

Key Points
Isogenic heterozygous GATA2 KO iPSC showed decreased differentiation of hematopoietic cells compared with wild-type controls.
Cell lineages profoundly affected in GATA2-deficient patients were preserved during differentiation from GATA2-deficient iPSC.

Abstract
GATA2 deficiency is an inherited or sporadic genetic disorder characterized by distinct cellular deficiency, bone marrow failure, various infections, lymphedema, pulmonary alveolar proteinosis, and predisposition to myeloid malignancies resulting from heterozygous loss-of-function mutations in the GATA2 gene. How heterozygous GATA2 mutations affect human hematopoietic development or cause characteristic cellular deficiency and eventual hypoplastic myelodysplastic syndrome or leukemia is not fully understood. We used induced pluripotent stem cells (iPSCs) to study hematopoietic development in the setting of GATA2 deficiency. We performed hematopoietic differentiation using iPSC derived from patients with GATA2 deficiency and examined their ability to commit to mesoderm, hemogenic endothelial precursors (HEPs), hematopoietic stem progenitor cells, and natural killer (NK) cells. Patient-derived iPSC, either derived from fibroblasts/marrow stromal cells or peripheral blood mononuclear cells, did not show significant defects in committing to mesoderm, HEP, hematopoietic stem progenitor, or NK cells. However, HEP derived from GATA2-mutant iPSC showed impaired maturation toward hematopoietic lineages. Hematopoietic differentiation was nearly abolished from homozygous GATA2 knockout (KO) iPSC lines and markedly reduced in heterozygous KO lines compared with isogenic controls. On the other hand, correction of the mutated GATA2 allele in patient-specific iPSC did not alter hematopoietic development consistently in our model. GATA2 deficiency usually manifests within the first decade of life. Newborn and infant hematopoiesis appears to be grossly intact; therefore, our iPSC model indeed may resemble the disease phenotype, suggesting that other genetic, epigenetic, or environmental factors may contribute to bone marrow failure in these patients following birth. However, heterogeneity of PSC-based models and limitations of in vitro differentiation protocol may limit the possibility to detect subtle cellular phenotypes.
Introduction
GATA2 deficiency is a rare, inherited, or sporadic genetic disorder characterized by variable onset of a pleomorphic constellation of immune, hematologic, and lymphatic abnormalities linked to heterozygous mutations in the GATA2 gene.1-6 Patients develop monocytopenia, B and natural killer (NK) lymphopenia, and dendritic cell deficiency as disease progresses, usually by late childhood/adolescence or young adulthood, leading to vulnerabilities to various infections, particularly to nontuberculous mycobacteria and human papillomavirus.5,6 Patients with GATA2 deficiency also frequently progress to bone marrow failure, myelodysplastic syndrome (MDS), and/or acute myelogenous leukemia, and may present with lymphedema or pulmonary alveolar proteinosis.7-9 A large fraction of children with MDS have been documented as having GATA2 deficiency.10,11 The incomplete penetrance and clinical heterogeneity in GATA2 deficiency is puzzling, as are the mechanisms by which stereotypical loss of circulating monocytes and dendritic, B, and/or NK cells occur.5 Acquisition of additional genomic aberrations, such as somatic mutations or chromosomal rearrangements, have been linked to disease progression in many patients with GATA2 deficiency.10,12-14
GATA proteins are transcription factors with central roles in early embryonic development and lineage specification. They exert their function by binding to the 6-nucleotide GATA motif.15 Whereas GATA1 is important in erythroid and megakaryocytic specification of hematopoietic stem cells (HSCs), GATA2 is a master regulator of hematopoiesis, implicated in the initial generation and maintenance of HSC, primarily studied in murine knockout (KO) models.16-20 In patients with GATA2 deficiency syndromes, predicted deleterious heterozygous point mutations, small insertions and deletions have been reported throughout the GATA2 gene in both exons and introns.5 The second zinc finger (ZF) DNA-binding domain appears to be the most frequently mutated region leading to pathology, presumably by affecting the capacity of the ZF to bind to target sequences.5,21 Haploinsufficiency is likely the reason for the majority of GATA2 deficiency phenotypes, but potential dominant negative effects have been also reported for some mutant GATA2 proteins.21-23 There is no clear link between specific GATA2 mutations and hematologic phenotype.5
Rodent models only partly recapitulate the human phenotype.1,3,24,25 Gata2-homozygous KO in mice is embryonically lethal resulting from a lack of hematopoietic development.24 Although the number of HSC is reduced in the aorta-gonad-mesonephros region in Gata2-heterozygous KO mice, adult animals do not display significant hematopoietic abnormalities and do not develop specific loss of monocytes, B cells, or NK cells.25 However, serial repopulation assays in mice indicated decreased engraftment potential of heterozygous KO HSC.25 Studies on primary hematopoietic stem and progenitor cells (HSPCs) from patients with GATA2 deficiency are ideal, but very challenging because of generally marked marrow hypocellularity, at least before the development of overt MDS/acute myelogenous leukemia.9
Induced pluripotent stem cell (iPSC) disease models have been widely used to overcome this lack of primary patient HSPC in inherited bone marrow failure syndromes.26 We hypothesized that human pluripotent stem cells, particularly patient-specific iPSC, could be used to study potential developmental defects in GATA2 deficiency. We derived iPSC from patients with GATA2 deficiency at different disease stages and quantitatively and qualitatively evaluated their hematopoietic developmental potential. Furthermore, we generated isogenic GATA2 KO lines from control iPSC and corrected the GATA2 mutations in patient-specific iPSC to investigate the phenotype independent of potential individual patient genetic or epigenetic confounding factors. Surprisingly, our iPSC hematopoietic differentiation model revealed limited differences between GATA2-deficient and normal control or corrected iPSC, implying that the role of GATA2 in embryonic hematopoietic pathways may be distinct from those in adult HSPC, and/or that additional cell intrinsic and extrinsic processes contribute to the phenotype associated with heterozygous GATA2 deficiency.
Methods
Study approval
All patients and healthy volunteers signed informed consent in accordance with the Declaration of Helsinki for institutional review board–approved protocols (04-H-0012, 07-H-0113, 10-H-0126, 01-I-0202, or 13-I-0157) at the National Institutes of Health.
Mesodermal and HSPC differentiation of iPSC
Mesodermal and hematopoietic differentiation of iPSC was performed under feeder-free, defined media conditions. At day 0, iPSCs were harvested with ACCUTASE (Millipore, Burlington, MA) and counted with Vi-Cell XR (Beckman Coulter, Indianapolis, IN). A total of 500 000 cells were resuspended in 5 mL of mesodermal induction media, consisting of vascular endothelial growth factor 30 ng/mL (R&D Systems, Minneapolis, MN), BMP4 30 ng/mL (R&D Systems), stem cell factor (SCF) 40 ng/mL (Miltenyi Biotec, Bergisch Gladbach, Germany), Activin A 50 ng/mL (R&D Systems) in STEMdiff APEL media (Stem Cell Technologies), and 50 μL per well was aliquoted onto a 96-well ultra-low attachment plate (Corning, Tewksbury, MA) using a multichannel pipette. A total of 10 mM Y27632 was added to the media at day 0. The plate was spun at 78g for 3 minutes and then moved to a tissue culture incubator at 37°C, 21% O2, and left undisturbed for 48 hours to form spin embryoid bodies (spin EBs). The mesodermal induction media were continued until day 4, when samples were taken for flow cytometry analysis. The remaining EB were cultured in hematopoietic specification media, consisting of SCF 300 ng/mL, FLT3L 300 ng/mL (Miltenyi Biotec), interleukin-3 (IL-3) 10 ng/mL (R&D Systems), IL-6 10 ng/mL (R&D Systems), G-CSF 50 ng/mL (Amgen, Thousand Oaks, CA), and BMP4 25 ng/mL in STEMdiff APEL media. The media were changed every 3 days until day 16. Subsequently, after single-cell dissociation using Trypsin-EDTA (0.25%) (Gibco, Gaithersburg, MD), CD34+CD45+ cells were enumerated and sorted by fluorescence-activated cell sorting (FACS).
For colony-forming unit (CFU) assays, 3000 sorted CD34+CD45+ cells at day 16 were mixed with 3 mL MethoCult H4435 Enriched (Stem Cell Technologies). One-third volume of MethoCult was dispensed into 35-mm dishes in duplicate. Colonies were counted 10 to 14 days later.
HEP OP9 coculture assay
Spin EB were made and mesodermal induction and hematopoietic specification was conducted in feeder-free defined conditions until day 9 as described previously. At day 9, VE-Cadherin+CD73−CD43−CD235a− hemogenic endothelial precursors (HEP) were isolated by FACS as described previously.27 Two thousand HEP were seeded onto OP9 stromal cells (ATCC; catalog no. CRL-2749) in a 12-well plate in 1 mL of α minimum essential media (Invitrogen) supplemented with 20% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), SCF 50 ng/mL, TPO 50 ng/mL (Miltenyi Biotec), IL-3 10 ng/mL, and IL-6 20 ng/mL and cultured for 5 days before analysis of hematopoietic cells by flow cytometry. HSPC were separated from OP9 stromal cells via gating of CD29−CD34+ cells.
Single cell RNA-seq
The 10X Genomics Chromium microfluidics platform (10X Genomics, Pleasanton, CA, USA) was used for droplet-based single cell partitioning. Single-cell RNA-sequencing (RNA-seq) libraries were prepared using version 2 of GemCode Single-Cell 3′ Gel Bead and Library Kit per the manufacturer’s protocol.28 The libraries were not multiplexed for sequencing. See supplemental Methods for details of the analysis pipeline.
Statistical analysis
Data are shown as mean ± the standard error of the mean unless specified otherwise. Statistical analysis was performed by Student t test or analysis of variance (ANOVA).
Results
Hematopoietic development of patient-derived GATA2 mutant compared with control iPSC
iPSC from fibroblasts (FBs), marrow stromal cells (MSCs), or peripheral blood mononuclear cells (PBMCs) from 4 subjects with GATA2 deficiency syndrome phenotypes and heterozygous mutations in either ZF domain locations R337X or R361H or within intron 5 (Int5) were derived (Table 1; supplemental Table 1). Stage-specific cellular phenotypes were analyzed at different time points during in vitro hematopoietic differentiation (Figure 1A; supplemental Figure 1). We compared patient FB/MSC-iPSC with control FB/MSC-iPSC, and patient PBMC-iPSC with control PBMC-iPSC to mitigate the potential effect of secondary genetic or epigenetic abnormalities in patient PBMC.
Clinical features of study subjects
| Subject no. | GATA2 mutation | Sex | Overall clinical manifestations | Age, y | BM diagnosis | BM cytogenetics | NK (126-729/μL) | B (61-321/μL) | Monocytes (0.24-0.86 K/μL) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | R361H | M | MDS, warts, frequent sinus/lung infections | 31 | Hypoplastic MDS | 46, XY [20] | 2 | 42 | 0.01 |
| 2 | R337X | F | MDS, lymphedema | 49 | Hypoplastic marrow | 46, XX [20] | 2 | 1 | 0.02 |
| 3 | Intron 5 | F | MDS, warts | 29 | Hypoplastic MDS | 46, XX, +1, der(1;14)(q10;p10) [5]/47, idem, +21 [5]/46, XX [10] | 10 | 7 | 0 |
| 4 | Intron 5 | M | Asymptomatic carrier | 60 | Normal BM | 46, XY [20] | 230 | 64 | 0.8 |
| Subject no. | GATA2 mutation | Sex | Overall clinical manifestations | Age, y | BM diagnosis | BM cytogenetics | NK (126-729/μL) | B (61-321/μL) | Monocytes (0.24-0.86 K/μL) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | R361H | M | MDS, warts, frequent sinus/lung infections | 31 | Hypoplastic MDS | 46, XY [20] | 2 | 42 | 0.01 |
| 2 | R337X | F | MDS, lymphedema | 49 | Hypoplastic marrow | 46, XX [20] | 2 | 1 | 0.02 |
| 3 | Intron 5 | F | MDS, warts | 29 | Hypoplastic MDS | 46, XX, +1, der(1;14)(q10;p10) [5]/47, idem, +21 [5]/46, XX [10] | 10 | 7 | 0 |
| 4 | Intron 5 | M | Asymptomatic carrier | 60 | Normal BM | 46, XY [20] | 230 | 64 | 0.8 |
Age, BM, diagnosis and cytogenetic results, peripheral blood NK, and B and monocyte counts at the time of PBMC collection for iPSC generation, except for subject 2, in which findings at the time of skin biopsy for fibroblast generation were listed. Reference ranges for counts are in parentheses.
BM, bone marrow; F, female; M, male.
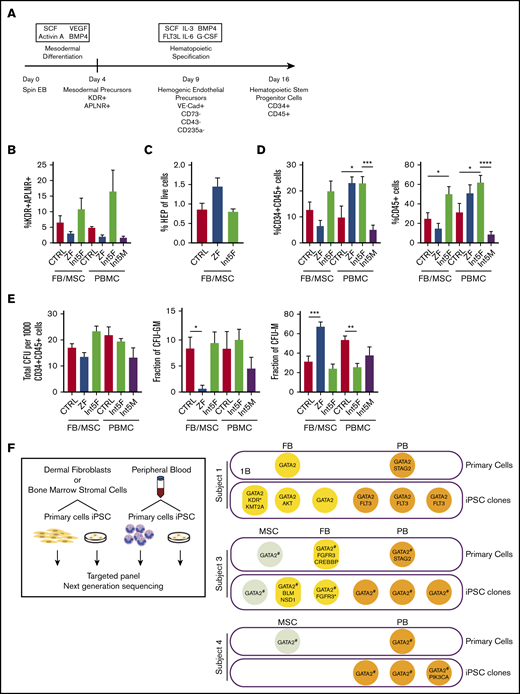
Hematopoietic differentiation of patient iPSC is grossly intact. (A) Hematopoietic differentiation scheme. Spin EB derived from iPSC underwent a multistep hematopoietic differentiation process (top). Mesodermal precursors were evaluated at day 4. HEP were assessed at day 9. At day 16 of hematopoietic differentiation, CD34+CD45+ cells were sorted for CFU assay and gene expression analysis. FB/MSC-ZF includes iPSC from subject 1 (GATA2 R361H) and subject 2 (GATA2 R337X). FB/MSC-Int5 includes iPSC from subject 3 (intron 5 proband). Patient PBMC-derived iPSC efficiency was compared with healthy volunteer PBMC-derived iPSC controls. PBMC-ZF includes 3 clones from subject 1 (R361H). PBMC-Int5F includes 3 clones from subject 3 (proband). PBMC-Int5M includes 3 clones from subject 4 (asymptomatic carrier). A minimum of 2 independent experiments were performed unless otherwise specified. (B) Percentage of mesodermal precursor cells on day 4 of differentiation. (FB/MSC-control: n = 10; ZF, n = 7; Int5, n = 3; PBMC-control: n = 4; ZF, n = 3; Int5, n = 6; Int5M, n = 3). One ZF2 clone (R337X-FB-1) and 3 Int5 clones among the FB/MSC-iPSC group and 3 R361H clones and 3 Int5M clones among the PBMC-iPSC group were tested in 1 experiment. (C) Fraction of HEP at day 9 derived from various control, ZF, or intron 5 GATA2-mutant iPSC (control: n = 10; ZF, n = 6; Int5, n = 3). Three Int5 clones were tested in 1 experiment. (D) Fraction of CD34+CD45+ cells and CD45+ cells at day 16 of hematopoietic differentiation (FB/MSC-control: n = 14; ZF, n = 11; Int5, n = 6; PBMC-control: n = 6; ZF, n = 3; Int5, n = 9; Int5M, n = 6). One ZF clone (R337X-FB-1) among the FB/MSC-iPSC group and 3 R361H clones among the PBMC-iPSC group were tested in 1 experiment. (E) Functional properties of iPSC-derived CD34+CD45+ cells as evaluated by CFU assay. The number and type of CFU colonies from 1000 sorted CD34+CD45+ cells were scored. Total number of colonies per 1000 sorted CD34+CD45+ (FB/MSC-control: n = 20; ZF, n = 10; Int5, n = 6; PBMC-control: n = 6, Int5F, n = 12; Int5M, n = 6). (F) Fraction of CFU subtype per total number of colonies, respective of mutation types. P values were determined by 1-way ANOVA followed by Dunnett multiple comparison test. Int5F and Int5M were compared independently by unpaired Student t test. *P < .05, **P < .01, and ***P < .005; ****P < .0001. (F) Targeted myeloid neoplasm panel next-generation sequencing was performed on patient’s primary cells (dermal fibroblasts, bone marrow stromal cells, or peripheral blood) and iPSC clones derived from those primary cells. *Two separate variants (supplemental Table 9). #GATA2 intronic variants were examined by Sanger sequencing because intronic regions were not covered in the targeted next-generation sequencing.
Hematopoietic differentiation of patient iPSC is grossly intact. (A) Hematopoietic differentiation scheme. Spin EB derived from iPSC underwent a multistep hematopoietic differentiation process (top). Mesodermal precursors were evaluated at day 4. HEP were assessed at day 9. At day 16 of hematopoietic differentiation, CD34+CD45+ cells were sorted for CFU assay and gene expression analysis. FB/MSC-ZF includes iPSC from subject 1 (GATA2 R361H) and subject 2 (GATA2 R337X). FB/MSC-Int5 includes iPSC from subject 3 (intron 5 proband). Patient PBMC-derived iPSC efficiency was compared with healthy volunteer PBMC-derived iPSC controls. PBMC-ZF includes 3 clones from subject 1 (R361H). PBMC-Int5F includes 3 clones from subject 3 (proband). PBMC-Int5M includes 3 clones from subject 4 (asymptomatic carrier). A minimum of 2 independent experiments were performed unless otherwise specified. (B) Percentage of mesodermal precursor cells on day 4 of differentiation. (FB/MSC-control: n = 10; ZF, n = 7; Int5, n = 3; PBMC-control: n = 4; ZF, n = 3; Int5, n = 6; Int5M, n = 3). One ZF2 clone (R337X-FB-1) and 3 Int5 clones among the FB/MSC-iPSC group and 3 R361H clones and 3 Int5M clones among the PBMC-iPSC group were tested in 1 experiment. (C) Fraction of HEP at day 9 derived from various control, ZF, or intron 5 GATA2-mutant iPSC (control: n = 10; ZF, n = 6; Int5, n = 3). Three Int5 clones were tested in 1 experiment. (D) Fraction of CD34+CD45+ cells and CD45+ cells at day 16 of hematopoietic differentiation (FB/MSC-control: n = 14; ZF, n = 11; Int5, n = 6; PBMC-control: n = 6; ZF, n = 3; Int5, n = 9; Int5M, n = 6). One ZF clone (R337X-FB-1) among the FB/MSC-iPSC group and 3 R361H clones among the PBMC-iPSC group were tested in 1 experiment. (E) Functional properties of iPSC-derived CD34+CD45+ cells as evaluated by CFU assay. The number and type of CFU colonies from 1000 sorted CD34+CD45+ cells were scored. Total number of colonies per 1000 sorted CD34+CD45+ (FB/MSC-control: n = 20; ZF, n = 10; Int5, n = 6; PBMC-control: n = 6, Int5F, n = 12; Int5M, n = 6). (F) Fraction of CFU subtype per total number of colonies, respective of mutation types. P values were determined by 1-way ANOVA followed by Dunnett multiple comparison test. Int5F and Int5M were compared independently by unpaired Student t test. *P < .05, **P < .01, and ***P < .005; ****P < .0001. (F) Targeted myeloid neoplasm panel next-generation sequencing was performed on patient’s primary cells (dermal fibroblasts, bone marrow stromal cells, or peripheral blood) and iPSC clones derived from those primary cells. *Two separate variants (supplemental Table 9). #GATA2 intronic variants were examined by Sanger sequencing because intronic regions were not covered in the targeted next-generation sequencing.
Mesodermal precursors with hematopoietic potential were defined as CD31−CD43−CD73−KDR+APLNR+ cells, detected at day 4 of spin EB differentiation.27 FB/MSC-derived ZF, Int5, and control iPSC produced similar numbers of mesodermal precursors (Figure 1B; supplemental Figure 2A). Similarly, the fraction of HEP, defined as live VE-Cadherin+CD73−CD43−CD235a− cells at day 9 of differentiation culture, was also not significantly different between the experimental groups (Figure 1C; supplemental Figure 2B). Finally, no differences in the number of CD34+CD45+ cells produced at day 16, considered to represent HSPC, were observed among FB/MSC-ZF, Int5, and control iPSC (Figure 1D; supplemental Figure 1C). On the other hand, when total CD45+ cells were analyzed, Int5 iPSC produced more CD45+ hematopoietic cells compared with control iPSC, but not ZF2 iPSC (Figure 1D; supplemental Figure 1D). PBMC-derived patient iPSC also did not exhibit significant differences in mesodermal induction (Figure 1B; supplemental Figure 2F), but increased CD34+CD45+ commitment was observed in Int5F iPSC (subject 3) compared with control PBMC or Int5M iPSC (subject 4) (Figure 1D; supplemental Figure 2G).
To further investigate the hematopoietic properties of GATA2-deficient patient CD34+CD45+ cells differentiated from iPSC, given the distinct lineage abnormalities observed in these patients, CFU assays were performed on sorted CD34+CD45+ cells. In the CFU assays using either FB/MSC-iPSC or PBMC-iPSC, the total number of colonies generated from 1000 sorted CD34+CD45+ cells was not different between ZF and control or Int5 and control (Figure 1E; supplemental Figure 2E-H). The number of CFU-granulocyte macrophage (CFU-GM) colonies representing committed progenitors was significantly reduced, whereas the frequency of CFU-macrophage (CFU-M), representing colonies of differentiated macrophages/monocytes, was higher in FB/MSC-ZF iPSC compared with control iPSC. However, these differences were not observed in FB/MSC-Int5 or PBMC-Int5 iPSC. Overall, the distribution of colony types did not recapitulate the specific monocyte deficiency observed in the peripheral blood of GATA2 deficiency patients, and there was no consistent reduction in CFU production of any type from overall GATA2-mutant iPSC compared with control iPSC.
To determine whether patients acquired somatic mutations in their HSPC that may have contributed to the differentiation phenotype of iPSC derived from patients’ PBMC, we performed targeted next-generation sequencing of 275 genes that are frequently mutated in myeloid neoplasm on primary cells (FB, MSC, and PB) and iPSC clones derived from those primary cells (Figure 1F; supplemental Tables 9 and 10). Two patients who had symptomatic bone marrow failure acquired STAG2 mutations in their blood, which was not captured in any of the PBMC-iPSC clones derived and analyzed.
GATA2 haploinsufficiency modeled via editing of isogenic iPSC reveals hematopoietic developmental defects
To dissect confounding factors such as clonal heterogeneity of iPSC resulting from different genetic29-31 or epigenetic backgrounds32-34 that might have accounted for the lack of phenotypic variation between control iPSC- and GATA2-deficient iPSC hematopoietic differentiation, we generated isogenic GATA2 heterozygous and homozygous KO iPSC lines starting from normal control iPSC, and corrected mutations in patient-specific iPSC using CRISPR/Cas9 technology to create additional sets of isogenic mutant and wild-type GATA2 iPSC (Figure 2A). The second ZF domain of the GATA2 gene was targeted by CRISPR/Cas9 to model mutations that frequently occur in patients. We successfully generated biallelic frameshift mutations (c.1085_1092del8) and monoallelic frameshift mutation (c.1085_1088del4ins9 or c.1085_1092del8) leading to premature stop codons (supplemental Figure 5A-B). The GATA2 c.1085_1092del8 8-bp deletion observed in our isogenic lines is predicted to have high impact on its function by Ensembl Variant Effect Predictor (supplemental Table 8) and to affect Zn binding and the structure stabilization of the GATA2/DNA complex (Figure 2B; supplemental Methods). Both GATA2+/− and GATA2−/− iPSC clones grew similarly to parental GATA2+/+ iPSC clones in culture, had characteristic iPSC morphology and immunohistochemistry staining, and formed teratomas (supplemental Figures 8 and 9). Homology-directed repair was used to correct GATA2 R361H and R337X patient lines (supplemental Figure 5C), creating additional isogenic and mutant sets of clones.
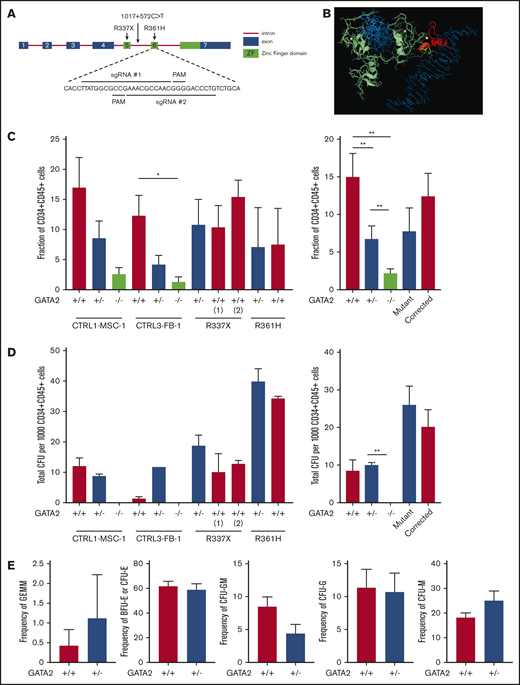
Reduced hematopoietic differentiation potential of GATA2-haploinsufficient isogenic iPSC. (A) Exon 6 of the GATA2 gene was targeted to generate knockout isogenic cell lines. (B) Structure of the wild-type GATA2/DNA complex modeled from the corresponding GATA3 structure: sequence RNA targeted for deletion by CRISPR (yellow); the 38-aa long C terminus sequence (red) is truncated and modified because of a premature stop codon; the resulting 13-aa segment has unknown, albeit predictable, conformation in solution; DNA molecule (blue), Zn ions (white), and unmodified GATA2 sequence (green). (C) Isogenic iPSC lines were differentiated into hematopoietic lineage as described previously. Fraction of CD34+CD45+ cells and the number and type of CFU were examined from independent GATA2 knockout isogenic lines and the 2 corrected patient-specific iPSC lines shown. A minimum of 2 independent experiments was performed. Fraction of CD34+CD45+ cells was grouped per cell line and GATA2 status. (Left: n = 4, 4, 4, 4, 4, 4, 4, 4, 3, 2, and 2, respectively; right: n = 8, 8, 8, 5, and 5, respectively). (D) Total number of colonies from 1000 sorted CD34+CD45+ cells per cell line and GATA2 status. (Left: n = 4, 4, 2, 2, 2, 2, 4, 4, 4, 2, and 2, respectively; right: n = 6, 6, 4, 6, and 6, respectively). (E) Fraction of CFU subtype per total number of colonies, respective of mutation types (GATA2+/+, n = 10; GATA2+/−, n = 10). Paired Student t test was performed to compare the effect of GATA2 gene status with its isogenic cell line. *P < .05; **P < .01. Nonsignificant P values are not shown.
Reduced hematopoietic differentiation potential of GATA2-haploinsufficient isogenic iPSC. (A) Exon 6 of the GATA2 gene was targeted to generate knockout isogenic cell lines. (B) Structure of the wild-type GATA2/DNA complex modeled from the corresponding GATA3 structure: sequence RNA targeted for deletion by CRISPR (yellow); the 38-aa long C terminus sequence (red) is truncated and modified because of a premature stop codon; the resulting 13-aa segment has unknown, albeit predictable, conformation in solution; DNA molecule (blue), Zn ions (white), and unmodified GATA2 sequence (green). (C) Isogenic iPSC lines were differentiated into hematopoietic lineage as described previously. Fraction of CD34+CD45+ cells and the number and type of CFU were examined from independent GATA2 knockout isogenic lines and the 2 corrected patient-specific iPSC lines shown. A minimum of 2 independent experiments was performed. Fraction of CD34+CD45+ cells was grouped per cell line and GATA2 status. (Left: n = 4, 4, 4, 4, 4, 4, 4, 4, 3, 2, and 2, respectively; right: n = 8, 8, 8, 5, and 5, respectively). (D) Total number of colonies from 1000 sorted CD34+CD45+ cells per cell line and GATA2 status. (Left: n = 4, 4, 2, 2, 2, 2, 4, 4, 4, 2, and 2, respectively; right: n = 6, 6, 4, 6, and 6, respectively). (E) Fraction of CFU subtype per total number of colonies, respective of mutation types (GATA2+/+, n = 10; GATA2+/−, n = 10). Paired Student t test was performed to compare the effect of GATA2 gene status with its isogenic cell line. *P < .05; **P < .01. Nonsignificant P values are not shown.
In contrast to the previous experiments using patient-derived iPSC, isogenic heterozygous GATA2 KO lines showed consistent decreases in CD34+CD45+ cells after 16 days of hematopoietic differentiation. Expectedly, hematopoietic differentiation from homozygous GATA2 KO IPSC was almost completely abolished (Figure 2C; supplemental Figure 4). The differentiation potential of the gene-corrected patient iPSC lines was comparable to the parental patient iPSC (Figure 2C). Thus the isogenic pairs of these particular mutations confirmed the prior results comparing patient lines with nonisogenic control lines.
CFU assays did not show a difference in hematopoietic differentiation potential in CD34+CD45+ cells derived from either GATA2+/+ or GATA2+/− iPSC (Figure 2D). Similar to the patient-derived iPSC, GATA2+/− iPSC showed a trend toward a higher frequency of CFU-M and lower frequency of CFU-GM, but these differences were statistically not significant. Overall, colony subtypes were not significantly different between GATA2+/+ and GATA2+/− iPSC (Figure 2E; supplemental Figure 3B,D).
Decreased hematopoietic potential of HEP in patient-derived GATA2-mutant iPSC
GATA2 has been proposed to be critical in endothelial-to-hematopoietic transition at the aorta-gonad-mesonephros region, a major source of definitive HSC during embryogenesis.19,35 Therefore, interrogating the effect of GATA2 haploinsufficiency in generation and functionality of hemogenic endothelium may uncover pathomechanisms of GATA2 deficiency. Choi et al27 established an in vitro OP9 coculture method to identify and characterize HEP. We took advantage of this method and characterized the functionality of HEP from our patient-specific iPSC models. HEP, isolated from day 9 of spin EB hematopoietic differentiation culture, were evaluated for hematopoietic potential by 5 days of in vitro coculture with OP9 stromal cells after sorting (Figure 3A). Hematopoietic phenotypes, assessed by acquisition of the hematopoietic markers CD43 or CD45, showed trend toward decreased hematopoietic potential in samples from GATA2 mutant clones compared with control iPSC, albeit with clonal heterogeneity (Figure 3B; supplemental Figure 2I).
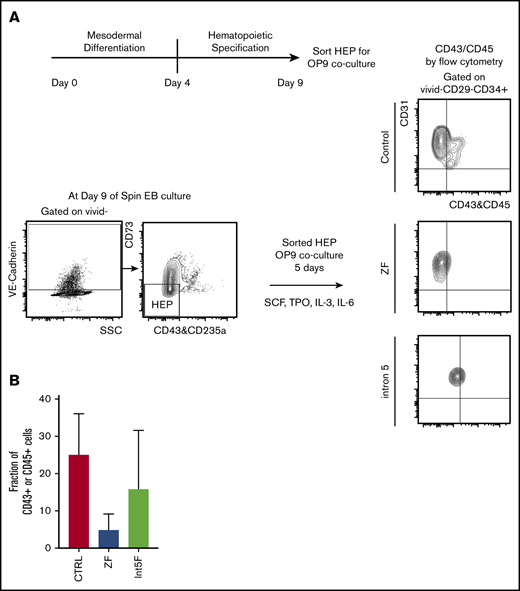
Hematopoietic potential of HEP from patient-specific iPSC may be reduced. (A) Experimental scheme. HEP (VE-Cad+CD73−CD43−CD235a− cells) were isolated by FACS on day 9 and cultured for additional 5 days on OP-9 mouse bone marrow stromal cells with addition of cytokines for 5 days, at which time hematopoietic potential was assessed by measuring CD43+ and CD45+ cells by flow cytometry. (B) Hematopoietic potential (%CD43+ and/or CD45+ cells) of HEP after 5 days of OP-9 coculture is shown. Minimum of 2 independent experiments were performed except for 3 Int5F clones that were differentiated in 1 experiment (control, n = 8; ZF, n = 6; Int5F, n = 3; P = nonsignificant (NS), unpaired Student t test was performed between control and ZF or control and Int5F). NS P values are not shown.
Hematopoietic potential of HEP from patient-specific iPSC may be reduced. (A) Experimental scheme. HEP (VE-Cad+CD73−CD43−CD235a− cells) were isolated by FACS on day 9 and cultured for additional 5 days on OP-9 mouse bone marrow stromal cells with addition of cytokines for 5 days, at which time hematopoietic potential was assessed by measuring CD43+ and CD45+ cells by flow cytometry. (B) Hematopoietic potential (%CD43+ and/or CD45+ cells) of HEP after 5 days of OP-9 coculture is shown. Minimum of 2 independent experiments were performed except for 3 Int5F clones that were differentiated in 1 experiment (control, n = 8; ZF, n = 6; Int5F, n = 3; P = nonsignificant (NS), unpaired Student t test was performed between control and ZF or control and Int5F). NS P values are not shown.
GATA2-deficient iPSC can be differentiated into NK cells
GATA2 deficiency often presents with severe NK lymphopenia, subjecting patients to increased risk of various viral infections and cancers.5 There is marked loss of putative immature CD56bright NK cells, with relative preservation of a subset of CD56dim defined as having an adaptive phenotype, even in patients with frank bone marrow failure.36,37 How GATA2 deficiency affects NK cell development and function remains poorly understood. We hypothesized that GATA2 haploinsufficiency may decrease NK cell differentiation capacity of iPSC-derived HSPC. Similar to the previously mentioned hematopoietic differentiation method, we differentiated iPSC into CD34+CD45+cells for 11 days using the spin EB method. On day 11, EBs were further differentiated toward NK cell lineages, defined here as CD45+CD3−CD56+ cells. Although we observed some heterogeneity among clones, both control and GATA2 patient-specific iPSC produced similar fractions of CD45+CD3−CD56+ cells (Figure 4A; supplemental Figure 2J). Similarly, GATA2 KO isogenic iPSC lines and gene corrected GATA2 patient-specific IPSC produced phenotypical NK cells. Although the fraction of live CD45+CD3−CD56+ cells was higher in GATA2+/− and GATA2−/− KO iPSC lines compared with unmanipulated counterparts, when total cell expansion of the whole culture was taken into consideration, absolute NK cell output was comparable between GATA2+/+ and GATA2+/− iPSCs, whereas NK output from GATA2−/− iPSC was the lowest. GATA2 patient-specific iPSC produced comparable NK cells to corrected isogenic counterparts (Figure 4B). In summary, the loss of the GATA2 gene did not abolish the ability of CD34+CD45+ cells to differentiate into NK cells. Despite GATA2−/− iPSC producing far fewer HSPC, the efficiency of NK cell differentiation from these HSPC was comparable to GATA2+/+ or GATA2+/− iPSC, although the absolute number of NK cells produced per starting iPSC culture was reduced because of the HSPC production deficiency (Figure 4A-B; supplemental Figure 10A).
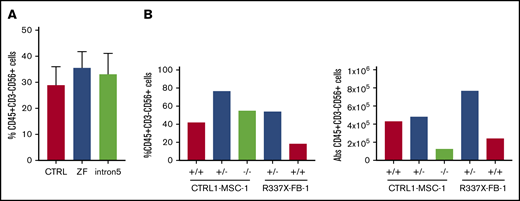
GATA2-deficient HSPCs derived from iPSC can commit to NK lineage differentiation. (A) NK cell output, defined as live CD45+CD3−CD56+ cells, upon 3 weeks of NK-specific differentiation. A minimum of 2 independent experiments were performed (control, n = 6; ZF, n = 8; Int5, n = 6; P = NS by ANOVA). (B) Percent NK output and absolute number of NK cells differentiated from isogenic lines. The experiment was performed once.
GATA2-deficient HSPCs derived from iPSC can commit to NK lineage differentiation. (A) NK cell output, defined as live CD45+CD3−CD56+ cells, upon 3 weeks of NK-specific differentiation. A minimum of 2 independent experiments were performed (control, n = 6; ZF, n = 8; Int5, n = 6; P = NS by ANOVA). (B) Percent NK output and absolute number of NK cells differentiated from isogenic lines. The experiment was performed once.
Single cell RNA-seq results
We sequenced a total of 6,357 sorted single CD34+CD45+ cells differentiated from iPSCs (2734 cells from GATA2 R337X iPSC and 215 cells from its corrected isogenic pair; 1985 cells from a healthy control iPSC line (CTRL1-MSC-1) and 1423 cells from its isogenic GATA2 heterozygous KO iPSC (CTRL1-GATA2+/−). The sequenced number of CD34+CD45+ cells from the corrected clone of GATA2 R337X was low because of low cell input.
We used Monocle to cluster the data and to impute cell types. Single cells from the healthy volunteer grouped into 12 clusters over a range of parameters, whereas those from the GATA2 R337X patient grouped into 11 clusters for equivalent parameters. We computed the cluster-specific differential expression of genes and imputed cell types for each cluster as described in the methods. Supervised cell type classification was unable to resolve T/B/NK lymphocyte progenitors; therefore, we generically labeled these as “lymphocytes.” Figure 5A-B shows the cell type identifications of each cluster. The proportion of cells in each type is depicted in Figure 5G.

Single-cell RNA-seq of sorted CD34+CD45+cells from GATA2 WT vs GATA2+/−iPSC reveal subtle differences in lineage commitment and gene expression profiles of GATA2 target genes. Imputed cell type classification for single-cell RNA-seq data from healthy volunteer pair (WT vs GATA2+/−) (A) and GATA2 R337X pair (patient mutant vs corrected) (B). The gene expression data have been dimensionally reduced via Uniform Manifold Approximation and Projection. (C) Pseudotemporally ordered cells derived from iPSC of healthy volunteer. Cells are colored by cell type. The center of each subplot represents the branch point between the myeloid and lymphoid lineages; myeloid lineages move to the right as they mature and lymphoid lineages move to the left. (D) Pseudotemporal ordering of cells derived from GATA2 WT iPSC are colored in blue; those from GATA2+/− iPSC are colored in red. (E) Pseudotemporally ordered cells derived from iPSC of GATA2 R337X pair. (F) Pseudotemporal ordering of cells derived from a corrected clone of GATA2 R337X iPSC are colored in blue; those from GATA2 R337X iPSC are colored in red. (G) Fraction of each cell type per GATA2 status. (H) Volcano plot of differentially expressed genes depending on GATA2 status. The 2 dotted vertical lines indicate log2-fold change; 2 dotted horizontal lines indicate P < .05. Ba-Prog, basophil progenitor; DC-Prog, dendritic cell progenitor; Eo-Prog, eosinophil progenitor; Er-Pro, erythroid progenitor; Lymph-Prog, lymphoid progenitor; Mk-Er-Prog, megakaryocyte-erythrocyte progenitor; Mk-Prog, megakaryocyte progenitor; Mo-Prog, monocyte progenitor; MPP, multipotent progenitor; Neu-Prog, neutrophil progenitor.
Single-cell RNA-seq of sorted CD34+CD45+cells from GATA2 WT vs GATA2+/−iPSC reveal subtle differences in lineage commitment and gene expression profiles of GATA2 target genes. Imputed cell type classification for single-cell RNA-seq data from healthy volunteer pair (WT vs GATA2+/−) (A) and GATA2 R337X pair (patient mutant vs corrected) (B). The gene expression data have been dimensionally reduced via Uniform Manifold Approximation and Projection. (C) Pseudotemporally ordered cells derived from iPSC of healthy volunteer. Cells are colored by cell type. The center of each subplot represents the branch point between the myeloid and lymphoid lineages; myeloid lineages move to the right as they mature and lymphoid lineages move to the left. (D) Pseudotemporal ordering of cells derived from GATA2 WT iPSC are colored in blue; those from GATA2+/− iPSC are colored in red. (E) Pseudotemporally ordered cells derived from iPSC of GATA2 R337X pair. (F) Pseudotemporal ordering of cells derived from a corrected clone of GATA2 R337X iPSC are colored in blue; those from GATA2 R337X iPSC are colored in red. (G) Fraction of each cell type per GATA2 status. (H) Volcano plot of differentially expressed genes depending on GATA2 status. The 2 dotted vertical lines indicate log2-fold change; 2 dotted horizontal lines indicate P < .05. Ba-Prog, basophil progenitor; DC-Prog, dendritic cell progenitor; Eo-Prog, eosinophil progenitor; Er-Pro, erythroid progenitor; Lymph-Prog, lymphoid progenitor; Mk-Er-Prog, megakaryocyte-erythrocyte progenitor; Mk-Prog, megakaryocyte progenitor; Mo-Prog, monocyte progenitor; MPP, multipotent progenitor; Neu-Prog, neutrophil progenitor.
To determine whether there was any association between GATA2 mutational status and the proportion of cell types, we performed a χ2 goodness-of-fit test. For GATA2+/+ vs GATA2+/− cells derived from the healthy volunteer, we found a χ2 of 125.14 with a P value of 10−5, whereas for the case of GATA2 R337X pair (mutant vs corrected), we found a χ2 of 329.4 with a P value of 10−5, suggesting there are statistically significant differences in the proportion of cell types depending on GATA2 status. Pairwise comparison of proportions38 with Benjamini-Hochberg corrections for multiple comparisons39 revealed a trend toward a statistically significant difference in the proportion of HSPC that was larger in the GATA2+/+ cells; adjusted P values were found to <.001 for the healthy volunteer, but only .29 for the GATA2 R337X patient (supplemental Figure 11).
Next we used Monocle to trace hematopoietic development by ordering the cells in pseudotime. Figure 5C-F depict the differentiation of HSC, located near the center of the curves and bifurcating toward either the myeloid or lymphoid lineage. Overall, we found a greater proportion of cells in the lymphoid branch; for the GATA2 R337X pair, the proportion of cells in the lymphoid branch was larger to a statistically significant degree in the GATA2+/− cells; adjusted P < .001. However, no obvious block in differentiation was observed in either pair.
Finally, we examined the differential expression of genes depending on GATA2 status (Figure 5H). We found that 43 of 102 target genes known to be regulated by GATA2 in the Harmonizome database40 were differentially regulated, with adjusted P < .05; however, the absolute values of the log2-fold changes were <1 in all cases. In addition, we found that genes important in megakaryocyte-erythrocyte progenitor development (STAT1, MEIS1, PLEK, GFI1B, GATA1, HBD, PPBP, and PF1) to be significantly overexpressed, whereas genes important in granulocyte-monocyte progenitor and neutrophil development (CD14, CD36, LYZ, MNDA, MS4A7, and S100A8) were significantly underexpressed in GATA2+/− compared with GATA2+/+ cells.
Discussion
Heterozygous germline mutations of GATA2 have been associated with variety of conditions, now grouped under the diagnostic moniker “GATA2 deficiency syndrome.” Despite the clear link between GATA2 loss-of-function mutations and clinical manifestations, the pathomechanisms linking GATA2 deficiency to heterogeneous and variably penetrant clinical phenotypes are poorly understood. To experimentally investigate the effect of GATA2 deficiency on hematopoiesis in human cells, we used an iPSC-based differentiation model to evaluate HSPC development and differentiation quantitatively and qualitatively.
Our results using patient-specific iPSC lines compared with normal volunteer control iPSC revealed no major developmental defects of hematopoiesis linked to GATA2 patient-specific mutations. Hematopoietic differentiation from 2 independent isogenic knockout lines showed stepwise decrease in GATA2+/− and GATA2−/− compared with isogenic GATA2+/+ iPSC in the numbers of CD34+CD45+ hematopoietic cells produced. However, similar to the results comparing patient iPSC with normal volunteer control iPSC, hematopoietic output from 2 independent patient-derived mutant iPSC vs corrected isogenic lines was similar, suggesting the possibility that the patient-specific GATA2 R337X or R361H mutations may retain enough residual GATA2 function to result in quantitatively normal hematopoietic development as assayed in vitro from embryonic-like iPSC.
However, this hypothesis raises a couple of questions. How much GATA2 is necessary for normal development, and are GATA2 requirements different during postnatal hematopoiesis, a stage potentially poorly modeled by hematopoietic differentiation from iPSC? Could differential “allelic regulation” explain variable penetration of the phenotype, similar to what have been reported for pluripotency genes during reprogramming?41 What are the potential other factors cooperating with an abnormal GATA2 protein promoting bone marrow failure and other manifestations of GATA2 deficiencies? We created both FB/MSC- and PBMC-derived iPSC from 2 patients and 1 asymptomatic carrier to ask if hematopoietic defects require acquisition of secondary mutations. However, our PBMC-iPSC failed to capture STAG2 mutations that were identified in the peripheral blood of 2 subjects who developed symptomatic bone marrow failure. This may explain the discrepancies between the iPSC and the clinical phenotypes in our model. Potentially, mutations in this gene results in decreased ability to derive iPSC. Overall, there were no marked differences in iPSC hematopoietic output or characteristics from these 2 starting cell sources. In fact, Int5F-PBMC iPSC (subject 3) made more HSPC compared with control PBMC iPSC, which is not likely because of intron 5 mutation itself given that Int5M-PBMC iPSC (subject 4) derived from the father of subject 3 showed limited hematopoietic potential. Overall, low and variable efficiency of control PBMC iPSC limits the interpretation of this result.
Our iPSC differentiation studies did not reproduce the specific lineage-differentiation abnormalities observed in GATA2 deficiency patients. Minimal differences were observed between control and GATA2-deficient differentiated hematopoietic cells plated in CFU assays. The total number of colonies formed per 1000 CD34+CD45+ cells was not different between normal control or patient iPSC derived from patients with ZF or Int5 mutations. There appeared to be a partial loss of multipotent GEMM (trend only), CFU-GM, and erythroid colonies (burst-forming unit-erythroid/CFU-erythroid) on a per-cell basis comparing plating of CD34+CD45+ cells derived from ZF-mutant patient iPSC with control iPSC. However, CFU-M were not decreased and were overall increased; thus, our model did not recapitulate the monocytopenia observed in these patients, but may suggest subtle differences in lineage differentiation potential.
NK cell differentiation was not impaired comparing isogenic wild-type to GATA2-haploinsufficient iPSC. Total NK output did correlate with HSPC output achieved before NK-specific culture condition was initiated; thus, total NK output was reduced from the GATA2-haploinsufficient iPSC on a per-cell basis.
These findings are in line with some observations in patients with GATA2 deficiency. Patients do have residual NK cells in their peripheral blood that decrease with time and overall disease progression, suggesting that NK cell differentiation is not grossly impaired, but that production of new NK cells decreases as HSPC progressively decline in the bone marrow of these patients. We hypothesize that residual CD56dim NK cells do not require ongoing production from HSPC, but instead self-renew independently, as supported by our previous observations of NK cell life histories in GATA2 patients, paroxysmal nocturnal hemoglobinuria patients, and a genetically barcoded nonhuman primate model.37,42,43 Our model is the first to investigate NK differentiation from GATA2-deficient iPSC.
Analysis of human monocyte, B cell, or NK differentiation in vivo from GATA2-deficient iPSC-derived HSPC would be ideal. However, current hematopoietic differentiation methods do not produce HSPC able to engraft in immunodeficient mice, a limitation in the current system, and further evidence that iPSC hematopoietic differentiation models may reflect primitive rather than definitive hematopoiesis.44
Our hematopoietic differentiation results are in line with previous reports on GATA2-deficient pluripotent stem cell models in that heterozygous GATA2-mutant iPSC can produce HSPC.45,46 Kotini et al45 reported a clonal evolution model using patient-derived iPSC at various stages of the development of myeloid malignancies and included 1 patient with a germline heterozygous GATA2 T357N mutation. CRISPR/Cas9-mediated inactivation of the second GATA2 allele in this patient line resulted in a modest reduction in hematopoietic differentiation and prolonged retention of CD90, a marker for long-term HSC.
Our iPSC differentiation results support previous observations from Gata2 KO studies in mice, suggesting a critical role of GATA2 for endothelial-to-hematopoietic transition (EHT),19,35,47-50 but GATA2 haploinsufficiency or loss-of-function mutations do not result in gross defects in ongoing fetal or adult hematopoiesis.25 These seemingly contradictory results could indicate that an EHT-independent developmental pathway, perhaps persisting from the yolk sac HSC compartment, might partially compensate for reduced output of definitive HSPC.47,48,51-54 Our HEP-OP9 coculture model showed that the ability to generate HEP was not different between groups, although there was a trend toward decreased hematopoietic potential of HEP in ZF or Int5 compared with controls. This observation is in line with a recent report demonstrating GATA2 is dispensable for generation of hemogenic endothelium, but plays a role in EHT.55 The use of Activin A during mesoderm induction in our hematopoietic differentiation protocol might have caused our in vitro differentiation to favor primitive hematopoiesis.52
Single-cell RNA-seq analysis of the cells from GATA2+/+ vs GATA2+/− iPSCs supports the conclusion that there are subtle differences in hematopoiesis. Our analysis shows a trend toward statistically different proportions of cells in progenitor lineages, consistent with the preservation of more primitive populations in GATA2+/+ iPSC compared with their isogenic GATA2-mutant counterparts. Pseudotemporal ordering found a trend toward a greater proportion of lymphoid progenitors in GATA2-mutant iPSC, particularly in the GATA2 R337X pair. However, no developmental blocks were evident that could account for the monocytopenia, B and NK lymphopenia, and dendritic cell deficiency. We found that genes involved in megakaryocyte-erythrocyte progenitor development were overexpressed in a statistically and biologically significant degree, whereas genes involved in granulocyte-monocyte progenitor and neutrophil development was underexpressed. These findings are consistent with the observation that hemoglobin and platelet counts are relatively preserved until later stages, whereas monocytopenia may be observed in early stages of BMF in GATA2-deficient patients. Unfortunately, the number of cells we were able to generate and sequence for this study was insufficient to fully resolve the identity of many cell types or to infer the exact ontogeny of the mature lineages.
To our knowledge, our work represents the first stepwise investigation of a GATA2 dosage effect in isogenic human cells (GATA2+/+, GATA2+/−, and GATA2−/−) and in corrected patient lines. Our finding of very subtle defects in HSPC during in vitro differentiation from GATA2-deficient iPSC may suggest that also HSC extrinsic factors (eg, infections, immunological imbalances, autoimmunity, bone marrow microenvironment) may play a role in the pathogenesis of bone marrow failures by further depleting or impacting HSPC that may be hypersensitive to these factors because of GATA2 deficiency.
Modeling hematopoietic dysfunction using iPSC is very challenging because of limitations in the current hematopoietic differentiation methods.26,44 The differentiation method we used mainly examines linear progression of differentiation from iPSC to mesoderm to embryonal HSPC. Unless multiple hematopoietic transcription factors are overexpressed,56 iPSC-derived HSPC do not achieve long-term multilineage engraftment in NSG mice; therefore, self-renewal or multilineage differentiation capacity cannot easily be assessed, which is likely to be key to understanding human GATA2 deficiency. The long latency and variable penetrance in these patients suggests that adult HSPC may be more affected by GATA2 haploinsufficiency than embryonal HSPC and that these 2 HSPC types may have major differences in differentiation capacity to various lineages. That iPSC-derived HSPC have features close to embryonal HSPC may explain why our iPSC model was not able to recapitulate specific lineage deficiencies seen in GATA2 deficiency.
Acknowledgments
The authors thank Michael N. Sack (National Heart, Lung, and Blood Institute [NHLBI]), who provided healthy volunteer peripheral blood mononuclear cell under his institutional review board–approved protocol; Harry L. Malech (National Institute of Allergy and Infectious Diseases) for providing the Cre-recombinase vector; and Keyvan Keyvanfar and NHLBI flow cytometry core for their help in the fluorescence-activated cell sorting experiments. M.J. thanks the Translational Research Training in Hematology program (American Society of Hematology/European Heart Association) for training and faculty mentorship.
This research was supported by the Division of Intramural Research, NHLBI, NIH. M.J. is supported in part by grants from NIH National Center for Advancing Translational Sciences, Clinical and Translational Science Award program (UL1 TR001866), and American Society of Hematology (Scholar Award).
Authorship
Contribution: M.J. designed experiments; collected, analyzed, and interpreted data; and wrote the manuscript; S.C. and X.G. analyzed the single-cell RNA-sequencing data and wrote the manuscript; J.Z. derived isogenic iPSC lines and wrote the manuscript; S.J.Y. collected and interpreted data; S.G.H. performed teratoma assays and interpreted data; V.D. collected and interpreted data; E.K. and F.S.D. derived patient-specific iPSC lines; S.A.H. performed computational modeling of mutant GATA2 protein; M.A. performed targeted next-generation sequencing and analyzed sequencing data; A.P.H., S.M.H., and D.D.H. referred patients and interpreted data; D.T. conceptualized the study and interpreted data; C.E.D. conceptualized the study, interpreted data, and wrote the manuscript; and T.W. conceptualized the study, collected, analyzed, and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thomas Winkler, Hematology Branch, NHLBI/NIH, 10 Center Dr, Bethesda, MD 20892; e-mail: winklert@nhlbi.nih.gov; and Cynthia E. Dunbar, Translational Stem Cell Biology Branch, NHLBI/NIH, 10 Center Dr, CRC/Building 10, Room 4E-5132, Bethesda, MD 20892; e-mail: dunbarc@nhlbi.nih.gov.

