces an iron deficiency response and enhances an anti-inflammatory M2 phenotype in murine macrophages. These studies could pave the way for new therapeutic approaches for modulating macrophage function in diverse contexts, including antimicrobial immunity, type 2 diabetes, and metabolic homeostasis.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Deliang Zhang for providing SIH and IRP1 polyclonal antibodies and also thank David Eric Anderson for performing mass spectrometry–based proteomics and analysis in the Advanced Mass Spectrometry Core Facility of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
This work was supported by the Intramural Research Program of the National Institutes of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, #ZIA HL006005-12.
Authorship
Contribution: W.A. planned and performed experiments, collected and analyzed data, and wrote the manuscript; J.L., M.C.G., T.A.R., C.K., J.Z., and K.C. performed experiments and reviewed the manuscript; and G.P.R. designed and supervised the study and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Griffin P. Rodgers, Molecular and Clinical Hematology Branch, National Institutes of Health, Building 10, Room 9N-113A, Bethesda, MD 20892-2560; e-mail: gr5n@nih.gov.

