

Key Points
Aged miR-146b KO mice developed hematopoietic malignancies with different cell morphologies than those in aged miR-146a KO mice.
The targets of miR-146b were identical to those of miR-146a.

Abstract
Excessive and constitutive activation of nuclear factor-κB (NF-κB) leads to abnormal cell proliferation and differentiation, leading to the development of malignant tumors, including lymphoma. MicroRNA 146a (miR-146a) and miR-146b, both of which carry an identical seed sequence, have been shown to contribute to inflammatory diseases and tumors by suppressing the expression of key molecules required for NF-κB activation. However, the functional and physiological differences between miR-146a and miR-146b in disease onset have not been fully elucidated. In this study, we generated miR-146b–knockout (KO) and miR-146a–KO mice by genome editing and found that both strains developed hematopoietic malignancies such as B-cell lymphoma and acute myeloid leukemia during aging. However, the B-cell lymphomas observed in miR-146a– and miR-146b–KO mice were histologically different in their morphology, and the malignancy rate is lower in miR-146b mice than miR-146a mice. Upon mitogenic stimulation, the expression of miR-146a and miR-146b was increased, but miR-146b expression was lower than that of miR-146a. Using a previously developed screening system for microRNA targets, we observed that miR-146a and miR-146b could target the same mRNAs, including TRAF6, and inhibit subsequent NF-κB activity. Consistent with these findings, both miR-146a– and miR-146b–KO B cells showed a high proliferative capacity. Taken together, sustained NF-κB activation in miR-146b KO mice could lead to the development of hematopoietic malignancy with aging.
Introduction
MicroRNAs (miRNAs) are small noncoding RNAs that bind partial complementary sequences of their target mRNAs and typically pair to the miRNA seed sequence (nucleotides 2-7), leading to their translation repression and/or degradation.1 In addition, pairing to the 3′ portion (particularly nucleotides 13-16) of miRNA plays supplementary or compensatory roles for the seed pairing to enhance binding specificity and affinity.1 Accumulating evidence has demonstrated the critical roles of miRNAs in diverse biological processes, including inflammation, cell differentiation, cell growth, and tumorigenesis.2-4
Abnormalities in cell proliferation and differentiation could be caused by malignancies and promote tumor development. Nuclear factor-κB (NF-κB) is a key transcription factor for development, proliferation, inflammation, and tumorigenesis. Constitutive NF-κB activation has been detected in various types of lymphoid and myeloid malignancies.5-7 Genetic mutations in TNFAIP3, CARD11, MYD88, and CD79A/B are frequently found in patients with lymphoma, especially in those with diffuse large B-cell lymphoma (DLBCL), and these mutations could lead to excessive activation of NF-κB.8-13 Therefore, elevated NF-κB activation is considered to be a driver of tumorigenesis in B cells.
miRNA 146a (miR-146a) was originally identified as an NF-κB–induced miRNA that could repress NF-κB activity through inhibition of tumor necrosis factor receptor–associated factor 6 (TRAF6) and interleukin-1 receptor–associated kinase 1 (IRAK1), both of which play critical roles in NF-κB activation.14 Thus, miR-146a could act as a feedback inhibitor for NF-κB activation. Indeed, miR-146a–deficient (knockout [KO]) mice showed autoimmunity such as T-cell–mediated multiorgan inflammation.15-17 Furthermore, aged miR-146a KO mice spontaneously developed lymphoid and myeloid malignancies,16,18 indicating that miR-146a functions as a tumor suppressor by negatively regulating NF-κB activity.
miR-146b, a homolog of miR-146a, has the same seed sequence as that of miR-146a and can also repress TRAF6 and IRAK1 expression.14 Therefore, miR-146b could also play a critical role in preventing tumor development. It was reported that reduced expression of miR-146b was observed in various types of tumors, such as breast cancer,19,20 glioma,21 gallbladder cancer,22 and large B-cell lymphoma.23 In addition, it had been reported that overexpression of miR-146b suppresses malignancy in lymphoma,23,24 leukemia,25 breast cancer,26 and gliomas.27,28 However, the physiological roles of miR-146b and the functional differences between miR-146a and miR-146b in the context of tumorigenesis remain elusive.
Here, we generated miR-146b KO mice by genome editing and observed that aged miR-146b KO mice, as well as miR-146a KO mice, spontaneously developed B-cell lymphoma and acute myeloid leukemia (AML). In addition, we found that miR-146b suppressed the genes targeted by miR-146a using a previously developed screening system for miRNA targets.
Materials and methods
Mice
miR-146a KO mice were generated as described previously.29 To generate miR-146b KO mice, we used a custom transcription activator-like effector nuclease (TALEN) access service (Cellectis Bioresearch, Paris, France) to design and construct plasmids encoding a cytomegalovirus promoter–driven TALEN specific to the miR-146b gene locus. The target sequences of TALENs were as follows: 5′-TTGGCCACCTGGCTCTGA-3′ and 5′-GGCTGT GAGCTCTAGCA-3′. In vitro transcription of TALEN mRNAs, purification of RNAs, and microinjection were performed as described previously.29 Both miR-146a and miR-146b KO mice (BDF-1:C57BL/6N × DBA2 background) were backcrossed to C57BL/6N (Sankyo Labo Service Corporation, Tokyo, Japan) 1 to 3 generations and were kept under specific-pathogen-free conditions. Supplemental Table 1 shows the generations of backcross of aging mice. We backcrossed the young mice for 3 generations. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at the Tokyo Medical and Dental University (approval no. A2017180).
Histology and immunohistochemistry
Histologic sections (3 μm thick) were cut from formalin-fixed and paraffin-embedded tissue samples and mounted on silane-coated slides (Muto Pure Chemicals, Tokyo, Japan). After sections were deparaffinized and rehydrated, they were subjected to an antigen activation treatment, as detailed in supplemental Table 2. Sections were then treated with 3% hydrogen peroxide in methanol for 10 minutes. Subsequently, sections were incubated overnight at 25°C with an appropriately diluted primary antibody obtained using Antibody Diluent (Dako, Glostrup, Denmark). Sections were then incubated with either EnVision + system-HRP, histofine simple stain MAX-PO (G), or a biotinylated antibody followed by incubation with streptavidin-peroxidase complex for 30 minutes each at room temperature in a humidified chamber. The primary and secondary antibodies are listed in supplemental Table 2. Before and after each step, sections were washed in phosphate-buffered saline containing 0.2% Tween-20. The signal was developed as a brown reaction product using peroxidase substrate diaminobenzidine (HistofineSimplestain DAB Solution; Nichirei Biosciences, Tokyo, Japan). All specimens were counterstained with Mayer hematoxylin. Adjacent sections were also examined with hematoxylin and eosin staining for conventional histopathologic examination. Images were taken using BX51 microscope equipped with a UPlanSApo 4× 0.16 and PlanFI 100× 0.95 objective lens and an Olympus DP72 digital camera (Olympus, Tokyo, Japan) and were observed using DP2-BSW software (version 2.1).
B-cell isolation and stimulation
Splenocytes were incubated with biotinylated anti-mouse CD19 (6D5; BioLegend, San Diego, CA) antibody and then enriched by streptavidin magnetic particles (IMag Streptavidin Particles Plus-DM; BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. Isolated B cells were stimulated with lipopolysaccharide (LPS; Escherichia coli 055:B5, Sigma-Aldrich, St. Louis, MO), anti-mouse-CD40 antibody (HM40-3, BioLegend), or anti-immunoglobulin μ chain (anti-IgM) F(ab′)2 fragment (115-006-075, Jackson ImmunoResearch Laboratories, West Grove, PA) for indicated periods.
Quantitative reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from liver or splenic B cells using ISOGEN (Nippongene, Tokyo, Japan), according to the manufacturer’s instructions. RNAs were reverse transcribed using specific primers (miR-146a; reverse transcription primer tube (RT): 000468, miR-146b; RT: 001097, SnoRNA202; RT: 001232, Thermo Fisher Scientific, Waltham, MA) with a TaqMan MicroRNA Reverse Transcription Kit. Quantitative PCR was performed with specific primers and probes (miR-146a; TaqMan assay tube (TM): 000468, miR-146b; TM: 001097, SnoRNA202; TM: 001232, Thermo Fisher Scientific) using Thunderbird Probe qPCR Mix (TOYOBO, Tokyo, Japan). Expression levels of miRNAs were normalized to snoRNA202-expression levels and calculated using the ΔCt method or ΔΔCt method.
Western blot analysis
Stimulated B cells were suspended in RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 1% sodium dodecyl sulfate, and 0.5 mM phenylmethylsulfonyl fluoride) and incubated on ice for 30 minutes. After centrifugation, the supernatant was collected and the protein content was measured using a DC Protein Assay Kit (Bio-Rad, Hercules, CA). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Merck Millipore, Billerica, MA). After blocking with Blocking One (Nacalai Tesque, Kyoto, Japan) for 1 hour, membranes were incubated with anti-TRAF6 antibody (1:1000, T2-1SC, BioLegend), or anti-β-actin antibody (AC-74, Sigma-Aldrich) at 4°C overnight and then incubated for 1 hour with a horseradish peroxidase–conjugated anti-rabbit IgG antibody (1:10 000, NA9340V; GE Healthcare, Buckinghamshire, United Kingdom) or anti-mouse IgG antibody (1:10 000, NA931V, GE Healthcare). Proteins were detected using a Pierce ECL Western Blotting Substrate (32106, Thermo Fisher Scientific).
Luciferase assay
The genomic regions of primary human miR-146a and miR-146b were amplified with PCR and cloned into a pcDNA3.1 vector (Invitrogen, Carlsbad, CA). Cell-based screening for the targets of miR-146a and miR-146b was performed using a reporter library as previously described.30 In brief, pcDNA-miR-146a, pcDNA-miR-146b, or pcDNA3.1 vectors (Invitrogen) and a pRL-SV40 (Promega, Madison, WI) Renilla luciferase construct were added to 384-well reporter library plates with Opti-MEM containing FuGENE HD (Promega) and incubated for 20 minutes. Next, HEK293FT cells (Invitrogen) in 10% fetal bovine serum/ Dulbecco’s modified Eagle medium were added into each well. Cells were cultured in a 5% CO2 incubator at 37°C for 24 hours. Wortmannin (final concentration of 100 nM, Sigma-Aldrich), a nonsense-mediated mRNA decay inhibitor, was then added into each well, and cells were cultured in a 5% CO2 incubator at 37°C for a further 5 hours. NF-κB activity was measured using a NF-κB-driven luciferase reporter. HEK293T cells were transfected with pcDNA-miR-146a, pcDNA-miR-146b or pcDNA3.1 vectors along with an NF-κB reporter vector and pRL-SV40 (both purchased from Promega) using Polyethylenimine MAX (Polysciences, Warrington, PA) and incubated for 48 hours. Cells were stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) for the last 16 hours. Luciferase activity was measured using a dual luciferase reporter assay system (Promega) and normalized to Renilla luciferase activity.
B-cell proliferation assay
Isolated B cells were stimulated with indicated concentration of LPS (E coli 055:B5, Sigma-Aldrich), anti-CD40 antibody (HM40-3, BioLegend), or anti-IgM F(ab')2 fragment (115-006-075, Jackson ImmunoResearch) for 48 hours. 1 µCi [3H]-labeled thymidine (NET-027A, PerkinElmer, Waltham, MA) was added to the culture medium for the last 16 hours. Cells were harvested, and [3H]-thymidine uptake was measured using a scintillation counter (MicroBeta 2450, PerkinElmer).
Statistical tests
All statistical analyses were performed using Excel (Microsoft) or Prism 5 (GraphPad Software, San Diego, CA) software. For animal survival curves, the P value was calculated using the Mantel-Cox log-rank test; for all other data, the 2-tailed Student t test was used.
Results
Generation of miR-146a and miR-146b KO mice
Genome editing methods, such as clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 or TALEN, can be used to introduce short genomic deletions in vivo and are useful in generating miRNA-deficient mice. We previously reported the successful generation of miR-146a KO mice by TALEN.29 To compare the functions of miR-146a and miR-146b in vivo, we also generated miR-146b KO mice by TALEN and obtained mice with short deletions in the miR-146b locus (Figure 1A).
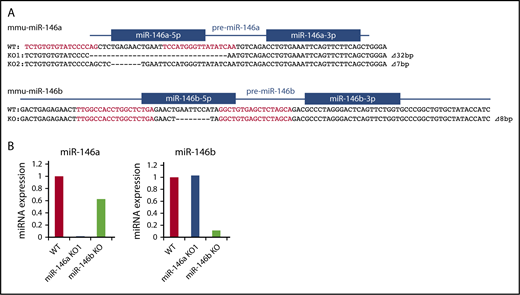
Production of miR-146a and miR-146b KO mice with TALEN. (A) Genetic deletion in miR-146a– and miR-146b–deficient mice generated by TALEN. The genomic sequence is shown at the top, with the target sequences of a pair of TALEN colored in red. Blue lines and boxes indicate pre-miRNA and mature miRNA sequences, respectively. Deleted nucleotides are indicated with dashes. The sizes of the deletions and insertions are shown to the right of the mutated alleles with Δ. WT indicates alleles without a mutation. (B) RT-PCR analysis of miR-146a and miR-146b in liver tissue derived from mutant mice. The genotype of each animal is indicated at the bottom. The expression level of miR-146a and miR-146b in the WT animal was adjusted to 1.
Production of miR-146a and miR-146b KO mice with TALEN. (A) Genetic deletion in miR-146a– and miR-146b–deficient mice generated by TALEN. The genomic sequence is shown at the top, with the target sequences of a pair of TALEN colored in red. Blue lines and boxes indicate pre-miRNA and mature miRNA sequences, respectively. Deleted nucleotides are indicated with dashes. The sizes of the deletions and insertions are shown to the right of the mutated alleles with Δ. WT indicates alleles without a mutation. (B) RT-PCR analysis of miR-146a and miR-146b in liver tissue derived from mutant mice. The genotype of each animal is indicated at the bottom. The expression level of miR-146a and miR-146b in the WT animal was adjusted to 1.
Kim et al indicated that a deletion greater than 3 bp in a mature miRNA sequence totally blocks miRNA processing.31 Thus, in all experiments, we used miR-146a/b KO mice, in which there is a ≧7-bp deletion in the miR-146a/b sequence. We confirmed expression of miR-146a/b by quantitative PCR. In these mice, expression of miR-146a or miR-146b was greatly decreased in the liver compared with that in wild-type (WT) mice (Figure 1B). Expression of mature-miR-146a/b should be diminished in the corresponding KO; however, low level expression of miR-146b was observed in miR-146b KO mice. This could be a result of potential cross-reactivity of miR-146a with the miR-146b primers, as reported previously.32
Next, to examine whether miR-146a and miR-146b are involved in lymphocyte development, we analyzed splenocytes and bone marrow cells derived from each miR-146 KO mouse. In vitro, we used miR-146a KO1 (miR-146a−/−) and miR-146b KO (miR-146b−/−) mice in all experiments. There were phenotypic changes in the populations of transitional type 1 and 2 B cells in the spleens of miR-146a and miR-146b KO mice, compared with those in WT mice. Furthermore, there was a change in the populations of Gr1+CD11b+ cells in the bone marrow of miR-146b KO mice, compared with those in miR-146a KO mice (supplemental Figures 1-3). Because miR-146a KO mice showed rapid mortality upon LPS administration, we examined LPS-induced endotoxin shock in miR-146a KO and miR-146b KO mice. Consistent with a previous report, all miR-146a KO mice that received a sublethal dose of LPS died within 30 hours.18 In addition, 7 out of 9 miR-146b KO mice died at the end of the experiment (supplemental Figure 4), indicating that both miR-146a and miR-146b are important for resistance to septic shock and play an important role in preventing systemic inflammation in vivo.
miR-146a and miR-146b KO mice develop hematopoietic malignancies in aging
It was reported that miR-146a KO mice on a mixed 129/Sv and C57BL/6 background or mice on a pure C57BL/6 genetic background develop hematopoietic malignancies.16,18 These results prompted us to examine the role of miR-146b in lymphomagenesis in aging. As a result, in mice aged 10 to 23 months, 38% (3/8), 33% (1/3), and 19% (4/21) of miR-146a KO1 mice, KO2 mice, and miR-146b KO mice, respectively, developed a hematopoietic malignancy (Figure 2A) with splenomegaly (Figure 2B). Interestingly, the malignancies observed in miR-146a and miR-146b KO mice were slightly different. Immunohistological analyses revealed that the lymphomas observed in the spleen and lymph nodes of miR-146a KO mice were characterized by the presence of medium-to-large atypical lymphoid cells with a nodular or diffuse growth pattern. In addition, the majority of these cells were negative for CD3 and positive for CD20, indicating that miR-146a KO mice had B-cell lymphoma. These characteristics resemble those of DLBCL in humans (Figure 2C). On the other hand, miR-146b KO mice also developed malignant tumors. However, their pathological features were distinct from those of miR-146a KO mice. In the spleen and lymph nodes of miR-146b KO mice (2 of 21), small-to-medium atypical lymphoid cells with a nodular growth pattern were observed. These cells were negative for CD3 and positive for CD20 and resembled those of follicular lymphoma in humans (Figure 2C). Ki67 is a marker for proliferative cells and correlates with the malignancy of cancer. The ratio of Ki67-positive cells in miR-146a KO mice was up to 70% and higher than that in miR-146b KO mice (30%) (Figure 2C; supplemental Figure 5). These results indicated that a deficiency of miR-146a or miR-146b was associated with B-cell lymphoma and that the cytological malignancy of B-cell lymphoma was higher in miR-146a KO mice than miR-146b KO mice.

Histological analysis of spleens and lymph nodes in WT mice and representative malignancies in miR-146a or miR-146b KO mice. (A) Increased rate of tumorigenesis in mice with miR-146a or miR-146b ablation. n = 11 (WT), 8 (miR-146a KO1), 3 (miR-146a KO2), and 21 (miR-146b KO). (B) Photograph of spleens isolated from WT and miR-146b KO mice at 16 months. (C) miR-146a and miR-146b KO mice developed B-cell lymphomas in the spleen and lymph nodes. Scale bars represent 500 µm (low magnification), 20 µm (high magnification), and 2.5 µm (insets). (D) miR-146b KO mice developed acute leukemia. Scale bars represent 500 µm (low magnification) and 20 µm (high magnification), and 2.5 µm (inset). HE, hematoxylin and eosin; FL, follicular lymphoma.
Histological analysis of spleens and lymph nodes in WT mice and representative malignancies in miR-146a or miR-146b KO mice. (A) Increased rate of tumorigenesis in mice with miR-146a or miR-146b ablation. n = 11 (WT), 8 (miR-146a KO1), 3 (miR-146a KO2), and 21 (miR-146b KO). (B) Photograph of spleens isolated from WT and miR-146b KO mice at 16 months. (C) miR-146a and miR-146b KO mice developed B-cell lymphomas in the spleen and lymph nodes. Scale bars represent 500 µm (low magnification), 20 µm (high magnification), and 2.5 µm (insets). (D) miR-146b KO mice developed acute leukemia. Scale bars represent 500 µm (low magnification) and 20 µm (high magnification), and 2.5 µm (inset). HE, hematoxylin and eosin; FL, follicular lymphoma.
Next, we performed additional immunohistochemistry for CD10, Bcl6, and MUM1 based on the Hans classifier.33 The results indicated that all the lymphomas developed in miR-146a KO mice were Bcl6 positive. Among them, 2 of 4 mice were CD10 positive, and 1 of 4 mice was MUM1 positive (supplemental Figure 6). Lymphomas developed in miR-146b KO mouse were positive for MUM1 and negative for both CD10 and Bcl6.
Interestingly, in 2 miR-146b KO mice without lymphoma, CD20 (−), CD3 (−), CD34 (+), and MPO (+) cells with nuclear atypia were found in the liver and lymph nodes. These cells were similar to cells seen in AML (Figure 2D).
Increased expression of miR-146a and miR-146b upon mitogenic stimulation of B cells
It has been reported that stimulation via antigen receptors and ligands for Toll-like receptors strongly induces NF-κB activation, leading to the induction of miR-146a and miR-146b.14,18,34-44 In B cells, signals via the B-cell receptor, TLR4 and CD40 could induce NF-κB activation. Therefore, we examined the expression of miR-146a and miR-146b during mitogenic stimulation in splenic B cells. Upon the stimulation of anti-IgM F(ab′)2 fragment, LPS, and anti-CD40 antibody, expression of both miR-146a and miR-146b was strongly induced within 24 hours and was sustained at a high level for up to 72 hours (Figure 3A-C). However, miR-146b expression was lower than that of miR-146a throughout the stimulation period.
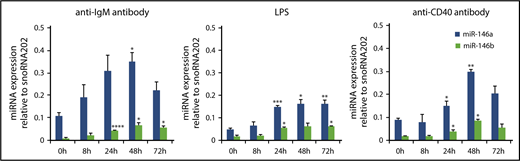
miR-146a and miR-146b induction in response to LPS, anti-CD40 antibody, and anti-IgM antibody treatment in splenic B cells. Quantitative RT-PCR analysis of mature miR-146a and miR-146b expression in WT splenic B cells in response to 10 μg/mL anti-IgM antibody (A), 1 μg/mL LPS (B), and 0.1 μg/mL anti-CD40 antibody (C). Mature miR-146a and miR-146b expression was normalized to that of snoRNA202. Data are presented as mean ± standard error of the mean (SEM); n = 3. *P < .05, **P < .01, ***P < .001, ****P < .0001.
miR-146a and miR-146b induction in response to LPS, anti-CD40 antibody, and anti-IgM antibody treatment in splenic B cells. Quantitative RT-PCR analysis of mature miR-146a and miR-146b expression in WT splenic B cells in response to 10 μg/mL anti-IgM antibody (A), 1 μg/mL LPS (B), and 0.1 μg/mL anti-CD40 antibody (C). Mature miR-146a and miR-146b expression was normalized to that of snoRNA202. Data are presented as mean ± standard error of the mean (SEM); n = 3. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Negative regulation of NF-κB activity by miR-146a and miR-146b
NF-κB induces miR-146a expression, and induced miR-146a suppresses NF-κB activity via downregulation of molecules that are required for NF-κB activation. Because miR-146a and miR-146b can be strongly induced by various stimuli, it is plausible that miR-146a and miR-146b act as negative-feedback regulators in B cells. We first confirmed whether miR-146a and miR-146b suppress NF-κB activity by luciferase assay. NF-κB-driven luciferase activity was decreased in a dose-dependent manner for miR-146a and miR-146b by PMA, which is a strong agonist of protein kinase C (Figure 4A). These results suggested that both miR-146a and miR-146b play an important role in the suppression of NF-κB activity.
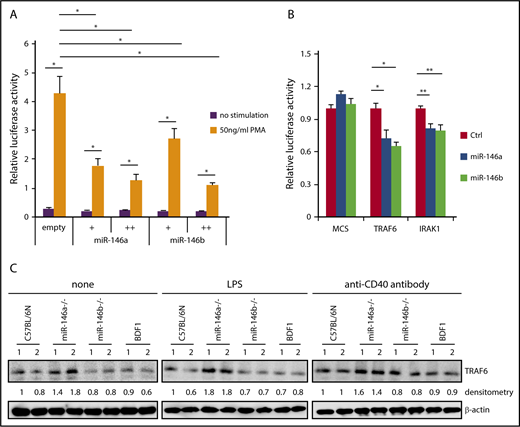
Suppression of NF-κB activity by miR-146a and miR-146b. (A) Luciferase reporter assay for miR-146a– or miR-146b–dependent regulation of NF-κB. Luciferase activity was measured in HEK293T cells transfected with a control empty vector, miR-146a, or miR-146b expression vector (with or without 50 ng/mL PMA). Data are presented as the mean ± SEM. Two independent experiments were performed, and similar results were obtained. +, 5 ng; ++, 75 ng. (B) Luciferase reporter assays in 293FT cells transfected with pcDNA-miR-146a (miR-146a), pcDNA-miR-146b (miR-146b), or an empty vector (Ctrl). MCS, pLuc2-KAP-MCS (multicloning site as an empty reporter). Data are presented as the mean ± SEM; n = 3. (C) Western blot analysis of TRAF6 protein expression in nonstimulated WT and miR-146a or miR-146b KO splenic B cells and those stimulated with LPS or anti-CD40 antibody. Two mice per genotype were analyzed; representative results are shown. *P < .05, **P < .01.
Suppression of NF-κB activity by miR-146a and miR-146b. (A) Luciferase reporter assay for miR-146a– or miR-146b–dependent regulation of NF-κB. Luciferase activity was measured in HEK293T cells transfected with a control empty vector, miR-146a, or miR-146b expression vector (with or without 50 ng/mL PMA). Data are presented as the mean ± SEM. Two independent experiments were performed, and similar results were obtained. +, 5 ng; ++, 75 ng. (B) Luciferase reporter assays in 293FT cells transfected with pcDNA-miR-146a (miR-146a), pcDNA-miR-146b (miR-146b), or an empty vector (Ctrl). MCS, pLuc2-KAP-MCS (multicloning site as an empty reporter). Data are presented as the mean ± SEM; n = 3. (C) Western blot analysis of TRAF6 protein expression in nonstimulated WT and miR-146a or miR-146b KO splenic B cells and those stimulated with LPS or anti-CD40 antibody. Two mice per genotype were analyzed; representative results are shown. *P < .05, **P < .01.
To examine the potential role of miR-146b in NF-κB activity, we evaluated individual mRNAs targeted by miR-146a or miR-146b using a reporter library system, which we previously developed as a screening system for miRNA targets.30 The reporter library includes the luciferase gene composed of 4891 full-length cDNAs in the 3′ untranslated region and can be used to evaluate targets through its full-length sequence at the translation level. The screening procedure is shown in supplemental Figure 7A. As a result, we identified 2 reported miR-146a and miR-146b targets (TRAF6 and IRAK1) and 3 previously unreported targets (Figure 4B; supplemental Figure 7B). Importantly, the identified targets of miR-146b were identical to those of miR-146a. This result suggested that miR-146a and miR-146b have redundant role in the suppression of NF-κB activity by regulating similar target genes, including TRAF6 and IRAK1. We examined the expression of 3 new potential targets for miR-146 (TSCOT, RABL2, and ABHD5) in B cells. The expression of TSCOT was not measurable in B cells. The expression of both RABL2 and ABHD5 in B cells of miR-146a/b KO mice was slightly increased compared with that in B cells of WT mice, although this difference was not statistically significant (supplemental Figure 7B-C). RABL2 is a member of the RAS GTPase superfamily.45-47 ABHD5 is lysophosphatidic acid acyltransferase, which functions in phosphatidic acid biosynthesis.48 Although the function of these genes in B cells is not clear, these genes could be involved in the miR-146a/b–dependent gene regulatory network.
We next examined the expression of TRAF6 in miR-146a and miR-146b KO B cells. Isolated B cells were stimulated with LPS or anti-CD40 antibody for 48 hours, and expression of TRAF6 was analyzed by immunoblotting. As shown in Figure 4C, expression of TRAF6 was elevated in miR-146a KO B cells stimulated with LPS or anti-CD40 antibody and nonstimulated miR-146a KO cells compared with WT cells. However, there was no visible change in TRAF6 expression in miR-146b KO B cells compared with WT cells. This discrepancy may reflect the difference between miR-146a and miR-146b expression levels in B cells.
Higher proliferative capacity of miR-146a and miR-146b KO B cells upon mitogenic stimulation
NF-κB promotes cell proliferation via progression of the cell cycle and induction of antiapoptotic factors.5,7,49,50 We showed that miR-146b exerts an inhibitory effect on NF-κB activity and that deficiency of miR-146b promotes the spontaneous development of B-cell lymphoma, suggesting that miR-146b KO B cells could enhance proliferative capacity. Thus, we performed a [3H]-labeled thymidine incorporation experiment upon stimulation with LPS, anti-CD40 antibody, and anti-IgM F(ab′)2 fragment in B cells derived from both types of KO mice and WT mice. As shown in Figure 5, all LPS, anti-CD40 antibody, and anti-IgM F(ab′)2 fragment stimulations promoted [3H]-thymidine uptake in miR-146a and miR-146b KO B cells compared with WT B cells (Figure 5). The elevated proliferative capacity was comparable between miR-146a and miR-146b KO B cells. This result was consistent with the malignancy observed in B cells of miR-146a and miR-146b KO mice, suggesting that miR-146b also plays a critical role in regulating the B-cell proliferation and preventing B-cell lymphoma in aging.
![Figure 5. Proliferation of WT, miR-146a KO, and miR-146b KO B cells in response to LPS, anti-CD40, and anti-IgM antibody. Splenic B cells (CD19+) were incubated for 48 hours in the presence of media only (none) or the indicated mitogens (0.1, 1, or 10 μg/mL LPS; 0.1, 1, or 5 μg/mL anti-CD40; and 10 μg/mL anti-IgM). Proliferation was measured by [3H]-thymidine incorporation. Data are presented as mean ± SEM. n = 6. *P < .05, **P < .01. cpm, counts per minute.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/23/10.1182_bloodadvances.2018017954/8/m_advances017954f5.png?Expires=1765168764&Signature=VffJEyo8X9LY4Ad0UJQWVsSZGBD79kHWTjw2iuOaAULMCajriFhdoLtcpevSCxfPPJr~rSUQa7RzeRFBCLZx0In2W7bufHf~d-zMdwwlVXoO29lEbuhMc1lNo-imEUjucmHtzeW~sMgVyf6MPP4MJ4~XA0sdhBZZ7OREPiVPV4EBmKEgEnXPaEGNeoAnBXZmv7qTFLQL4MGTNjA1pTHNab8qp5vdqjjmCQA2racxwqk2B1hqn5We1uTueW6oxaX0EaHeoK2WGwjZy1c5GRem8t7g2v2NfW9N5dEMJI05gGXLJ-l9XIxdLoq1hGeXrKYOmP-F39sPna3FQqNAmpjwmg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proliferation of WT, miR-146a KO, and miR-146b KO B cells in response to LPS, anti-CD40, and anti-IgM antibody. Splenic B cells (CD19+) were incubated for 48 hours in the presence of media only (none) or the indicated mitogens (0.1, 1, or 10 μg/mL LPS; 0.1, 1, or 5 μg/mL anti-CD40; and 10 μg/mL anti-IgM). Proliferation was measured by [3H]-thymidine incorporation. Data are presented as mean ± SEM. n = 6. *P < .05, **P < .01. cpm, counts per minute.
Proliferation of WT, miR-146a KO, and miR-146b KO B cells in response to LPS, anti-CD40, and anti-IgM antibody. Splenic B cells (CD19+) were incubated for 48 hours in the presence of media only (none) or the indicated mitogens (0.1, 1, or 10 μg/mL LPS; 0.1, 1, or 5 μg/mL anti-CD40; and 10 μg/mL anti-IgM). Proliferation was measured by [3H]-thymidine incorporation. Data are presented as mean ± SEM. n = 6. *P < .05, **P < .01. cpm, counts per minute.
Discussion
In this study, we showed that miR-146b KO mice and miR-146a KO mice spontaneously developed a hematopoietic malignancy with age. Our miR-146a KO mice developed lymphomas. It has been reported that myeloid tumors occur frequently in miR-146a KO mice on a pure C57BL/6 background16 and that lymphomas develop in miR-146a KO mice on mixed backgrounds.18 Our data are consistent with the previous data reported for mice on mixed backgrounds. B-cell lymphomas that developed in miR-146b KO mice had a smaller cell morphology and lower Ki67 staining ratio than those in miR-146a KO mice (Figure 2A,C; supplemental Figure 5). Lymphomas in miR-146b KO mice corresponded to a low-grade follicular lymphoma, whereas those in miR-146a KO mice corresponded to a high-grade follicular lymphoma and DLBCL. Consistent with the findings in miR-146a and miR-146b KO mice, proliferation of B cells upon stimulation by B-cell receptor, CD40, and TLR4 was significantly promoted in both miR-146a and miR-146b KO B cells (Figure 5).
In detailed immunophenotype analysis, CD10 and Bcl6 are expressed in lymphocyte precursor cells and germinal center B cells, respectively, and MUM1 is expressed in later stages of B-cell differentiation after the expression of CD10 and Bcl6, suggesting that the immunophenotype of lymphomas in miR-146b KO mice could be different from those that developed in miR-146a KO mice (supplemental Figure 6).
Although both aged miR-146a KO and miR-146b KO mice developed B-cell lymphomas, different pathological features were observed between miR-146a and miR-146b KO mice, raising the possibility that they target distinct mRNAs. However, miR-146a and miR-146b have identical 5′ seed and 3′ supplementary sequences. Indeed, our screening system for miRNA targets with a high-throughput assay revealed that miR-146a and miR-146b targeted the identical mRNA, and there was no specific target of miR-146b (Figure 4B; supplemental Figure 7B). Although in LPS- and anti-CD40 antibody–stimulated primary splenic B cells, elevated expression of TRAF6 was not clearly observed in miR-146b KO B cells (Figure 4C). We confirmed that forced expression of miR-146b suppressed the expression of a luciferase gene harboring TRAF6 and IRAK1 sequences (Figure 4B).
While both miR-146a and miR-146b were expressed and increased by mitogenic stimulation in B cells, the expression level of miR-146b was relatively lower than that of miR-146a (Figure 3). There were no significant changes in the expression of miR-146b target genes, such as TRAF6, at the protein level in primary B cells. However, enhanced cell proliferation upon mitogenic stimulation was observed in miR-146b KO B cells, which was sufficient for miR-146a expression (Figures 3 and 5). Therefore, weak and sustained elevation of NF-κB activation may lead to the development of hematological malignancies with age in miR-146b KO mice. These findings reflect the histological differences in B-cell lymphoma observed in miR-146a and miR-146b KO mice. Interestingly, it has been reported that tumorigenesis in Eμ-Myc transgenic miR-146a KO mice is not caused by the TRAF6 axis,51 suggesting the potential for a TRAF6-independent pathway in miR-146a/b tumor-suppressive function.
The excessive and constitutive activation of NF-κB has been shown to promote B-cell lymphoma via cell-cycle progression and the suppression of apoptosis.8,52 In patients with DLBCL, genetic mutations in TNFAIP3,8,9,12 CARD11,10,12 MYD88,11,12 and CD79A/B12,13 are frequently observed and are associated with the activation of NF-κB. We found that PMA-induced NF-κB–driven luciferase activity was inhibited by miR-146a and miR-146b (Figure 4A). It has been suggested that the chronic activation of NF-κB is associated with myeloproliferative diseases and that AML was present in miR-146a KO mice.16 In this study, miR-146b KO mice developed AML (Figure 2D). Taken together, these results suggest that miR-146b contributes to the prevention of tumor development with aging.
In addition to NF-κB, several other transcription factors such as IRF 3/7, c-Myc, c-Fos, STAT3/6, and C/EBPβ have been reported to regulate miR-146a and/or miR-146b. In hematopoietic cells, PU.1 regulates miR-146a in hematopoietic progenitors and mature bone marrow cells,53 and GATA1 regulates miR-146b in erythropoiesis and megakaryocyte development.54 It has been reported that both miR-146a and miR-146b were expressed and cooperatively act together in germ center B and T cells, whereas the expression level of miR-146b is much lower than that of miR-146a.55 Consisted with these findings, we also observed low expression of miR-146b compared with miR-146a, which may result in lower malignancies of miR-146b aging mice.
Intriguingly, it was demonstrated that the deletion of a genetic region including the miR-146a locus has been found in myelodysplastic syndrome associated with chromosome 5q deficiency and that miR-146a is the gene responsible for the development of this disease.56 Thus, it would be of interest to examine whether the genetic alteration of miR-146b in humans could be associated with lymphatic or myeloid disease.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank S. Takada and M. Tamano (National Institute of Child Health, Tokyo, Japan) for supporting the generation of KO mice and M. Mori and H. Suzuki for discussions.
This research was supported by the Japan Agency for Medical Research and Development (Core Research for Evolutional Science and Technology grants JP15gm0410001 and JP17gm0810008), the Naito Foundation, the Daiichi Sankyo Foundation of Life Science, the Japan Society for the Promotion of Science (Kakenhi grants 26113008, 15H02560, and 15K15544), and National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (grants AR050631 and AR065379) (H.A.).
Authorship
Contribution: T. Mitsumura, Y.I., T.C., and H.A. designed the study; T. Mitsumura, Y.I., and T.C. analyzed the data; Y.I. and T.K. generated the KO mice; T. Mitsumura, Y.I., R.K., K.U., T.I., and K.Y. performed pathological analyses; T. Mitsumura, Y.I., T.C., R.K., T. Matsushima, Y.T., Y.E., M.K., Y.M., N.I., and H.A. summarized and interpreted the findings; T. Mitsumura, Y.I., and T.C. drafted the manuscript; H.A. critically reviewed the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hiroshi Asahara, Department of Systems BioMedicine, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University (TMDU), 1-5-45, Yushima, Bunkyo-ku, Tokyo 113-8510, Japan; e-mail: asahara@scripps.edu or asahara.syst@tmd.ac.jp.

