

Key Points
Distinct sphingolipid metabolism of AML with MDS-related changes defines unique sensitivity to nanoliposomal C6-ceramide.
Vinblastine alters sphingolipid metabolism to enhance the sensitivity of AML to nanoliposomal C6-ceramide.
Introduction
Therapeutic advances for the treatment of acute myeloid leukemia (AML) have been limited in part because of an incomplete understanding of its underlying biology.1-4 AML that arises out of myelodysplastic syndrome (MDS) highlights this challenge and represents a disease subcategory with a decidedly poor clinical outlook.1-6 We studied sphingolipid biology in different forms of AML and evaluated the anti-AML efficacy of nanoliposomal C6-ceramide (Lip-C6). Sphingolipids are an extensive classification of lipids that play roles in cell survival and proliferation as well as stress and death.7-12 The most well-studied sphingolipid is ceramide, a proapoptotic sphingolipid, which serves as a hypothetical center of sphingolipid metabolism.7-12 Lip-C6 delivers the short-chain C6-ceramide analog and has entered phase 1 clinical trials for patients with solid tumors (ClinicalTrials.gov identifier: NCT02834611).13 Lip-C6 exerts anticancer efficacy across a spectrum of malignancies.7,13-17 This study evaluated the preclinical efficacy of Lip-C6 for AML with MDS-related changes (AML-MRC), which was uniquely sensitive to Lip-C6 because of its propensity to convert C6-ceramide to proapoptotic sphingolipid metabolites. In contrast, resistance to Lip-C6 by de novo AML (DN-AML) was overcome by using cotreatment with vinblastine, which restored this proapoptotic sphingolipid phenotype.
Methods
Patient samples
Patients were classified as having either AML-MRC according to World Health Organization (WHO) criteria1,2 or as having DN-AML if they did not have MDS-related changes or an antecedent hematologic disorder or dysplasia (supplemental Table 1). MDS-related changes were noted if there was a history of prior MDS or cytogenetic abnormalities associated with MDS as defined by Vardiman et al3 in an update to the WHO classification. We also performed next-generation mutational profiling as previously described,4,15 to subclassify many patients based on the occurrence of MDS-related mutations per Papaemmanuil et al.1 All clinical samples and information were collected at the Penn State College of Medicine under Institutional Review Board–approved informed consent. Additional sample information and clinical data can be found in the supplemental Methods.
Murine AML-MRC and DN-AML samples
The Nup98-HoxD13 transgenic mouse model fully recapitulates the cellular features of MDS and predictably evolves to AML.18 Therefore, we used this mouse as a model of AML-MRC. In contrast, we used the MLL-AF9 and Flt3-ITD transgenic mice as models of DN-AML. In all cases, transgenic mouse leukemia was confirmed by the investigative team and the New Hampshire Veterinary Diagnostic Laboratory.
Additional methods
Liposome generation,14-17 apoptosis assays,16 colony-forming assays,16 lipidomics,19 and animal therapeutic studies15,20 were carried out as previously described. Additional study-specific information for these methods, as well as details on cell culture and statistical analyses can be found in the supplemental Methods. For original data, please contact the corresponding authors. All animal studies were approved by the Institutional Animal Care and Use Committees of the University of New Hampshire and the Penn State College of Medicine.
Results and discussion
Nanoliposomal ceramide exerts distinct efficacy toward AML-MRC
Apoptosis was evaluated in patient AML samples exposed to 20 µM Lip-C6 for 48 hours. AML-MRC samples were uniquely sensitive to Lip-C6 (Figure 1A; supplemental Table 2). In contrast, variable sensitivity to Lip-C6 was observed from patient DN-AML samples (Figure 1B; supplemental Table 2). Lip-C6 substantially interfered with the colony-forming capacity of isolated bone marrow cells from transgenic AML-MRC mice,18 but not from transgenic DN-AML mice and wild-type controls (Figure 1C; supplemental Figures 1-3). Similarly, in vivo treatment with Lip-C6 significantly reduced detectable leukemia in AML-MRC but not DN-AML transgenic mice (Figure 1D; supplemental Figures 4 and 5). In addition, the colony-forming capacity for human AML-MRC patient samples was significantly lower in the presence of Lip-C6 than for DN-AML patient samples (Figure 1E; supplemental Table 2; supplemental Figures 1-3). Overall, these results showed that samples of AML-MRC are uniquely sensitive to Lip-C6 monotherapy.
![Figure 1. AML-MRC is uniquely sensitive to Lip-C6. Human patient samples of AML-MRC (n = 15) (A) or DN-AML (n = 15) (B) were exposed for 48 hours to 20 µM Lip-C6 or control nanoliposomes without ceramide (Lip-Ghost). Apoptosis within the leukemia stem cell (CD34+CD38–) and bulk leukemia fractions were quantified by flow cytometry (1-way analysis of variance [ANOVA] with Tukey’s post hoc comparison). *P ≤ .0042 compared with controls, **P < .0001 compared with controls. (C) Colony-forming assays evaluating dose-response of Lip-C6 were performed using bone marrow samples harvested from wild-type, transgenic AML-MRC, or transgenic DN-AML mice. *P ≤ .0071, **P ≤ .0001, and ***P ≤ .0468 for mouse AML-MRC compared with both wild-type and DN-AML (2-way ANOVA with Tukey’s post hoc comparison). Error bars represent the standard error of the mean (SEM). Mouse wild-type, n = 3; mouse DN-AML, n = 4; and mouse AML-MRC, n = 4. (D) Therapeutic efficacy of Lip-C6 as a standalone treatment was evaluated using transgenic mouse models of AML-MRC (Nup98-HoxD13) and DN-AML (Flt3-ITD). Mice were treated for 10 days with daily injections of Lip-C6 (11.6 mg/kg) or Lip-Ghost (volume-matched). Mice were then euthanized, and bone marrow was isolated and prepared for flow cytometry in which Gr-1+CD11b+ cells were evaluated and quantified as representative of leukemia burden. *P = .045 (n = 4 per group; unpaired Student t test with Welch’s correction) comparing the leukemia burden in the bone marrow of AML-MRC (Nup98-HoxD13) transgenic mice treated with Lip-C6 with those treated with control nanoliposomes without ceramide (Ghost). See supplemental Figures 4 and 5 for representative blood smears and bone marrow flow cytometry histograms. (E) Colony-forming assays evaluating a dose-response of Lip-C6 were performed using human mononuclear cells obtained from patients with AML-MRC or DN-AML. #P = .047 and ##P ≤ .003 for human AML-MRC compared with DN-AML (2-way ANOVA with Sidek’s post hoc comparison). Error bars represent the SEM; human DN-AML, n = 6; human AML-MRC, n = 5. (F-G) Patient samples were exposed for 24 hours in vitro to 10 µM Lip-C6, and the metabolism of C6-ceramide was evaluated by lipidomic analysis. (F) Comparing the ratios of C6-ceramide prodeath to neutral prosurvival metabolites in AML-MRC and DN-AML (unpaired Student t test with Welch’s correction; *P = .0081). Error bars represent the SEM; n = 4. (G) Patient AML-MRC preferentially converts C6-ceramide to the prodeath metabolite sphingosine as well as endogenous/physiological ceramides rather than neutral or prosurvival metabolites (S1P, C6-sphingomyelin [SM], C6-cerebrosides [includes both C6-glucosylceramide and C6-galatosylceramide]) (1-way ANOVA with Tukey’s post hoc comparison; *P < .0001 compared with all other metabolites; **P ≤ .0294 compared with endogenous ceramides, C6-SM, and C6-cerebrosides; #P < .0001 compared with C6-SM; ##P < .0001 compared with all other metabolites; $P = .028 compared with C6-cerebrosides; $$P ≤ .0219 compared with S1P, C6-SM, and C6-cerebrosides; &P ≤ .0014 compared with S1P and C6-cerebrosides; 0026amp;&P ≤ .0057 compared with C6-SM and C6-cerebrosides). Error bars represent the SEM; n ≤ 4. (H) Metabolism of C6-ceramide to proapoptotic or neutral or prosurvival metabolites. AC, acid ceramidase; CERS, ceramide synthases; GCS, glucosylceramide synthase; SMS, sphingomyelin synthases; SPHK, sphingosine kinase.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/17/10.1182_bloodadvances.2018021295/4/m_advances021295f1.png?Expires=1767822102&Signature=k1CYtyEaI99cWf6PK3OnDAMiL5HFSDtCUpdUS~hocYPNGr9AsKLzs1Y8PyTqUwqUKtCimCeVzq6u-6mdaor-MSWRsP7vnXx9lpwBaoAkrH8HZAs6Saq88EPyCDtVHNwcvOt2VPVIMDT71hWzkpiBxxZQuoixT4TESbKwP8bmR8WVxdp2H2uQ2SiSpC7ZdwP01Bx~kqgmq~uDsF4xAs4GqSzO31-e5oIhuWFCId6odfE9C3lgalMeSkRTFcGSpbAdL8RopaZ29iIT37AHYP1cnC5Hj9wRifyPGq8xhss5DE3Vl5weICCv5V6ODPLf2c58fYODyCyJf7B9P9WJ3hsSQA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AML-MRC is uniquely sensitive to Lip-C6. Human patient samples of AML-MRC (n = 15) (A) or DN-AML (n = 15) (B) were exposed for 48 hours to 20 µM Lip-C6 or control nanoliposomes without ceramide (Lip-Ghost). Apoptosis within the leukemia stem cell (CD34+CD38–) and bulk leukemia fractions were quantified by flow cytometry (1-way analysis of variance [ANOVA] with Tukey’s post hoc comparison). *P ≤ .0042 compared with controls, **P < .0001 compared with controls. (C) Colony-forming assays evaluating dose-response of Lip-C6 were performed using bone marrow samples harvested from wild-type, transgenic AML-MRC, or transgenic DN-AML mice. *P ≤ .0071, **P ≤ .0001, and ***P ≤ .0468 for mouse AML-MRC compared with both wild-type and DN-AML (2-way ANOVA with Tukey’s post hoc comparison). Error bars represent the standard error of the mean (SEM). Mouse wild-type, n = 3; mouse DN-AML, n = 4; and mouse AML-MRC, n = 4. (D) Therapeutic efficacy of Lip-C6 as a standalone treatment was evaluated using transgenic mouse models of AML-MRC (Nup98-HoxD13) and DN-AML (Flt3-ITD). Mice were treated for 10 days with daily injections of Lip-C6 (11.6 mg/kg) or Lip-Ghost (volume-matched). Mice were then euthanized, and bone marrow was isolated and prepared for flow cytometry in which Gr-1+CD11b+ cells were evaluated and quantified as representative of leukemia burden. *P = .045 (n = 4 per group; unpaired Student t test with Welch’s correction) comparing the leukemia burden in the bone marrow of AML-MRC (Nup98-HoxD13) transgenic mice treated with Lip-C6 with those treated with control nanoliposomes without ceramide (Ghost). See supplemental Figures 4 and 5 for representative blood smears and bone marrow flow cytometry histograms. (E) Colony-forming assays evaluating a dose-response of Lip-C6 were performed using human mononuclear cells obtained from patients with AML-MRC or DN-AML. #P = .047 and ##P ≤ .003 for human AML-MRC compared with DN-AML (2-way ANOVA with Sidek’s post hoc comparison). Error bars represent the SEM; human DN-AML, n = 6; human AML-MRC, n = 5. (F-G) Patient samples were exposed for 24 hours in vitro to 10 µM Lip-C6, and the metabolism of C6-ceramide was evaluated by lipidomic analysis. (F) Comparing the ratios of C6-ceramide prodeath to neutral prosurvival metabolites in AML-MRC and DN-AML (unpaired Student t test with Welch’s correction; *P = .0081). Error bars represent the SEM; n = 4. (G) Patient AML-MRC preferentially converts C6-ceramide to the prodeath metabolite sphingosine as well as endogenous/physiological ceramides rather than neutral or prosurvival metabolites (S1P, C6-sphingomyelin [SM], C6-cerebrosides [includes both C6-glucosylceramide and C6-galatosylceramide]) (1-way ANOVA with Tukey’s post hoc comparison; *P < .0001 compared with all other metabolites; **P ≤ .0294 compared with endogenous ceramides, C6-SM, and C6-cerebrosides; #P < .0001 compared with C6-SM; ##P < .0001 compared with all other metabolites; $P = .028 compared with C6-cerebrosides; $$P ≤ .0219 compared with S1P, C6-SM, and C6-cerebrosides; &P ≤ .0014 compared with S1P and C6-cerebrosides; 0026amp;&P ≤ .0057 compared with C6-SM and C6-cerebrosides). Error bars represent the SEM; n ≤ 4. (H) Metabolism of C6-ceramide to proapoptotic or neutral or prosurvival metabolites. AC, acid ceramidase; CERS, ceramide synthases; GCS, glucosylceramide synthase; SMS, sphingomyelin synthases; SPHK, sphingosine kinase.
AML-MRC is uniquely sensitive to Lip-C6. Human patient samples of AML-MRC (n = 15) (A) or DN-AML (n = 15) (B) were exposed for 48 hours to 20 µM Lip-C6 or control nanoliposomes without ceramide (Lip-Ghost). Apoptosis within the leukemia stem cell (CD34+CD38–) and bulk leukemia fractions were quantified by flow cytometry (1-way analysis of variance [ANOVA] with Tukey’s post hoc comparison). *P ≤ .0042 compared with controls, **P < .0001 compared with controls. (C) Colony-forming assays evaluating dose-response of Lip-C6 were performed using bone marrow samples harvested from wild-type, transgenic AML-MRC, or transgenic DN-AML mice. *P ≤ .0071, **P ≤ .0001, and ***P ≤ .0468 for mouse AML-MRC compared with both wild-type and DN-AML (2-way ANOVA with Tukey’s post hoc comparison). Error bars represent the standard error of the mean (SEM). Mouse wild-type, n = 3; mouse DN-AML, n = 4; and mouse AML-MRC, n = 4. (D) Therapeutic efficacy of Lip-C6 as a standalone treatment was evaluated using transgenic mouse models of AML-MRC (Nup98-HoxD13) and DN-AML (Flt3-ITD). Mice were treated for 10 days with daily injections of Lip-C6 (11.6 mg/kg) or Lip-Ghost (volume-matched). Mice were then euthanized, and bone marrow was isolated and prepared for flow cytometry in which Gr-1+CD11b+ cells were evaluated and quantified as representative of leukemia burden. *P = .045 (n = 4 per group; unpaired Student t test with Welch’s correction) comparing the leukemia burden in the bone marrow of AML-MRC (Nup98-HoxD13) transgenic mice treated with Lip-C6 with those treated with control nanoliposomes without ceramide (Ghost). See supplemental Figures 4 and 5 for representative blood smears and bone marrow flow cytometry histograms. (E) Colony-forming assays evaluating a dose-response of Lip-C6 were performed using human mononuclear cells obtained from patients with AML-MRC or DN-AML. #P = .047 and ##P ≤ .003 for human AML-MRC compared with DN-AML (2-way ANOVA with Sidek’s post hoc comparison). Error bars represent the SEM; human DN-AML, n = 6; human AML-MRC, n = 5. (F-G) Patient samples were exposed for 24 hours in vitro to 10 µM Lip-C6, and the metabolism of C6-ceramide was evaluated by lipidomic analysis. (F) Comparing the ratios of C6-ceramide prodeath to neutral prosurvival metabolites in AML-MRC and DN-AML (unpaired Student t test with Welch’s correction; *P = .0081). Error bars represent the SEM; n = 4. (G) Patient AML-MRC preferentially converts C6-ceramide to the prodeath metabolite sphingosine as well as endogenous/physiological ceramides rather than neutral or prosurvival metabolites (S1P, C6-sphingomyelin [SM], C6-cerebrosides [includes both C6-glucosylceramide and C6-galatosylceramide]) (1-way ANOVA with Tukey’s post hoc comparison; *P < .0001 compared with all other metabolites; **P ≤ .0294 compared with endogenous ceramides, C6-SM, and C6-cerebrosides; #P < .0001 compared with C6-SM; ##P < .0001 compared with all other metabolites; $P = .028 compared with C6-cerebrosides; $$P ≤ .0219 compared with S1P, C6-SM, and C6-cerebrosides; &P ≤ .0014 compared with S1P and C6-cerebrosides; 0026amp;&P ≤ .0057 compared with C6-SM and C6-cerebrosides). Error bars represent the SEM; n ≤ 4. (H) Metabolism of C6-ceramide to proapoptotic or neutral or prosurvival metabolites. AC, acid ceramidase; CERS, ceramide synthases; GCS, glucosylceramide synthase; SMS, sphingomyelin synthases; SPHK, sphingosine kinase.
C6-ceramide is converted to proapoptotic sphingolipid metabolites by AML-MRC
Sphingolipids such as sphingosine-1-phosphate (S1P) and glucosylceramide oppose the proapoptotic activity of ceramide by stimulating proliferation and survival.7-12 Therefore, AML resistance to Lip-C6 was anticipated to follow C6-ceramide metabolism to neutral/prosurvival sphingolipids (Figure 1F-H). Patient AML samples were exposed to 10 µM Lip-C6 for 24 hours before lipidomic analysis.19 Patient AML-MRC primarily converted C6-ceramide to proapoptotic metabolites, with sphingosine being predominately elevated (Figure 1F-G; supplemental Figures 6 and 7). In contrast, patient DN-AML converted C6-ceramide to a wide range of both proapoptotic and neutral or prosurvival metabolites (Figure 1F; supplemental Figures 6-8).
Vinblastine promotes the accumulation of proapoptotic sphingolipids in AML
We previously demonstrated that vinblastine can synergize with Lip-C6 in models of colorectal cancer and hepatocellular carcinoma.14 As a microtubule destabilizing agent, vinblastine interferes with vesicular transport between subcellular compartments and may therefore impede sphingolipid metabolism. We used lipidomics to evaluate patient AML samples exposed to a combination of 10 µM Lip-C6 and 2.5 nM vinblastine for 24 hours.19 Vinblastine significantly directed the metabolism of C6-ceramide to the proapoptotic metabolites sphingosine and physiological/endogenous-ceramides (Figure 2A; supplemental Figure 7). We corroborated these findings using human AML cell lines (supplemental Figure 9). Vinblastine shifts sphingolipid metabolism so that C6-ceramide is converted to proapoptotic metabolites.
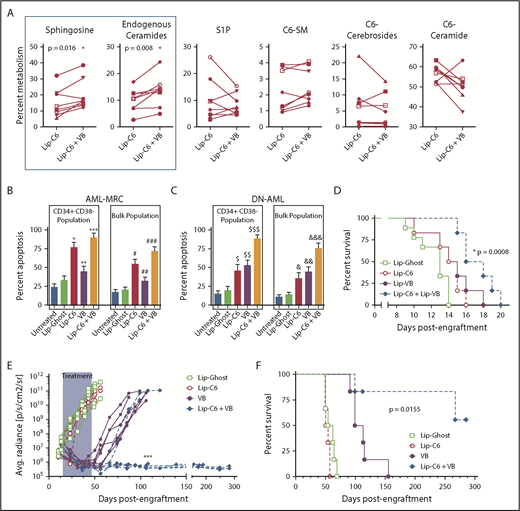
Combinatorial anti-AML efficacy for Lip-C6 and vinblastine. (A) Eight human AML patient samples were exposed for 24 hours in vitro to 10 µM Lip-C6 alone or in combination with 2.5 nM vinblastine (VB), and the metabolism of C6-ceramide was evaluated by lipidomic analysis. Vinblastine cotreatment significantly shifted C6-ceramide metabolism to sphingosine and endogenous/physiological ceramides, both proapoptotic metabolites. Unpaired Student t test with Welch’s correction; *P ≤ .02 (specific values indicated). Data points are averages of biological replicates (n ≥ 3). AML-MRC (n = 15) (B) or DN-AML (n = 15) (C) human patient samples were exposed for 48 hours to 20 µM Lip-C6, Lip-Ghost, 5 nM VB, or the combination of Lip-C6 and VB, and apoptosis within the leukemia stem cell (CD34+CD38–) and bulk leukemia fractions was quantified by flow cytometry (1-way ANOVA with Tukey’s post hoc comparison; *P < .0001, #P < .0001, $P ≤ .0072, or &P ≤ .0375 compared with untreated and Lip-Ghost; **P = .0006 or ##P = .0036 compared with Lip-C6; $$P ≤ .0003 or &&P ≤ .0375 compared with untreated and Lip-Ghost; ***P < .0001 or ###P < .0001 compared with untreated, Lip-Ghost, and VB; $$$P ≤ .0001 or &&&P < .001 compared with all other treatments). (D) Survival of C57BL/6J mice engrafted with murine C1498 AML cells and then treated with intravenous injections of anionic formulations of Lip-Ghost control, Lip-C6, liposomal vinblastine (Lip-VB), or the combination of Lip-C6 and Lip-VB. Mantel-Cox log rank test *P = .008 (n ≥ 6). (E) Leukemia burden was quantified after bioluminescent imaging of NSG mice engrafted with MV4-11-LUC-YFP AML cells. Mice were treated with intravenous injections of either the Lip-Ghost control, Lip-C6, VB (not formulated in a liposome), or the combination of both Lip-C6 and VB. Shaded area corresponds to the treatment duration. ****P < .0001 comparing all the Lip-C6 + VB treatment mice with all those that received VB treatment alone at day 106 (2-way ANOVA with Tukey’s post hoc comparison). Each line represents the leukemia burden of an individual mouse. (F) Survival of NSG mice engrafted with human MV4-11-LUC-YFP AML cells that were treated with intravenous injections of Lip-Ghost control, Lip-C6, VB (not formulated in a liposome), or Lip-C6 and VB combined. Mantel-Cox log rank test *P = .0159 (n = 6).
Combinatorial anti-AML efficacy for Lip-C6 and vinblastine. (A) Eight human AML patient samples were exposed for 24 hours in vitro to 10 µM Lip-C6 alone or in combination with 2.5 nM vinblastine (VB), and the metabolism of C6-ceramide was evaluated by lipidomic analysis. Vinblastine cotreatment significantly shifted C6-ceramide metabolism to sphingosine and endogenous/physiological ceramides, both proapoptotic metabolites. Unpaired Student t test with Welch’s correction; *P ≤ .02 (specific values indicated). Data points are averages of biological replicates (n ≥ 3). AML-MRC (n = 15) (B) or DN-AML (n = 15) (C) human patient samples were exposed for 48 hours to 20 µM Lip-C6, Lip-Ghost, 5 nM VB, or the combination of Lip-C6 and VB, and apoptosis within the leukemia stem cell (CD34+CD38–) and bulk leukemia fractions was quantified by flow cytometry (1-way ANOVA with Tukey’s post hoc comparison; *P < .0001, #P < .0001, $P ≤ .0072, or &P ≤ .0375 compared with untreated and Lip-Ghost; **P = .0006 or ##P = .0036 compared with Lip-C6; $$P ≤ .0003 or &&P ≤ .0375 compared with untreated and Lip-Ghost; ***P < .0001 or ###P < .0001 compared with untreated, Lip-Ghost, and VB; $$$P ≤ .0001 or &&&P < .001 compared with all other treatments). (D) Survival of C57BL/6J mice engrafted with murine C1498 AML cells and then treated with intravenous injections of anionic formulations of Lip-Ghost control, Lip-C6, liposomal vinblastine (Lip-VB), or the combination of Lip-C6 and Lip-VB. Mantel-Cox log rank test *P = .008 (n ≥ 6). (E) Leukemia burden was quantified after bioluminescent imaging of NSG mice engrafted with MV4-11-LUC-YFP AML cells. Mice were treated with intravenous injections of either the Lip-Ghost control, Lip-C6, VB (not formulated in a liposome), or the combination of both Lip-C6 and VB. Shaded area corresponds to the treatment duration. ****P < .0001 comparing all the Lip-C6 + VB treatment mice with all those that received VB treatment alone at day 106 (2-way ANOVA with Tukey’s post hoc comparison). Each line represents the leukemia burden of an individual mouse. (F) Survival of NSG mice engrafted with human MV4-11-LUC-YFP AML cells that were treated with intravenous injections of Lip-Ghost control, Lip-C6, VB (not formulated in a liposome), or Lip-C6 and VB combined. Mantel-Cox log rank test *P = .0159 (n = 6).
Vinblastine and nanoliposomal ceramide exert combinatorial anti-AML efficacy
Patient AML samples were exposed to 20 µM Lip-C6, 5 nM vinblastine for 48 hours, or both, and apoptosis was evaluated. Lip-C6 was much more effective for AML-MRC than for DN-AML, but the combination of Lip-C6 with vinblastine induced robust apoptosis of either AML-MRC or DN-AML (Figure 2B-C). In addition, the combination of Lip-C6 and liposomal vinblastine significantly prolonged the lifespan of C57BL/6J mice engrafted with C1498 cells, a rapidly lethal model of AML (Figure 2D).20 Likewise, in NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice xenografted with MV4-11-LUC-YFP human AML cells, vinblastine monotherapy mediated a transient cytoreductive effect, which was overcome, whereas Lip-C6 exerted no standalone efficacy in this model of FLT3-ITD AML (Figure 2E). In contrast, the combination of vinblastine with Lip-C6 abrogated detectable leukemia and induced a complete remission in 50% of the mice lasting >150 days after discontinuation of therapy (Figure 2E-F; supplemental Figures 10 and 11). Altogether, these studies demonstrated robust anti-AML efficacy for the combination of Lip-C6 with vinblastine.
This study has identified varied ceramide metabolism in MDS and AML, informing the development of a novel application of therapeutic Lip-C6 and vinblastine. AML-MRC is uniquely sensitive to Lip-C6 because C6-ceramide is preferentially converted to proapoptotic metabolites, including sphingosine and endogenous ceramides. In contrast, DN-AML was less sensitive to Lip-C6 because of an increased propensity to metabolize C6-ceramide to neutral or prosurvival metabolites. Combining Lip-C6 with vinblastine shifted the metabolism of C6-ceramide back to sphingosine and endogenous ceramides. By disrupting intracellular trafficking, vinblastine may restrict C6-ceramide delivered by Lip-C6 to plasma membrane-localized metabolic pathways. Ceramide species with endogenous fatty acid chain lengths may be more cytotoxic to leukemia cells.7-12,21 Dany et al21 recently showed that elevations of C18:0 ceramide can lead to lethal mitophagy in AML. Furthermore, ceramide accumulation may counterbalance S1P-mediated Mcl-1 expression, which is a pathway that can be targeted in certain cases of AML.22,23 Alternatively, sphingosine buildup, and subsequent lysosomal instability, may be responsible for the therapeutic efficacy observed in this study. Intriguingly, AML progenitor cells have been shown to be sensitive to lysosomal disruption.24 Finally, we recently showed that sphingosine-based molecules could disrupt endosomal-lysosomal membrane trafficking leading to a disruption in lysosomal biogenesis and autophagy.25 Overall, this study demonstrated that nanoliposomal delivery strategies for ceramide and vinblastine in combination that target sphingolipid metabolism have the potential to robustly treat the heterogeneous nature of AML.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Don Wojchowski of the University of New Hampshire for his review of the manuscript and helpful recommendations. In addition, the authors would like to acknowledge the Drug Discovery, Development, and Delivery core laboratory at the Penn State College of Medicine for helping to prepare Lip-C6 for this study.
This work was funded in part by grants from the National Institutes of Health, National Cancer Institute (P01 CA171983 [M.K., T.P.L., and H.-G.W.] and K22 CA190674 [B.M.B.]), a Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA0087480) (R.L.L.), a National Institute of General Medical Sciences, University of New Hampshire COBRE Pilot Project Grant (P20 GM113131) (B.M.B.), the Bess Family Charitable Fund (T.P.L.), a generous anonymous donor (T.P.L.), the Penn State University Kiesendahl Family Endowed Leukemia Research Fund and the Kenneth Noel Memorial Fund (D.F.C.), and Pennsylvania Tobacco Settlement funds (M.K. and T.P.L.).
Authorship
Contributions: B.M.B. designed and supervised the study, supervised, performed, and analyzed data generated from cellular and animal experiments, analyzed all sphingolipidomic data, performed statistical analysis, generated figures, and wrote the manuscript; W.W. and P.T.T. performed experiments using transgenic mouse models and C1498-engrafted mouse models, conducted colony-forming assays using transgenic murine models, helped analyze respective data, and generated figures; T.E.F. generated raw sphingolipidomic data using LC-MS3; C.A., N.R.K., and V.G.D. performed cellular and NSG mouse experiments, helped analyze respective data, and generated respective figures; D.B.N. performed histopathologic work after the animal experiments; R.M.O. performed cellular experiments; T.J.B. performed cellular experiments and revised the manuscript; E.C.S., A.L.C., and V.P. performed experiments using transgenic mouse models; S.-F.T. performed cellular experiments and extractions for lipidomics; S.S.S. developed and prepared nanoliposomes; S.T.S. helped develop anionic nanoliposomes; T.G.D. developed and prepared nanoliposomes and performed cellular lipidomic experiments; J.Z. and J.L. analyzed data and performed statistical analyses; A.D.V. and R.L.L. performed mutational profiling and advised the study; H.-G.W. provided modified MV4-11 cells and advised NSG animal experiments; D.J.F. and T.P.L. advised and revised the manuscript; A.S. designed, supervised, and performed cellular and NSG animal experiments, analyzed respective data, and helped generate respective figures; M.K. supervised the development of nanoliposomes, designed and supervised the study, and revised the manuscript; and D.F.C. designed and supervised the study, acquired patient materials, and revised the manuscript.
Conflict-of-interest disclosure: Several patents have been issued and licensed from the Penn State Research Foundation to Keystone Nano, Inc. (State College, PA) for proprietary nanoscale formulations that encapsulate therapeutic bioactive lipids, such as the ceramide nanoliposome. M.K. is the chief medical officer and cofounder of Keystone Nano, Inc. T.P.L. is on the scientific advisory board and has stock options for both Keystone Nano, Inc. and Bioniz Therapeutics. R.L.L. is on the supervisory board of Qiagen; is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis, which each include an equity interest; receives research support from and has consulted for Celgene and Roche; has received research support from Prelude Therapeutics; has consulted for Incyte, Novartis, Astellas Pharma, Morphosys, and Janssen Pharmaceutica; and has received honoraria from Eli Lilly and Amgen for invited lectures and from Gilead Sciences for grant reviews. D.F.C. has received research support for clinical studies conducted by Daiichi Sankyo, Ambit Biosciences, Astellas Pharma, Novartis, Incyte, Cyclacel Pharmaceuticals, Celgene, MedImmune, Merck, and Gilead Sciences. The remaining authors declare no competing financial interests.
Correspondence: Brian M. Barth, Department of Molecular, Cellular and Biomedical Sciences, University of New Hampshire, 46 College Rd, Durham, NH 03824; e-mail: brian.barth@unh.edu; and David F. Claxton, Penn State Hershey Cancer Institute, Division of Hematology and Oncology, Department of Medicine, Pennsylvania State University College of Medicine, 500 University Dr, Hershey, PA 17033; e-mail: dclaxton@pennstatehealth.psu.edu.

