

Key Points
The bone marrow microenvironment provides protection against FLT3 TKIs.
Stromal CYP3A4 contributes to bone marrow microenvironment–mediated FLT3 inhibitor resistance in FLT3/ITD AML.

Abstract
An intriguing aspect of the clinical activity of FMS-like tyrosine kinase 3 inhibitors (FLT3 TKIs) is their apparent higher activity against peripheral blasts from FLT3/internal tandem duplication (ITD) acute myeloid leukemia than marrow disease in the same patients. Accordingly, studies showed that the bone marrow microenvironment plays a role in FLT3 TKI resistance, although the underlying mechanisms are unclear. We recently identified a previously undescribed mechanism by which the bone marrow microenvironment can contribute to drug resistance: expression of cytochrome P450 enzymes (CYPs). In fact, bone marrow stromal cells (BMSCs) expressed most CYPs, including CYP3A4. Because hepatic CYP3A4 plays a role in the inactivation of several FLT3 TKIs, we explored the potential role of CYP3A4 in bone marrow microenvironment–mediated FLT3 TKI resistance. We found that CYP3A4 plays a major role in BMSC-mediated inhibition in the activity of 3 different FLT3 TKIs (sorafenib, quizartinib, and gilteritinib) against FLT3/ITD acute myeloid leukemia (AML). Furthermore, clarithromycin, a clinically active CYP3A4 inhibitor, significantly reversed the protective effects of BMSCs. We show, for the first time, that bone marrow stromal CYP3A4 contributes to FLT3 TKI resistance in the bone marrow. These results suggest that combining FLT3 TKIs with CYP3A4 inhibitors could be a promising strategy toward improving the activity of FLT3 TKIs.
Introduction
FMS-like tyrosine kinase 3 (FLT3) mutations are among the most frequent genetic alterations in acute myeloid leukemia (AML), occurring in up to one-third of cases.1 Activating FLT3 mutations in AML occur in 2 major forms: internal tandem duplications (ITDs) and tyrosine kinase domain (TKD) point mutations. Whereas TKD mutations are less common and their prognostic relevance is controversial,2-5 the more frequent ITD mutations generally convey an unfavorable prognosis.6-10 Of note, ITD mutations have little adverse impact on initial rates of complete remission but rather are associated with a high risk of relapse and lower overall survival. Moreover, although FLT3/ITD may only be present in a fraction of the AML cells at diagnosis, the FLT3/ITD clone generally predominates at relapse. FLT3/ITD therefore represents a crucial target in the war against AML.
Since the early 2000s, several small molecule FLT3 tyrosine kinase inhibitors (FLT3 TKIs) have been introduced into clinical trials to target AMLs with mutant FLT3.11-13 In general, FLT3 TKIs as single agents are well tolerated and display encouraging activities in killing circulating blasts. However, FLT3 TKIs fail to induce durable responses,11,13-17 suggesting that minimal residual disease responsible for relapse exhibits resistance to FLT3 TKIs. Many mechanisms of resistance to FLT3 TKIs have been proposed, and most seem to play important roles in at least some patients. These mechanisms include the following: (1) plasma protein binding that renders free FLT3 TKI levels insufficient to bind to targets11,18-20 ; (2) high intrinsic FLT3 ligand levels that interfere with FLT3 inhibition21 ; (3) the emergence of TKD point mutations that interfere with the binding of specific FLT3 TKIs with FLT322-24 ; (4) the activation of FLT3 downstream or alternative signaling pathways that can maintain the survival and growth of blasts25,26 ; and (5) the loss of FLT3/ITD due to clonal evolution.27,28
There is increasing evidence that specialized microenvironments or niches also play important roles in drug resistance.29-31 Consistent with data showing that FLT3 TKIs seem to be more effective in clearing peripheral blood blasts than bone marrow disease, several studies have found that the bone marrow microenvironment can protect FLT3/ITD AML from FLT3 TKIs.32-35 Both physical interactions and soluble factors have been implicated in niche-mediated chemoprotection, although the exact mechanisms remain unclear.
We previously reported that the bone marrow microenvironment expresses a variety of cytochrome P450 enzymes (CYPs), which seem to play important roles in the local metabolism of endogenous factors such as retinoids as well as chemotherapeutic agents.36-39 Moreover, we found that CYP3A4, which is responsible for the hepatic metabolism of many of the drugs currently in use,40-42 contributes to the chemoprotection of both non-FLT3 AML and multiple myeloma by the bone marrow microenvironment.38 Because hepatic CYP3A4 has also been shown to inactivate essentially all TKIs, including FLT3 TKIs,43-47 we hypothesized that CYP3A4 plays a role in protecting FLT3/ITD AML against FLT3 TKIs in the bone marrow microenvironment. The present study found that CYP3A4 apparently contributes to microenvironment-mediated resistance to FLT3 TKIs. Importantly, inhibiting CYP3A4 can overcome this component of stromal-mediated resistance to FLT3 tyrosine kinase inhibition.
Methods
Cell lines
F/STRO48 cells were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA) with 10% fetal bovine serum (FBS; MilliporeSigma, Burlington, MA), 2 mM L-glutamine (Thermo Fisher Scientific), and 100 U/mL penicillin with 100 μg/mL streptomycin (Thermo Fisher Scientific) at 33°C in 5% carbon dioxide (CO2). For clonogenic assay and conditioned media experiments, F/STRO were subjected to 20 Gy of irradiation before seeding on gelatinized well plates. Human FLT3/ITD+ AML cell lines, MV4-11 and Molm14, were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). Both MV4-11 and Molm14 were cultured in RPMI 1640 (Thermo Fisher Scientific) with 10% FBS, 2 mM L-glutamine, and 100 U/mL penicillin with 100 μg/mL streptomycin at 37°C in 5% CO2.
Primary bone marrow stroma
Human primary bone marrow stromal cells (BMSCs) were derived from normal bone marrow transplant donor harvests and processed as previously described.39 All normal donors provided informed consent under a Johns Hopkins Medicine Institutional Review Board–approved protocol according to the Declaration of Helsinki. Human primary BMSCs were cultured in gelatinized plates with IMDM (Thermo Fisher Scientific) containing 15% FBS, 5% horse serum (MilliporeSigma), 100 U/mL penicillin with 100 μg/mL streptomycin, 10−4 M β-mercaptoethanol (MilliporeSigma), and 10−5 M hydrocortisone 21-hemisuccinate (MilliporeSigma) at 33°C in 5% CO2.
Drugs
Sorafenib and quizartinib were obtained from LC Laboratories (Woburn, MA). Gilteritinib was obtained from Astellas Pharma US, Inc. (Northbrook, IL). Clarithromycin was purchased from MilliporeSigma. All compounds were dissolved in dimethyl sulfoxide (DMSO) as stock solutions. Working solutions were prepared by diluting stock solutions in RPMI 1640. The final concentration of DMSO in experiments was no more than 0.2%.
CYP3A4 knockdown
As previously published,38 lentiviral vectors expressing CYP3A4-targeting short hairpin RNA (shRNA; The RNAi Consortium, Broad Institute, Cambridge, MA), empty lentiviral vectors pGIPZ (Open Biosystems, Lafayette, CO), or nontargeting control lentiviral vectors pLKO.1-puro eGFP shRNA (obtained through the Johns Hopkins Genomics Resources in the HiT Center from Open Biosystems, Huntsville, AL) were transfected together with pCMV-dR8.9 and vesicular stomatitis virus G–expressing plasmids into 293T cells using Lipofectamine 2000 (Thermo Fisher Scientific) for lentiviral supernatant production. BMSCs were incubated with the viral supernatant and 8 µg/mL polybrene (MilliporeSigma) for transduction. After at least 48 hours, cells were treated with 6 µg/mL of puromycin (MilliporeSigma) to select for positive clones. The knockdown of CYP3A4 was confirmed.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (PCR) was performed as previously described.38 In brief, total RNA was extracted by using the RNeasy Mini Kit (Qiagen, Valencia, CA), and complementary DNA was synthesized by using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Quantitative real-time PCR was conducted by using sequence-specific primers (CYP3A4 forward: 5′-GCCTGGTGCTCCTCTATCTA-3′ and reverse: 5′-GGCTGTTGACCATCATAAAAG-3′; GAPDH forward: 5′- ACCCAGAAGACTGTGGATGG-3′ and reverse: 5′-TCTAGACGGCAGGTCAGGTC-3′), the Radiant Green Lo-ROX qPCR Kit (Alkali Scientific, Fort Lauderdale, FL), and the CFX96 real-time PCR detection system (Bio-Rad).
Clonogenic assays
Clonogenic assays were performed as previously described.37,38 Briefly, after 72 hours of drug treatment, cells were collected, counted, and resuspended at a density of 500 cells/mL in methylcellulose-based media containing 1.41% methylcellulose (MilliporeSigma), 30% FBS (MilliporeSigma), 1% bovine serum albumin (MilliporeSigma), 10% RPMI 1640, 2 mM L-glutamine, 100 U/mL penicillin with 100 μg/mL streptomycin, and 10−4 M β-mercaptoethanol in IMDM. The cell suspensions were then plated in 35 mm culture dishes in triplicates at 500 cells/dish. After 10 to 14 days of incubation at 37°C in 5% CO2, the recovery of colony-forming units was determined by colony counting under bright-field microscopy. A cell aggregate composed of >50 cells was defined as a colony.
FLT3 phosphorylation
FLT3/ITD AML cells were incubated with 1 mL of FLT3 TKI–containing medium that was conditioned by BMSCs at room temperature for 1 hour. Protein lysates of the cells were obtained and immunoprecipitated with anti-FLT3 antibody (sc-480; Santa Cruz Biotechnology, Dallas, TX) and Protein A agarose beads (MilliporeSigma). The immunoprecipitants were separated on an 8% sodium dodecyl sulfate polyacrylamide gel and transferred to an Immobilon membrane (MilliporeSigma). The membranes were incubated with anti-phosphotyrosine antibody (4G10; MilliporeSigma), then stripped and reprobed with anti-FLT3 antibody (Santa Cruz Biotechnology) to measure total FLT3. Proteins were visualized by using chemiluminescence (Clarity Western ECL Substrate; Bio-Rad), exposed on autoradiographic films (LabScientific, Highlands, NJ), and scanned by using a densitometer (Bio-Rad). Quantification of band volumes was conducted by using Image Lab software (Bio-Rad).
Xenograft mouse model
For generating luciferase-expressing Molm14, pLenti-CMV-LUC-Puro lentiviral vectors (Addgene, Cambridge, MA) were transfected into 293T cells as described earlier for lentiviral supernatant production. Molm14 cells were then incubated with the lentiviral supernatant and 8 µg/mL polybrene for transduction. After at least 48 hours, cells were selected by using 0.5 µg/mL puromycin. The correspondence of cell number to bioluminescence signal was verified.
For each tumor, 2.5 × 105 luciferase+ Molm14 cells with 5 × 105 control or shCYP3A4 primary human BMSCs were resuspended within RPMI 1640 (Thermo Fisher Scientific) containing 50% Matrigel Matrix (Corning, Corning, NY). Before injection, NOD/SCID/IL2γ−/− (NSG) male mice (The Jackson Laboratory, Bar Harbor, ME) at least 8 weeks old were anesthetized and shaved on both sides of the flank region. For each mouse, the cell suspensions containing control and shCYP3A4 BMSCs were injected subcutaneously into the left and right flank, respectively, to establish xenograft tumors.
After the tumors engrafted, mice were given 10 mg/kg sorafenib (LC Laboratories) by intraperitoneal injection 3 times per week. IVIS Spectrum In Vivo Imaging System (PerkinElmer, Waltham, MA) was used to obtain tumor bioluminescence signals. Before imaging, mice were anesthetized with isoflurane and injected intraperitoneally with 150 mg/kg D-luciferin (PerkinElmer). Signals were quantified by using Living Image Software (PerkinElmer). All animal studies were conducted under a protocol approved by the Johns Hopkins University Animal Care and Use Committee.
Flow cytometry
F/STRO cells were trypsinized and washed with cold phosphate-buffered saline. Cells were then fixed and permeabilized in Cytofix/Cytoperm Solution (BD Biosciences, San Jose, CA) for 20 minutes at 4°C. After being washed twice with Perm/Wash Buffer (BD Biosciences), cells were stained with rabbit anti-human CYP3A4 antibody (ab135813; Abcam, Cambridge, MA) or normal rabbit immunoglobulin G (sc-3888; Santa Cruz Biotechnology) as isotype control for 1 hour 25 minutes at 4°C. After the primary antibody staining, cells were washed with Perm/Wash Buffer and stained with phycoerythrin-conjugated anti-rabbit immunoglobulin G antibody (sc-3739; Santa Cruz Biotechnology) for 40 minutes at 4°C in the dark. Cells were then washed twice, resuspended in Perm/Wash Buffer, and analyzed by using a FACSCalibur flow cytometry (BD Biosciences). Data were analyzed by using FlowJo software (FlowJo, Ashland, OR).
Quantification of quizartinib
Culture media (RPMI + 10% FBS + 2 mM L-glutamine + 100 U/mL penicillin with 100 μg/mL streptomycin) supplemented with 100 nM quizartinib were incubated with F/STRO monolayer at 37°C in 5% CO2. The conditioned media were then collected and stored at −80°C until analysis. Samples were analyzed for quizartinib by using liquid chromatography with tandem mass spectrometry by the Sidney Kimmel Comprehensive Cancer Center Analytical Pharmacology Core. The analyte was extracted from 25 µL of cell culture media with 100 µL of acetonitrile containing internal standard (sunitinib-d10). Samples were centrifuged, and the top layer was transferred to an autosampler vial for liquid chromatography with tandem mass spectrometry analysis. Chromatographic separation was achieved with a Cortecs C18 analytical column (2.1 × 50 mm, 2.7 µm; Waters Corporation, Milford, MA) at 40°C with a gradient. Mobile phase A was water containing 0.1% formic acid, and mobile phase B was acetonitrile containing 0.1% formic acid. The gradient started with mobile phase B held at 20% for 0.5 minute and increased to 100% over 0.5 minute; 100% mobile phase B was held for 2 minutes and then returned back to 20% mobile phase B and allowed to equilibrate for 2 minutes. The total run time was 4 minutes, with a flow rate of 0.4 mL/min. The column effluent was monitored by using a Sciex 4500 triple quadrupole mass spectrometer with electrospray ionization operating in positive mode. The mass spectrometer was programmed to monitor the following multiple reaction monitoring transitions: 561.3→421.0 for quizartinib and 409.1→283.2 for sunitinib-d10. The calibration curve was computed by using area ratio peak of the analyte to the internal standard by using a quadratic equation with a 1/x2 weighting function over the range of 10 to 1000 ng/mL.
Statistical analyses
Statistical analyses were performed by using GraphPad Prism (GraphPad Software, La Jolla, CA). For mouse xenograft experiments, statistical significance was calculated by using the paired 2-tailed Student t test. Otherwise, an unpaired 2-tailed Student t test was performed to evaluate statistical significance.
Results
CYP3A4 contributes to BMSC-mediated FLT3 TKI resistance in vitro
To investigate if CYP3A4 plays any role in the previously described32,33 protection of FLT3/ITD AML cells by BMSCs, the activity of 3 different clinically active FLT3 TKIs (sorafenib, quizartinib, and gilteritinib) was studied against FLT3/ITD AML cells cultured with CYP3A4 knockdown (shCYP3A4) or control (empty lentiviral vectors pGIPZ) BMSCs. The 50% inhibitory concentration (20 nM sorafenib, 0.5 nM quizartinib, and 20 nM gilteritinib) were chosen because they gave sufficient kill to allow manipulations to show differences in activity. The effective knockdown of CYP3A4 in BMSCs was confirmed (supplemental Figures 1 and 2). In addition, CYP3A4 expression was nondetectable in FLT3/ITD AML cell lines (supplemental Figure 3). As expected, all 3 TKIs inhibited the clonogenic growth of both FLT3/ITD AML lines but had little activity when cocultured with BMSCs (Figure 1; supplemental Figure 4). Moreover, the BMSC-mediated resistance was largely abrogated by CYP3A4 knockdown of BMSCs.
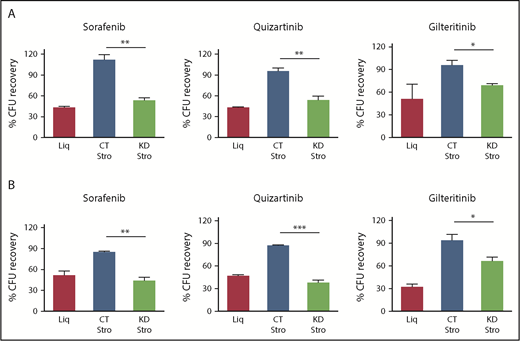
Knockdown of CYP3A4 in BMSCs reversed the stromal-mediated FLT3 TKI resistance of FLT3/ITD AML cells. Clonogenic recovery of MV4-11 (A) and Molm14 (B) cells when cultured without F/STRO (Liq), with control F/STRO (CT Stro), or with shRNA CYP3A4 knockdown F/STRO (KD Stro) after treatment with 3 different FLT3 TKIs (20 nM sorafenib, 0.5 nM quizartinib, and 20 nM gilteritinib) for 72 hours. Data represent mean ± standard error of the mean (SEM) of 3 to 4 independent experiments. *P < .05, **P < .01, ***P < .001. CFU, colony-forming unit.
Knockdown of CYP3A4 in BMSCs reversed the stromal-mediated FLT3 TKI resistance of FLT3/ITD AML cells. Clonogenic recovery of MV4-11 (A) and Molm14 (B) cells when cultured without F/STRO (Liq), with control F/STRO (CT Stro), or with shRNA CYP3A4 knockdown F/STRO (KD Stro) after treatment with 3 different FLT3 TKIs (20 nM sorafenib, 0.5 nM quizartinib, and 20 nM gilteritinib) for 72 hours. Data represent mean ± standard error of the mean (SEM) of 3 to 4 independent experiments. *P < .05, **P < .01, ***P < .001. CFU, colony-forming unit.
To further study the role of CYP3A4 in the protection against FLT3 TKIs by BMSCs, sorafenib, quizartinib, and gilteritinib were incubated with CYP3A4 knockdown BMSCs, control (empty lentiviral vectors pGIPZ) BMSCs, or just medium for 72 hours. The conditioned medium was then removed and incubated with FLT3/ITD AML Molm14 cells for 1 hour, and FLT3 phosphorylation was assessed in AML cells. In this setting, any effect of BMSCs on FLT3 TKI activity would have to be a direct effect on drug activity. Whereas the control BMSCs inhibited the activity of all 3 FLT3 TKIs, CYP3A4 knockdown eliminated the protection provided by the BMSCs (Figure 2). Moreover, CYP3A4 knockdown limited the FLT3 TKI clearance by BMSCs (supplemental Figure 5).
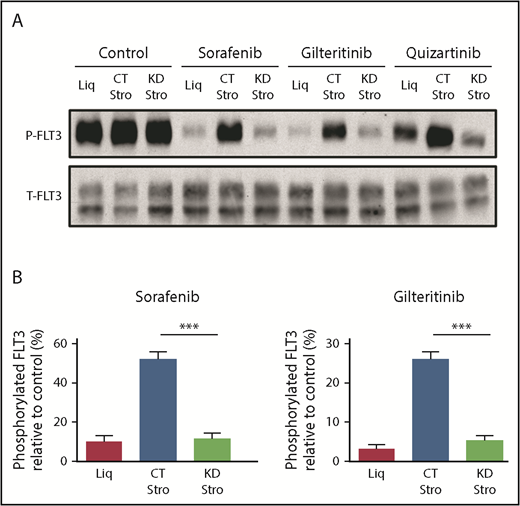
CYP3A4 in BMSCs blocked the inhibitory activity of FLT3 TKIs in FLT3/ITD AML in the absence of AML–stromal cellular contact. (A) Expression of phosphorylated FLT3 (P-FLT3) and total FLT3 (T-FLT3) in Molm14 cells by western blotting after 1 hour of incubation with 3 different FLT3 TKIs (20 nM sorafenib, 20 nM gilteritinib, and 3.5 nM quizartinib) that had been incubated with or without bone marrow stroma for 72 hours. The left 3 control lanes are conditioned media without TKIs (just DMSO). (B) Quantification of phosphorylated FLT3 in Molm14 cells. Data are the mean ± SEM of 4 (sorafenib) and 5 (gilteritinib) independent experiments. Quantification of phosphorylated FLT3 with quizartinib treatment is not presented because the experiment was performed once. ***P < .001. CT Stro, control F/STRO; KD Stro, shRNA CYP3A4 knockdown F/STRO; Liq, no F/STRO.
CYP3A4 in BMSCs blocked the inhibitory activity of FLT3 TKIs in FLT3/ITD AML in the absence of AML–stromal cellular contact. (A) Expression of phosphorylated FLT3 (P-FLT3) and total FLT3 (T-FLT3) in Molm14 cells by western blotting after 1 hour of incubation with 3 different FLT3 TKIs (20 nM sorafenib, 20 nM gilteritinib, and 3.5 nM quizartinib) that had been incubated with or without bone marrow stroma for 72 hours. The left 3 control lanes are conditioned media without TKIs (just DMSO). (B) Quantification of phosphorylated FLT3 in Molm14 cells. Data are the mean ± SEM of 4 (sorafenib) and 5 (gilteritinib) independent experiments. Quantification of phosphorylated FLT3 with quizartinib treatment is not presented because the experiment was performed once. ***P < .001. CT Stro, control F/STRO; KD Stro, shRNA CYP3A4 knockdown F/STRO; Liq, no F/STRO.
CYP3A4 contributes to stromal-mediated FLT3 TKI resistance in vivo
To further study the role of microenvironment CYP3A4 in FLT3 TKI resistance in an in vivo model, we developed a xenograft model of human FLT3/ITD AML–BMSC interactions, which allowed us to show the local effect of BMSCs on FLT3/ITD AML cells in vivo. Human primary BMSCs were transduced with lentivirus encoding shRNA that targets CYP3A4 or control vectors. The effective knockdown of CYP3A4 in primary BMSCs was confirmed by using quantitative real-time PCR (supplemental Figure 6). Luciferase+ FLT3/ITD AML Molm14 cells were combined with the control BMSCs or CYP3A4 knockdown BMSCs, and the AML/BMSC tumors were implanted subcutaneously into NSG mice. Each mouse carried FLT3/ITD AML cells + control stroma in the left flank and AML + CYP3A4 knockdown stroma in the right flank. After 4 days, the mice were given an intraperitoneal sorafenib injection (10 mg/kg) 3 times a week for 4 weeks. There was no difference in tumor growth before initiation of sorafenib. However, while the FLT3/ITD AML continued to grow exponentially with the control stroma, the AML with CYP3A4 knockdown stroma exhibited a significant response to treatment (Figure 3; supplemental Figure 7). This finding suggested that bone marrow stromal CYP3A4 also protected FLT3/ITD AML from FLT3 TKIs in vivo.
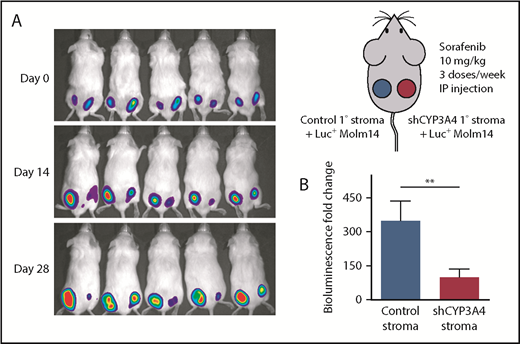
Knockdown of CYP3A4 in BMSCs reduced the stromal-mediated FLT3 TKI resistance of FLT3/ITD AML in the mouse xenograft model. (A) The right panel depicts the mouse xenograft model. Tumors on the left flanks were composed of luciferase-positive (Luc+) Molm14 cells and control human primary BMSCs (Control 1° stroma); tumors on the right flanks were composed of Luc+ Molm14 cells and human primary BMSCs with shRNA knockdown of CYP3A4 (shCYP3A4 1° stroma). Mice with xenograft tumors were treated with 10 mg/kg sorafenib 3 times per week for 4 weeks. The left panel shows the bioluminescent images representing tumor burden of 5 representative xenograft mice on day 0, day 14, and day 28 after the treatment started. (B) Fold change in the bioluminescent intensity of xenograft tumors with control or shCYP3A4 1° stroma on day 28 relative to day 0 during sorafenib treatment. Data represent the mean ± SEM of 10 independent xenografts. **P < .01. IP, intraperitoneal.
Knockdown of CYP3A4 in BMSCs reduced the stromal-mediated FLT3 TKI resistance of FLT3/ITD AML in the mouse xenograft model. (A) The right panel depicts the mouse xenograft model. Tumors on the left flanks were composed of luciferase-positive (Luc+) Molm14 cells and control human primary BMSCs (Control 1° stroma); tumors on the right flanks were composed of Luc+ Molm14 cells and human primary BMSCs with shRNA knockdown of CYP3A4 (shCYP3A4 1° stroma). Mice with xenograft tumors were treated with 10 mg/kg sorafenib 3 times per week for 4 weeks. The left panel shows the bioluminescent images representing tumor burden of 5 representative xenograft mice on day 0, day 14, and day 28 after the treatment started. (B) Fold change in the bioluminescent intensity of xenograft tumors with control or shCYP3A4 1° stroma on day 28 relative to day 0 during sorafenib treatment. Data represent the mean ± SEM of 10 independent xenografts. **P < .01. IP, intraperitoneal.
Clinically applicable CYP3A4 inhibition overcomes stromal-mediated FLT3 TKI resistance
We next examined whether a CYP3A4-inhibiting agent is able to block CYP3A4-mediated chemoprotection. Clarithromycin (Biaxin), a macrolide antibiotic, also acts as a potent CYP3A4 inhibitor by forming a metabolic intermediate that can covalently link to CYP3A4 irreversibly.49 We previously showed that it can overcome stromal-mediated resistance by inhibiting CYP3A4 in vitro, the mechanism potentially responsible for the clinical activity of the BiRd (Biaxin, lenalidomide [Revlimid], and dexamethasone) regimen in multiple myeloma.38 MV4-11 and Molm14 FLT3/ITD AML cells were cultured in the presence or absence of F/STRO BMSCs, with or without clarithromycin and the 3 different FLT3 TKIs (sorafenib, quizartinib, and gilteritinib) for 72 hours. Inhibition of CYP3A4 by clarithromycin reversed the stromal-mediated protection of both MV4-11 and Molm14 FLT3/ITD AML cells to all 3 FLT3 TKIs (Figure 4). Clarithromycin had no effect on the TKIs' anti-FLT3/ITD AML activity in the absence of stroma.
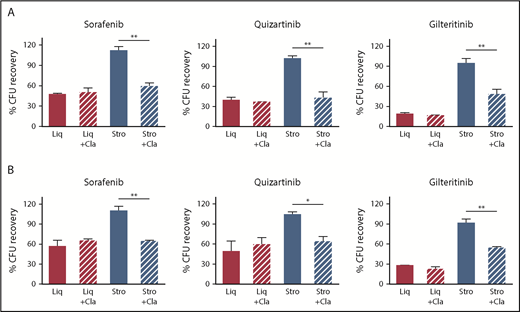
The CYP3A4 inhibitor clarithromycin reversed the stromal-mediated FLT3 TKI resistance of FLT3/ITD AML cells. Clonogenic recovery of MV4-11 (A) and Molm14 (B) cells when cultured without (Liq) or with F/STRO (Stro) and without or with clarithromycin (+Cla) after treatment with 3 different FLT3 TKIs (20 nM sorafenib, 0.5 nM quizartinib, and 20 nM gilteritinib) for 72 hours. Data are the mean ± SEM of 3 to 4 independent experiments. *P < .05, **P < .01.
The CYP3A4 inhibitor clarithromycin reversed the stromal-mediated FLT3 TKI resistance of FLT3/ITD AML cells. Clonogenic recovery of MV4-11 (A) and Molm14 (B) cells when cultured without (Liq) or with F/STRO (Stro) and without or with clarithromycin (+Cla) after treatment with 3 different FLT3 TKIs (20 nM sorafenib, 0.5 nM quizartinib, and 20 nM gilteritinib) for 72 hours. Data are the mean ± SEM of 3 to 4 independent experiments. *P < .05, **P < .01.
Discussion
As previously discussed, the basis for the limited activity of FLT3 TKIs seems to be multifactorial.11,18-26 Although substantial preclinical data suggest a role for the bone marrow microenvironment in clinical FLT3 TKI resistance, the mechanisms responsible for this role are unclear and also likely multifactorial. In this study, we confirmed that BMSCs are able to protect FLT3/ITD AML from FLT3 TKIs. Furthermore, we showed, for the first time, that CYP3A4 in the AML microenvironment plays an important role in this process (Figures 1-4).
CYP3A4 inactivates a wide range of exogenous compounds, including many of the most commonly used chemotherapy drugs. The contributions of CYP3A4 expression to systemic (by hepatic inactivation)50-53 and, in cancer cells, intrinsic drug resistance have been well studied.54-57 More recently, we showed that bone marrow stromal CYP3A4 protected AML and multiple myeloma from standard chemotherapy in both in vitro and in vivo models.38 Given the apparent multifaceted role of CYP3A4 in therapeutic resistance, interfering with CYP3A4 activity has the potential for improving the activity of a variety of anticancer agents, including FLT3 TKIs. Although FLT3/ITD mutations may not be the first leukemogenic hit, the FLT3/ITD clone tends to dominate at relapse.58,59 Thus, being able to better eliminate FLT3/ITD clones, perhaps by overcoming the protective properties of the microenvironment against FLT3 TKIs, might help prevent the persistence of these clones.
Drug-metabolizing enzymes expressed in the tumor microenvironment could represent biochemical barriers between plasma and unique malignant cell niches, resulting in potential sanctuary sites from drugs. Pharmacokinetic parameters such as affinity and velocity constants may allow CYPs to play complementary roles in the systemic (hepatic) and local (microenvironmental) inactivation of drugs.60 It is noteworthy that in addition to the bone marrow, the liver and intestine are the major organs expressing CYP3A4. Clearly, a major role of CYP3A4 in these organs is to provide systemic protection against toxic insults. In this study, we developed a xenograft model of AML-BMSC interactions, which plainly showed that stromal CYP3A4 contributes to drug resistance in vivo. A limitation of our xenograft mouse model is that it does not represent the human CYP3A4 system. When hepatic and intestinal CYP3A4 are involved, it is unclear how substantial the effect of bone marrow stromal CYP3A4 is in relation to hepatic and intestinal CYP3A4. A genetically modified mouse that expresses human CYP3A4 and recapitulates the human CYP3A4 system could be valuable for investigating the contribution of bone marrow stromal CYP3A4 on drug resistance in a humanized CYP3A4 system.61,62 However, the advantage of our xenograft model is that the overall effect of mouse liver CYP drug inactivation will be the same on both tumors with control and CYP3A4 knockdown stroma, allowing the specific study of the human CYP3A4 in the stroma.
Although FLT3 TKIs have an excellent therapeutic ratio, it is still a concern that the blockade of CYP3A4 in the liver and intestine will lead to both higher drug exposure locally and systemically with higher degrees of toxicity. In fact, inhibition of systemic CYPs has been studied clinically, with expected increased systemic drug levels.45,63,64 Pharmacologically adjusting drug doses to maintain safe systemic concentrations should control for inhibition of these enzymes in the liver, while using CYP inhibitors to remove barriers to therapeutic drug levels in the tumor microenvironment.37,38
The full-text version of this article contains a data supplement.
Acknowledgments
The human mesenchymal stromal cell line, F/STRO, was a generous gift from Pierre Marie.48 The authors also thank Jonathan Ling for providing pLKO.1-puro eGFP shRNA control plasmids.
This work was supported by National Institutes of Health, National Cancer Institute grants P01 CA15396 (R.J.J.) and P30 CA006973 (including the Analytic Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins) and by a Leukemia & Lymphoma Society’s Translational Research Program grant (R.J.J. and G.G.).
Authorship
Contribution: Y.-T.C., R.J.J., and G.G. designed research; Y.-T.C., D.H., S.A., M.G., and M.S. performed experiments; Y.-T.C., R.J.J., G.G., and M.J.L. analyzed and interpreted data; Y.-T.C. and R.J.J. wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: M.J.L. serves as a consultant for Astellas, FujiFilm, Novartis, and Daiichi-Sankyo and receives research funding from those companies. The remaining authors declare no competing financial interests.
Correspondence: Richard J. Jones, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, The Bunting-Blaustein Cancer Research Building, Room 244, 1650 Orleans St, Baltimore, MD 21231; e-mail: rjjones@jhmi.edu.

