

Key Points
CH may be associated with broader ill health (worse performance status, increased and potentially novel comorbidities).
Serum interleukin-6 is elevated in people with CH and genetic subtypes, providing a view of the human systemic inflammatory landscape of CH.

Introduction
Clonal hematopoiesis (CH) of indeterminate potential (CHIP)/age-related CH is a new phenomenon of aging that affects a large proportion (>10%) of older adults. CH arises upon the expansion of a somatically mutated hematopoietic stem or progenitor cell in an otherwise hematological malignancy-free person.1,2 Pathologic somatic mutations repeatedly fall into hematological cancer-associated genes, particularly the cytosine methylation regulators DNMT3A and TET2.3-5 The current consensus for CHIP is a variant allele frequency (VAF) ≥0.02, corresponding to at least 4% affected peripheral cells.
CH is associated with some diseases of aging; groups have described increased risk of subsequent hematological cancer,3,4 atherosclerotic cardiovascular disease and stroke,3,4 and all-cause mortality.3,6 Self-reported asthma and chronic obstructive pulmonary disease were also associated with TET2-based CH,7 and whole-genome sequencing–based detection of CH was associated with chronic pulmonary disease.8 However, a comprehensive landscape of potential associations between CH and other comorbidities of aging has not yet been solidified. Furthermore, animal and in vitro models of CH suggest that inflammatory processes involving cytokines like interleukin-6 (IL-6) link the expanding mutant clone to observable disease.6,9-13 However, the role of inflammation in human CH pathogenesis remains largely unexplored.
This analytical cross-sectional study was designed before the known associations between CHIP and aging and selected clinical outcomes.3,4 Our study aims were to
Screen older adults who reside at a retirement/nursing home or attend outpatient family practice clinics for CH prevalence, identifying the proportions with unexplained anemia or abnormal laboratory parameters suggestive of an underlying stem cell disorder.14,15
Identify the proportions of patients with CH and coincident or preexisting comorbid diseases, and elevated peripheral blood cytokines.
We hypothesized that the intersection of CH and associated ill health is broader than initially described. Moreover, the preclinical connection between CH and inflammation and the contributions of inflammation to aging and hematopoietic compartment regulation16 suggest that people with CH have an aberrant systemic inflammatory milieu compared with those without CH.
Methods
Cross-sectional study participants and comorbidities
Three hundred fifty-nine older adults ≥65 years of age were recruited at Baycrest Health Sciences and the Family Practice Unit at Sunnybrook Health Sciences Centres (Toronto, ON, Canada) in 2014 to 2017. Exclusion criteria included history of established myeloid cancer, other cancer in the past 2 years, prior chemotherapy, bleeding or surgery in the past 3 months, acute infection, inflammatory condition within the last month, and being confined to a bed. All participants gave informed consent, and the study was approved by ethics boards at Baycrest Hospital, Sunnybrook Hospital, and Queen’s University.
CH determination, cytokine, and blood laboratory quantification
Peripheral blood DNA was isolated and sequenced using a 48-gene panel (see supplemental Methods and supplemental Table 1 for gene list) on the Ion Proton (Thermo Fisher Scientific). Variants were excluded if they did not meet criteria for identification of candidate driver mutations.19 A blood serum sample subset (N = 297) was run on a 42-plex cytokine array (Eve Technologies) to quantify cytokine/chemokine levels. Laboratory blood tests were performed at Sunnybrook.
Statistics
The study was originally designed to detect odds ratio (OR) = 4 for unexplained anemia with CH vs no-CH (see supplemental Methods for details).
Post hoc analysis of CH status identified relationships between genetic variants of CH (VAF >0.1 [CHVAF>0.1] vs VAF ≤0.1/no mutation, >1 mutation [CHmut>1] vs 1/no mutation, TET2 mutation [CHTET2] vs no mutation, and DNMT3A mutation [CHDNMT3A] vs no mutation) and comorbidities, and inflammatory cytokines. Binary logistic regressions determined prevalence ORs (as preferred for chronic disease/factors) and were adjusted for age if significantly related to the categorical groups (see supplemental Table 2). Serum cytokine levels were min-max normalized (0-100) to control for batch effects. The Benjamini and Hochberg’s adaptive linear step-up method and the Bonferroni adjustment were applied,20 and adjusted P < .05 was considered statistically significant. Analyses were performed using SASS or SPSS.
Results and discussion
We called 102 somatic variants (listed in supplemental Table 3, characteristics summarized in supplemental Figure 1) in 83 subjects (23.1%) from the 359-person cohort (Figure 1A-B). Mutations in TET2 and DNMT3A were present in 75.9% of persons with CH (Figure 1C).
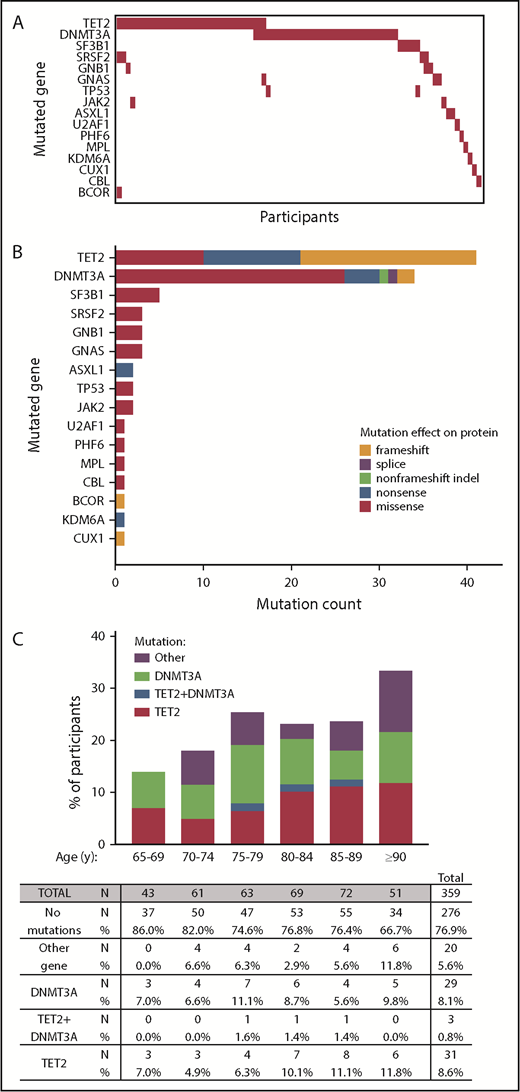
Detection of CH using 48-myeloid cancer gene panel. (A) Mutation profiles by participants. (B) The types of mutations and their frequencies in CH-associated genes (orange = frameshift; purple = splice site; green = non–frameshift indel; blue = nonsense; red = missense). (C) Percentage of participants with at least 1 mutation in TET2, in both TET2+DNMT3A, in DNMT3A, or only in another gene.
Detection of CH using 48-myeloid cancer gene panel. (A) Mutation profiles by participants. (B) The types of mutations and their frequencies in CH-associated genes (orange = frameshift; purple = splice site; green = non–frameshift indel; blue = nonsense; red = missense). (C) Percentage of participants with at least 1 mutation in TET2, in both TET2+DNMT3A, in DNMT3A, or only in another gene.
Blood laboratory values for the cohort (raw values in supplemental Table 4) did not differ between the CH vs no mutations group (ORs in supplemental Table 5). Fifty-two (14.4%) participants had unexplained anemia (as defined in supplemental Methods). Although this was more common in subjects with CH (61.5%) vs those without CH (52.9%), the result was not significant (Fisher’s exact test, P = .49). In examining CH subtypes, CHmut>1 was associated with lower white blood cell and lymphocyte counts, and CHVAF>0.1 and CHTET2 were associated with higher thyroid stimulating hormone levels (supplemental Table 5). These associations may have arisen from multiple testing and are strictly exploratory. Future studies may investigate whether such potential associations indicate early stages of bone marrow dysfunction in those with greater mutational burden and possible thyroid dysfunction (related to thyroiditis and CH monocytic cells, for example).
Broadly assessing the health of people with CH revealed higher ECOG performance status scores (poorer functioning) for those with CH (OR = 2.4, P < .01 for CHVAF>0.1, OR = 2.0, P = .03 for CHTET2; Figure 2A). The CCI (a higher score gives higher predicted odds of 10-year mortality) did not differ between those with or without CH, although patients with CHDNMT3A had a higher number of comorbidities (OR = 1.4, P = .01). Specific comorbidities, including cardiovascular disease, did not associate with any CH (supplemental Table 6). However, CHmut>1 and CHDNMT3A were associated with chronic pulmonary disease; CHmut>1 (and borderline CHVAF>0.1 and CHDNMT3A) was associated with diabetes with end organ damage, and CHDNMT3A was associated with gastroesophageal reflux disease (GERD). These are in accordance with previous comorbid associations reported in humans,3,4,6-8,21 with the novel addition of GERD. However, future studies should test if this GERD association is the spurious result from multiple testing or, for example, related to CH inflammatory responses or parallel mutations in the esophageal epithelium.

ECOG performance status score, CCI, comorbidity count, and cytokine associations with CH. (A) Univariate logistic regression ORs with 95% confidence intervals (CIs) and Benjamini-Hochberg false discovery rate (FDR)–adjusted P values or Bonferroni-adjusted P values, after necessary age adjustment for CH N = 83 (vs no CH N = 276), and CH subtypes (CHVAF>0.1, N = 36 vs 323; CHTET2, N = 34 vs 325; CHDNMT3A, N = 32 vs 327; CHmut>1, N = 11 vs 348). ECOG performance status score (where a higher score means poorer functioning); CCI (where a higher score means lower predicted 10-year survival); Any comorbidity count, among myocardial infarction, angina/coronary artery bypass graft, congestive heart failure, peripheral vascular disease, cerebrovascular disease, valvular heart disease, arrhythmia, deep vein thrombosis/pulmonary embolism, peptic ulcer disease, GERD, diabetes with/without end organ damage, renal disease, liver disease, chronic pulmonary disease (symptomatic/asymptomatic), rheumatic disease, arthritis. (B) IL-6, IL-8, and TNF-α cytokine levels quantified by multiplex cytokine array on blood serum, min-max normalized (0-100), and plotted with medians ± interquartile range by CH status. P values from Mann-Whitney U tests.
ECOG performance status score, CCI, comorbidity count, and cytokine associations with CH. (A) Univariate logistic regression ORs with 95% confidence intervals (CIs) and Benjamini-Hochberg false discovery rate (FDR)–adjusted P values or Bonferroni-adjusted P values, after necessary age adjustment for CH N = 83 (vs no CH N = 276), and CH subtypes (CHVAF>0.1, N = 36 vs 323; CHTET2, N = 34 vs 325; CHDNMT3A, N = 32 vs 327; CHmut>1, N = 11 vs 348). ECOG performance status score (where a higher score means poorer functioning); CCI (where a higher score means lower predicted 10-year survival); Any comorbidity count, among myocardial infarction, angina/coronary artery bypass graft, congestive heart failure, peripheral vascular disease, cerebrovascular disease, valvular heart disease, arrhythmia, deep vein thrombosis/pulmonary embolism, peptic ulcer disease, GERD, diabetes with/without end organ damage, renal disease, liver disease, chronic pulmonary disease (symptomatic/asymptomatic), rheumatic disease, arthritis. (B) IL-6, IL-8, and TNF-α cytokine levels quantified by multiplex cytokine array on blood serum, min-max normalized (0-100), and plotted with medians ± interquartile range by CH status. P values from Mann-Whitney U tests.
In investigating the inflammatory backdrop of CH, no serum cytokine level was increased in the overall CH group by stringent Bonferroni-adjusted univariate analysis, although higher serum levels of IL-6 (pleiotropic proinflammatory driver22 ), MCP1 (CCL2) (monocyte chemoattractant protein), and tumor necrosis factor-α (TNF-α) were associated with CH with FDR-adjusted P < .1 (supplemental Table 7). Notably, normalized IL-6 values (nonadjusted for age) were about twofold higher in CH and nearly all CH subtypes compared with people without mutations (Figure 2B); raw mean and median serum IL-6 concentrations in CH were 12.9 pg/mL and 2.3 pg/mL vs 3.8 and 1.5 pg/mL without CH (not shown). This trend aligns with preclinical studies where proinflammatory cytokines, including IL-6, were elevated in mouse Dnmt3a-deficient13 or Tet2-deficient9,10,13 macrophages and human TET2mut macrophages.11 IL-8 and TNF-α are also graphed (Figure 2B) from the cytokine list that had associations with CH subtypes. Of note, Jaiswal et al6 originally examined plasma IL-8 levels among controls in the PROMIS study and found the 12 participants with TET2-mutant CHIP had significantly higher IL-8 levels than those without TET2 mutations. This is the only other study to show differential cytokine levels in people with CH. The lack of information about temporality (ie, whether higher cytokine levels or comorbidities preceded, were coincident with, or followed CH) is a concern in our analytical cross-sectional study. Although this antecedent-consequent bias weakens the reported relationships, our study is hypothesis stimulating about CH mutation vs comorbidity causation.
Although our published prevalence of ASXL1 mutations (∼32%) in MDS and CMML using the same technology23 is in keeping with the literature,24,25 our DNMT3A:TET2:ASXL1 variant ratio in CH differs from studies using Illumina next-generation sequencing technology.3-5 This is potentially related to our broader TET2 exon coverage, but likely decreased sensitivity for, and underrepresentation of, mutations in the CG-rich ASXL1 (missing frameshifts), similar to another Ion Torrent platform study.7 Furthermore, given the association of CHIP with mortality and our specific exclusion of subjects with previous or concurrent hematologic cancers, our cross-sectional study may have reduced mutation prevalence. These limitations may dilute our associations.
The lack of the expected association with atherosclerotic cardiovascular disease (eg, myocardial infarct) may be due to a lack of power to detect it. We further acknowledge that it may relate to survival bias in this cross-sectional study (prestudy cardiovascular events/deaths in individuals with CH may have preempted their enrollment and DNA sample collection, removing an association between CH and cardiovascular disease in our study). Attenuating effects of ubiquitously used treatments in the last decade, such as statins, may also weaken the expected association.
In summary, by examining CH and subtypes thereof, we show novel associations with poor overall functioning (poorer ECOG scores for CH, and higher comorbidity counts for CHDNMT3A). Furthermore, we show that CH subtypes associate with shared and unique comorbidities and suggest potentially new associations for further investigation (eg, thyroid stimulating hormone, GERD). Last, we report consistently higher serum IL-6 levels in CH, providing the first view into the systemic inflammatory landscape of CH in humans by profiling several cytokines of relevance.
Presented in part at the 59th annual meeting of the American Society for Hematology, Atlanta, GA, 10 December 2017.
For original data, please contact rena.buckstein@sunnybrook.ca and rauhm@queensu.ca.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank James Kennedy, Steven Chan, Harriet Feilotter, Mark Ormiston, Peter Greer, and David Lillicrap for helpful discussions and the contributions of anonymous reviewers at the Blood journal for their time and helpful suggestions.
This study was supported by a Canadian Cancer Society Research Institute Innovation grant (702518) and funds from the Ontario Institute for Cancer Research and the Southeastern Ontario Academic Medical Organization. This study was supported by a Vanier Scholarship (E.K.C.).
Authorship
Contribution: E.K.C. performed DNA library generation, genetic analyses, validations, some statistical analyses, and wrote the manuscript; L.Z. performed the statistical analyses; M.J.R. and C.K.F. performed genetic analyses; T.I., G.R., S.Y., J.F., and B.M. performed participant recruitment and oversaw clinical data collection; M.J. performed Sunnybrook MDS scoring; B.S., D.J., E.B., J.H., and J.B. assisted with DNA library processing; A.J.M.M. and X.L. performed Ion Proton sequencing; and R.B. and M.J.R. conceived, designed, and implemented the study, received operating grant funding, and contributed to the writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rena Buckstein, Odette Cancer Centre, Sunnybrook Health Sciences Centre, 2075 Bayview Ave, Toronto, ON M4N 3M5, Canada; e-mail: rena.buckstein@sunnybrook.ca; and Michael Rauh, Department of Pathology and Molecular Medicine, Queen's University, 88 Stuart St, Kingston, ON K7L 3N6, Canada; e-mail: rauhm@queensu.ca.

