

Key Points
Leukemia-associated mutations can be detected many years before the onset of secondary leukemias in myeloma patients.
Stem and progenitor cells can act as reservoirs of mutations before the onset of secondary MDS and AML after treatment of myeloma.

Abstract
Therapy-related acute myeloid leukemia and myelodysplastic syndromes (t-AML/t-MDS) are secondary hematologic malignancies associated with poor prognosis, warranting insights into their predisposing conditions and cells of origin. We identified patients with myeloma who developed t-AML/t-MDS and analyzed their stem and progenitor cells collected years before the onset of secondary disease. We demonstrate that aberrant stem cells with high CD123 expression can be detected long before the onset of overt leukemia. Rigorous sorting, followed by targeted sequencing, resulted in ultradeep functional depth of sequencing and revealed preexisting mutant hematopoietic stem cell (HSC) clones, mainly harboring TP53 mutations, that became the dominant population at the time of leukemic presentation. Taken together, these data show that HSCs can act as reservoirs for leukemia-initiating cells many years before the onset of myeloid leukemia.
Introduction
Therapy-related acute myeloid leukemia and myelodysplastic syndrome (t-AML/t-MDS) are secondary malignancies that occur in patients receiving chemotherapy and/or radiation for the treatment of cancer.1 MDS-associated cytogenetic abnormalities as well as clinical t-MDS and t-AML have been reported to be relatively high in myeloma patients.2 As these secondary malignancies are associated with a poor prognosis, it is critical to identify risk factors and predispositions that can lead to their development.1,3 Clonal hematopoiesis is defined by the presence of somatic mutations in the blood and/or bone marrow in the absence of cytopenias or other evidence of hematologic malignancy.4-7 These mutations are generally detected at low variant allele frequencies; have been associated with increased age, exposure to radiation, and tobacco use8,9 ; and have been recognized as a risk factor for developing myeloid neoplasms.4,5,8,10-13
Others and we have shown that hematopoietic stem cell (HSC) subclones can harbor mutations in MDS and AML.14-16 Past studies examining longitudinally sampled pre- and postchemotherapy specimen showed that leukemia-associated mutations can exist in pretreatment samples.17,18 In the present study we attempted to determine whether stem and progenitor cells are the reservoirs of leukemia associated mutations in subjects that develop these secondary malignancies after myeloma treatment.
Methods
Details of the methods used are provided in the supplemental Data.
Patients and samples
The study protocol was approved by the University of Arkansas for Medical Sciences (UAMS) Institutional Review Board. Specimens were obtained in accordance with the Declaration of Helsinki. Six patients with multiple myeloma (MM) treated with autologous hemapoietic stem cell transplantation at the University of Arkansas for Medical Sciences (UAMS) from 2002 through 2015, who subsequently developed t-AML/t-MDS, were identified (supplemental Figure 1; supplemental Methods). CD34+ HSCs were retrieved from collection vials at the time point of stem cell harvest and were processed as below, whereas CD138+ MM cells were collected at the time point of MM relapse, as indicated. Sequencing of myeloma CD138+ cells for 2 patients was performed at Foundation Medicine. Sequencing of AML samples was performed at UAMS at the time of presentation.
Fluorescence-activated cell sorting of stem and progenitor cells
Specimens were separated into CD34+ and CD34− samples, by using MACS MicroBeads (Miltenyi Biotech). CD34+ samples were sorted into normal HSCs (Lin−CD34+CD38−), aberrant leukemia stem cells (LSCs; phenotypic HSCs that were CD123+, CD45RA, or IL1RAP+), granulocyte-macrophage progenitors (GMPs), and megakaryocyte-erythroid progenitors (MEPs). CD34− samples were sorted into granulocytes and T and B cells.
Sequencing of sorted cell populations
Genomic DNA was extracted from sorted cell populations and sent for targeted sequencing for panel of 236 Genes at a depth ranging from 500× to +5000×. Targeted genomic regions were sequenced using next-generation sequencing by Genoptix.
Results and discussion
Identification and genotyping of patients with MM, who developed secondary AML/MDS months and years after treatment
A total of 6 patients with MM were identified that developed secondary AML/MDS (sAML/sMDS) after autologous transplantation and had unused stem cell samples that had been collected at the time of transplantation (supplemental Table 1). Except for 1 patient who developed leukemia 3 months after autologous transplantation (patient 3), all the others developed leukemia after more than 2 years, with 1 case occurring after 10 years (mean time to sAML/sMDS, 5 years; median, 3.9 years). Commercially available DNA sequencing at the time of t-AML/t-MDS diagnosis showed that 5 of 6 patients had TP53 mutations in their AML samples, and the sixth patient had a RUNX1 mutation. (supplemental Table 1). All patients responded well to anti-MM therapy, and cause of death for all patients was the transformation to t-AML/t-MDS.
HSCs collected before the onset of disease reveal a higher number of CD123+ LSCs
We used a set of well-characterized LSC surface markers, comprising CD123, IL1RAP, CD99, and CD45Rα,16,19-21 in conjunction with established stem cell markers to differentiate between phenotypically normal and abnormal HSCs. Unused specimen from 6 patients with MM were purified by fluorescence-activated cell sorting (FACS) for phenotypically normal HSCs (Lin−CD34+CD38− IL1RAP−CD123−CD99−CD45Rα−) and aberrant stem cells (Lin−CD34+CD38− positive for at least 1 of the LSC markers IL1RAP, CD123, or CD45Rα). We also purified myeloid and erythroid progenitors (common myeloid progenitor [CMP], GMP, and MEP cells), as well as mature granulocytes and T and B cells (Figure 1; sorting scheme in Figure 2A). We found significantly lower HSC frequencies in patients with myeloma when compared with mobilized healthy control specimens (mean of 19% for HSCs vs 10% for patient samples; P = .04; Figure 1 A-B). We also observed a decreased number of CMPs in patient samples (Figure 1C-D; P = .01). Within the stem cell compartments, we observed a lower frequency of normal HSCs with the absence of LSC markers (Figure 1E-F; P = .02). Our data also showed a trend toward higher frequencies of aberrant stem cells with increased expression of CD123 in patients who developed secondary leukemia, when compared with healthy controls (Figure 1G-H; P = .09).
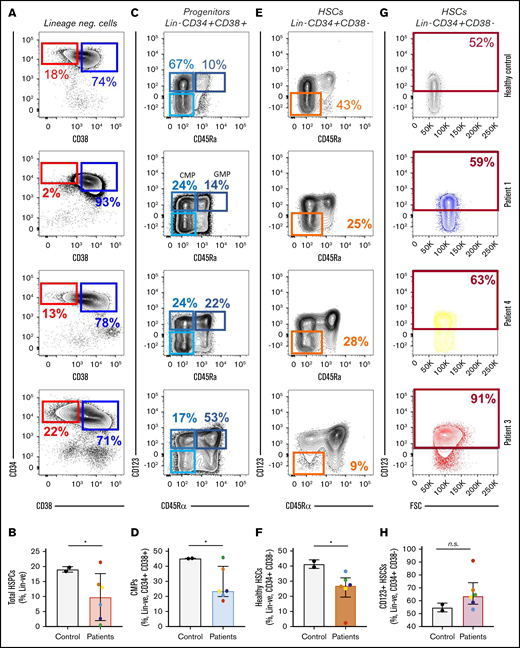
Aberrant phenotypic LSCs can be detected before the onset of sAML/sMDS. Flow cytometric analysis was conducted on collected stem and progenitor cells from patients who later developed sAML/sMDS and from mobilized controls. Patient samples demonstrated a decrease in total HSPCs (Lin− CD34+ CD38−) (A-B), a decrease in CMPs (Lin−, CD34+, CD38+, CD123+, CD45ra−) (C-D), a decrease in healthy HSPCs (Lin−, CD34+, CD38−, CD123−, CD45ra−) (E-F), and a trend toward an increase in CD123+ HSPCs (Lin−, CD34+, CD38−) (G-H). These effects were most pronounced in patient 3, who developed sAML shortly after specimen collection. Means and standard deviations for stem and progenitor populations are depicted as frequencies of parental population (top of FACS plots: panels A, C, E, and G; and y-axes: panels B, D, F, and H). Student t test: *P < .05. n.s., not significant.
Aberrant phenotypic LSCs can be detected before the onset of sAML/sMDS. Flow cytometric analysis was conducted on collected stem and progenitor cells from patients who later developed sAML/sMDS and from mobilized controls. Patient samples demonstrated a decrease in total HSPCs (Lin− CD34+ CD38−) (A-B), a decrease in CMPs (Lin−, CD34+, CD38+, CD123+, CD45ra−) (C-D), a decrease in healthy HSPCs (Lin−, CD34+, CD38−, CD123−, CD45ra−) (E-F), and a trend toward an increase in CD123+ HSPCs (Lin−, CD34+, CD38−) (G-H). These effects were most pronounced in patient 3, who developed sAML shortly after specimen collection. Means and standard deviations for stem and progenitor populations are depicted as frequencies of parental population (top of FACS plots: panels A, C, E, and G; and y-axes: panels B, D, F, and H). Student t test: *P < .05. n.s., not significant.
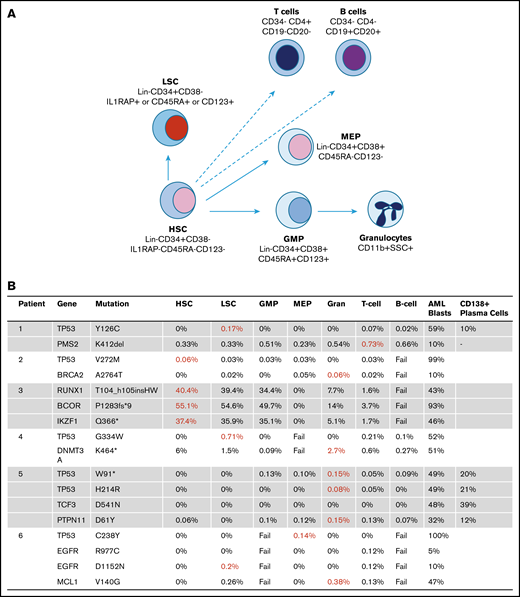
Frequency of t-AML/t-MDS mutations in stem cell collection samples prior to MM auto-HSCT. (A) Stem and progenitor populations, granulocytes, and T and B cells were FACS sorted and sequenced. (B) The AML samples and myeloma plasma cell samples were sequenced at different times at the respective clinical presentations. The variant allele frequencies (VAFs) of mutations in the different cell populations are shown. Populations with the highest VAFs are shown in red.
Frequency of t-AML/t-MDS mutations in stem cell collection samples prior to MM auto-HSCT. (A) Stem and progenitor populations, granulocytes, and T and B cells were FACS sorted and sequenced. (B) The AML samples and myeloma plasma cell samples were sequenced at different times at the respective clinical presentations. The variant allele frequencies (VAFs) of mutations in the different cell populations are shown. Populations with the highest VAFs are shown in red.
Patient 3 developed t-AML a few months after the autologous stem cell transplantation. Even though he had no obvious signs of myeloid malignancy at the time of collection, an analysis of his stem cell collection revealed a greatly increased number of aberrant stem cells, with the highest CD123 expression (Figure 1F), demonstrating leukemialike stem and progenitor alterations before the onset of clinically evident disease.
Preleukemic HSCs contain somatic mutations many years before the onset of secondary AML/MDS
Next, stem cell collections were sorted for phenotypic normal and leukemic stem cells, progenitors (GMPs and MEPs) and differentiated granulocytes and lymphocytes. Sorted cell populations were used for DNA isolation, and sufficient DNA for sequencing was extracted from 43 of 48 hematopoietic subpopulations. Deep-targeted sequencing was conducted for a total of 18 mutations that overlapped between panels (Figure 2B). Strikingly, in all 6 patients, the exact same driver mutation (TP53 or RUNX1) observed in sAML/sMDS samples were detected in stem or progenitor cells collected at the time of myeloma treatment.
In 50% (3 of 6) of the cases, the highest enrichment for the driver mutation was seen in phenotypically aberrant stem cells with LSC markers, whereas another subject had the highest mutational burden in the phenotypically normal HSC compartment. In the other 2 cases, the highest variant allele frequency was seen in myeloid cells (GMPs or granulocytes).
In the patient who developed secondary leukemia 3 months after stem cell collection, a high mutational load was observed in the aberrant stem cells (Figure 2B). A high mutational load was also seen in the HSCs, LSCs, and GMPs but not in the differentiated granulocytes, suggesting the presence of a preleukemic stage with significant stem and progenitor stage involvement.
In most of the patients in whom the stem cells were analyzed many years before leukemic presentation, we observed a mutational variant allele frequency of between 0.03% and 0.71% in the stem cell compartments. The differentiated populations did not reveal a mutational load for the TP53 mutations in most cases, suggesting that stem cells are the reservoir of these mutant subclones.
We had sequencing data from myeloma samples in 2 cases that were commercially sequenced at the time of myeloma presentation (patients 1 and 5; supplemental Table 1). The same TP53 driver mutations found in stem and progenitor compartments were also detected in 2 of the CD138+ MM samples. The CD138+ myeloma cells were separated by immunomagnetic beads, and it is possible that there was contamination by stem and progenitor cells, though the mutations were found at a high variant allele frequency (10%-21%; Figure 2). Both patients had stable myeloma (details in supplemental Methods) but died of complications of secondary myeloid neoplasms.
Our results demonstrate that disease-associated driver mutations can be detected many years before the development of t-AML and t-MDS. By using rigorous flow cytometry analysis, we also demonstrated that stem and progenitor cells act as reservoirs of these mutant subclones. We also observed that phenotypic aberrant stem cells with expression of LSC markers (Lin−, CD34+, CD38−, and CD123+) were detectable in these samples many years before the onset of clinical AML/MDS. Previous studies have shown that the presence of similar phenotypic LSCs (with CD123 positivity) is associated with a higher risk of MDS and AML.19 Because the patients had undergone chemotherapy and growth factor mobilization at the time of collection, it is possible that high CD123 expression on the stem cells was a result of these treatments. Future studies will test whether genotoxic or proliferative stress can result in aberrant CD123 expression on stem cells and whether the presence and magnitude of phenotypic LSCs can also be a potential biomarker for future development of myeloid neoplasms.
Acknowledgments
This work was supported by the Leukemia and Lymphoma Society, the Evans Foundation, and National Institutes of Health, National Heart, Lung and Blood Institute grant R01HL139487 (A.V.).
Authorship
Contribution: A. Sridharan designed and performed the research, analyzed the data, and wrote the manuscript; C.D.S. designed the research and provided the samples; G.G., M.D.S.F., V.T., I.M., T.D.B., G.S.C., S.A., and J.C. performed the research; K.P. analyzed the data; Y.X., M.P., G.S., C.K.B., R.B., L.K., A.G., and F.A.F., performed the sequencing studies and analysis; C.H. and A. Shastri analyzed the data; D.A., N.W., S.K.J., A.W., B.B., and G.J.M., provided samples for analysis; and U.S., B.W., and A.V., designed research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: A.V. has received research funding from GSK, Incyte, Medpacto, and Eli Lilly and is a scientific advisor for Stelexis, Novartis, Acceleron, and Celgene. The remaining authors declare no competing financial interests.
Correspondence: Amit Verma, Montefiore Medical Center, Albert Einstein College of Medicine, 1300 Morris Park Ave, Bronx, NY 10461; e-mail: amit.verma@einstein.yu.edu; Britta Will, Albert Einstein College of Medicine, Montefiore Medical Center, 1300 Morris Park Ave, Bronx, NY 10461; e-mail: britta.will@einstein.yu.edu; and Ulrich Steidl, Albert Einstein College of Medicine, Montefiore Medical Center, 1300 Morris Park Ave, Bronx, NY 10461; e-mail: ulrich.steidl@einstein.yu.edu.

