

Abstract
Clonally restricted hematopoiesis is a common aging-associated biological state that predisposes to subsequent development of a hematological malignancy or cardiovascular death. Clonal expansion driven by leukemia-associated somatic mutations, such as DNMT3A, ASXL1, or TET2, is best characterized, but oligoclonality can also emerge without recognized leukemia-driver mutations, perhaps as a result of stochastic neutral drift. Murine models provide compelling evidence that a major mechanism of increased cardiovascular mortality in the context of clonal hematopoiesis is accelerated atherogenesis driven by inflammasome-mediated endothelial injury, resulting from proinflammatory interactions between endothelium and macrophages derived from circulating clonal monocytes. Altered inflammation likely influences other biological processes as well. The rate of development of overt neoplasia in patients with clonal hematopoiesis of indeterminate potential (CHIP), as currently defined, is 0.5% to 1% per year. Contributing factors to clonal progression other than acquisition of secondary mutations in hematopoietic cells (ie, stronger leukemia drivers) are incompletely understood. Disordered endogenous immunity in the context of increased proliferative pressure, short telomeres leading to chromosomal instability, an unhealthy marrow microenvironment that favors expansion of clonal stem cells and acquisition of new mutations while failing to support healthy hematopoiesis, and aging-associated changes in hematopoietic stem cells, including altered DNA damage response, an altered transcriptional program, and consequences of epigenetic alterations, are all potential contributors to clonal progression. Clinical management of patients with CHIP includes monitoring for hematological changes and reduction of modifiable cardiovascular risk factors; eventually, it will also likely include anti-inflammatory therapies and targeted approaches to prune emergent dangerous clones.
Introduction: understanding clonal hematopoiesis in the context of hematological malignancy precursor conditions
A precursor condition is an indolent pathologic state that frequently progresses over time to overt disease without any external intervention.1 Well-defined cancer precursor states, such as adenomatous colon polyps, oral leukoplakia, and high-grade dysplasia of the uterine cervix, have illuminated the multi-step process of oncogenesis. In some cases, clinical recognition of cancer precursors led to successful early detection screening programs that improved public health, because resection of certain high-risk precancerous lesions from the body decreases the incidence of subsequent diagnosis of invasive cancer.
Hematological malignancy precursor conditions do not have anatomical correlates of other clonal states like polyps or hairy leukoplakia.2 In fact, blood counts and marrow morphology in patients with hematological precursor states may be entirely within normal limits. Instead, these states are defined solely by markers of clonality (ie, expansion of genetically identical cells that differ in some detectable way from the germline configuration and that represent the product of an outsized contribution to hematopoiesis by a single hematopoietic stem cell [HSC]).3
Clonality is a hallmark of cancer but is not, by itself, neoplasia defining. Well-recognized hematological malignancy precursor states that are not cancers include monoclonal gammopathy of undetermined significance (MGUS), recognized since the late 1950s and defined by disproportionate production of a single antibody idiotype or fragment by clonal plasma cells, as well as monoclonal B cell lymphocytosis (MBL), recognized since the 1980s and characterized by expansion of a clonal mature lymphocyte population with a specific immunophenotype.
The most recently described high-incidence hematological neoplasia precursor state, which has been termed clonal hematopoiesis of indeterminate potential (CHIP),4-6 is the subject of this article. CHIP is not defined by a homogenous cellular product as in MGUS or by a distinct immunophenotype like MBL; instead, it is defined by somatic mutations in leukemia-associated driver genes resulting in expansion of a genetically identical clone of marrow and blood cells. Clonal hematopoiesis likely can also occur by stochastic neutral drift acting on a small active population of HSCs, and this may have distinct implications compared with CHIP defined by leukemia-driver mutations (ie, mutations conferring a growth or survival advantage).7 In neutral drift, all stem cells have equal proliferative potential, but some die out over time as a random event, prompting the remaining cells, which may bear mutations that are of no proliferative advantage but make the cell progeny identifiable, to expand to replace the lost cells. Just as advances in diagnostic technology led to the definition of MGUS (ie, growing clinical use of serum protein electrophoresis beginning in the 1930s) and recognition of MBL (ie, development of monoclonal antibodies and clinical application of multicolor flow cytometry beginning in the 1970s), the widespread availability of high-throughput DNA sequencing technology in the 21st century created the conditions for recognition of CHIP and clonal hematopoiesis.
The distinction between precursor states and neoplasms has been surprisingly difficult to define. Cancer has historically been defined by its biological features (eg, clonality, uncontrolled proliferation, invasiveness), as well as by a natural history that includes a risk for death from disease-associated complications.
In some respects, several of the hematological disorders classified by the World Health Organization (WHO) as neoplasms8 could also themselves be considered precursor states. For instance, myelodysplastic syndromes (MDSs) or primary myelofibrosis could be considered precursor conditions for acute myeloid leukemia (AML), because ≥25% of patients with MDS and 10% to 20% of those with myelofibrosis will develop AML. Likewise, smoldering myeloma can become active multiple myeloma, which, in turn, can become plasma cell leukemia, and a proportion of patients with early-stage chronic lymphoid leukemia or low-grade follicular lymphoma will transform to more aggressive lymphoid neoplasms.
MBL and MGUS rarely result in death prior to progression to lymphoma or myeloma, unless a rare paraneoplastic manifestation or an uncommon complication such as light-chain amyloidosis supervenes. However, CHIP can directly contribute to death while it is still in the precursor phase.9-11
Description of CHIP
Before considering how CHIP can progress to hematological malignancy or cause cardiovascular events, it is important to try to achieve some semantic clarity, because terminology is rapidly evolving and may cause confusion. Terminological clarity requires an understanding of the history of how CHIP was described and defined.
Clonality can be inferred by assessment of age-related skewing of hematopoiesis, and this was how Philip Fialkow (1934-1996) and others demonstrated that conditions such as essential thrombocythemia and polycythemia vera are clonal neoplasms,12,13 an important factor in the eventual reclassification of William Dameshek’s myeloproliferative “disorders” as myeloproliferative “neoplasms.” Clonal skewing is defined by imbalanced expression of polymorphic X-linked genes such as G6PD, 1 copy of which normally is unexpressed due to lyonization/chromosome condensation in each female cell during fetal life. Because lyonization is random, expression of each X-linked polymorphic gene should be roughly equivalent if all HSCs are contributing approximately equally to blood cell production. If there is a >3:1 imbalance in the relative expression of polymorphic X-linked genes, that implies clonally restricted hematopoiesis.
Lambert Busque in Québec, Canada, had long been interested in the hematology of elderly women with clonal skewing, which provided evidence that clonally restricted hematopoiesis could occur with aging in the absence of disease.14 In 2012, he and his colleagues reported acquired TET2 mutations in 5% of these older women but not in any of the study subjects without clonal skewing.15 TET2 mutations had been described as leukemia-associated events in 2009 and are present in up to 25% of patients with myeloid neoplasms.16 It has long been recognized that some individuals have mutations in blood or marrow cells that are associated with hematological neoplasia, yet the individual does not meet any other diagnostic criteria for neoplasia. Usually, these mutations are only transiently detected; for instance, healthy people who have low levels of BCR-ABL1 fusion will rarely have this fusion detectable again a year later. The 2012 report was the first time a clonally restricted mutation had been seen with such a high frequency in hematopoietic cells in an apparently healthy population.
Also in 2012, other investigators analyzed genomic array data from >50 000 people and found that acquired chromosomal mosaicism, including aneuploidy and large segmental deletions involving chromosomal regions where genes important in myeloid neoplasia pathogenesis are localized, is rare prior to age 50 years but is detectable in 2% to 3% of older people.17,18 In those studies, chromosomal mosaicism reduced survival and conferred a 10-fold increased risk for developing a hematological neoplasm.
Two years later, Liran Shlush and colleagues in Israel reported the important observation that DNMT3A mutations are common in preleukemic stem cells.19 Additionally, 3 groups analyzed DNA sequencing results from many thousands of people who had enrolled in large genome-wide association studies designed to assess inherited genetic risk for nonhematological conditions or blood samples from patients with nonhematological malignancies sampled as part of The Cancer Genome Atlas. They found that mutations in known leukemia-driver genes, such as DNMT3A, TET2, and ASXL1, were common even though study subjects’ blood counts were typically normal or only mildly perturbed (eg, increased red cell distribution width).10,11,20 The prevalence of these mutations increased with aging. Such mutations were noted to be associated with an increased risk for a subsequent diagnosis of hematological neoplasia (mostly myeloid cancers, but sometimes lymphoid, too, reflecting the fact that the mutations occur in multipotent progenitors/HSCs) and with increased all-cause mortality. It is not understood why there is a myeloid bias in subsequent progression or at what level mutations occur (eg, always HSCs vs more committed progenitors).
Definition of CHIP
In 2015, recognizing that we needed a term to describe this emerging precursor condition, several colleagues and I proposed the term CHIP.4 Our definition proposal included the presence of a clonal blood cell population associated with a recognized hematological neoplasm driver mutation at a variant allele frequency (VAF) ≥ 2%, in the absence of severe cytopenias or a WHO-defined disorder.
The proposed VAF of 2% was arbitrary and may need to be adjusted as further information accumulates. The threshold of mutation detection depends, to some extent, on the analytical technique used. Clonally restricted mutations can be detected at a VAF well below 2% in a high proportion of middle-aged and older people21 using error-corrected methods and barcoding technologies, and those tiny clones may be less consequential. VAFs ∼ 2% are commonly used as minimal reporting thresholds by clinical laboratories using next-generation sequencing platforms.
Although we included a list of 19 genes in our original proposal, 3 genes that encode epigenetic modifiers (DNMT3A, TET2, and ASXL1) account for the lion’s share of acquired clonal hematopoiesis associated with recognized leukemia drivers (Table 1). Haplodeficiency of certain critical transcription factors results in leukemia stem cell formation and a risk for progression to MDS, suggesting that acquisition of somatic mutations in leukemia drivers is only 1 mechanism by which CHIP can evolve.22 The “indeterminate potential” part of the name reflects the uncertainty about how CHIP will progress in an individual person. CHIP is the subject of active investigation by many groups, and it may turn out to predispose to other conditions, such as autoimmune disorders, or to influence the risk for developing neurodegenerative changes or of response to immunotherapy for other neoplasms. Yet, in many individuals, clonal hematopoiesis likely has no consequence.
Mutations observed in blood of healthy older persons, separated by frequency of detection of variants
| Common (>50) variants, n | Less common (10-49) variants, n | Uncommon (5-9) variants, n* | |||
|---|---|---|---|---|---|
| DNMT3A | 403 | TP53 | 33 | GNAS | 8 |
| TET2 | 72† | JAK2 | 31 | BRCC3 | 6 |
| ASXL1 | 62† | SF3B1 | 27 | CREBBP | 6 |
| GNB1 | 22 | NRAS | 6 | ||
| CBL | 12 | RAD21 | 6 | ||
| SRSF2 | 11 | SETDB1 | 6 | ||
| PPM1D | † | U2AF1 | 5 | ||
| SETD2 | 5 | ||||
| Common (>50) variants, n | Less common (10-49) variants, n | Uncommon (5-9) variants, n* | |||
|---|---|---|---|---|---|
| DNMT3A | 403 | TP53 | 33 | GNAS | 8 |
| TET2 | 72† | JAK2 | 31 | BRCC3 | 6 |
| ASXL1 | 62† | SF3B1 | 27 | CREBBP | 6 |
| GNB1 | 22 | NRAS | 6 | ||
| CBL | 12 | RAD21 | 6 | ||
| SRSF2 | 11 | SETDB1 | 6 | ||
| PPM1D | † | U2AF1 | 5 | ||
| SETD2 | 5 | ||||
Most patients had only 1 mutation. In most series, DNMT3A accounts for >50% of all CHIP. ASXL1 and TET2 are likely underrepresented in this series as a result of poor coverage depth of ASXL1 exon 12 and sequencing of only exon 3 of TET2. PPM1D was not included in the series but would be expected to have a frequency less than TP53 but moderately common. This table is certainly incomplete, and other recurrent mutations will be discovered.
Variants found in <5 people included BCL11B, BCOR, BCORL1, BIRC3, BRAF, CARD11, CD58, CD79B, CNOT3, CUX1, DDX3X, EP300, ETV6, EZH2, FAM46C, FBXW7, FLT3, FOXP1, HIST1H1C, IDH2, IZKF1, JAK2, JARID2, KMT2D (MLL2), KDM6A, KIT, KLHL6, KRAS, LUC7L2, MPL, MYD88, NOTCH1, NOTCH2, PDSS2, PHF6, PIK3CA, PRDM1, PRPB40B, PTPN11, RIT1, RPS15, SETDB1, SF3A1, SMC1A, SMC3, STAG1, STAG2, STAT3, SUZ12, TBL1XR1, TET1, TNFAIP3, TNFRSF14, and ZRSR2.
Variant frequency is based on supplemental Table 2 of Jaiswal et al,10 which is derived from whole-exome sequencing of peripheral blood of 17 182 people with a median age of 58 y unselected for hematological phenotype, focused on 160 genes known to be recurrently mutated in hematological cancers. Frequency of mutations in other series might vary, especially because technical differences between targeted sequencing and whole-exome or whole-genome sequencing may yield different results; because marrow aspiration was not done, some patients may have had early undiagnosed hematological neoplasms meeting WHO criteria.
Clonal hematopoiesis vs CHIP
CHIP, as defined by point mutations or small insertion-deletion mutations in known leukemia-driver genes, represents only a subset of clonal hematopoiesis. Other groups have used the comparably euphonious term “age-related clonal hematopoiesis” (ARCH) to describe similar phenomena.23 Although CHIP is certainly more common in the elderly than in people younger than 50 years, detection of clones meeting the CHIP definition is not inevitable with aging; when CHIP is present in younger people it confers a cardiovascular and neoplasia progression risk similar to in older persons.
It may also be useful to distinguish CHIP from aging-associated clonal hematopoiesis not known to be associated with leukemia-driver gene mutations, although the latter also appears to predispose to increased mortality; therefore, that distinction may be artificial.7 The fact that our catalog of known leukemia-driver mutations is undoubtedly incomplete may account for the mortality increase even when clonality is present without a recognized driver mutation. Also, large chromosome structural variations, such as those observed in the 2012 somatic mosaicism analyses, were associated with increased mortality. Although whole-exome sequencing looking for novel leukemia-driver mutations is ongoing, this seems unlikely to be the sole explanation for the high frequency of aging-related clonal hematopoiesis.24 It is possible that nonexonic mutations, epigenetic alterations without coding variation, or loss of clonal diversity due to neutral drift with aging-associated stem cell attrition account for other cases of non-CHIP clonal hematopoiesis, and they may have distinct implications.
Finally, CHIP was defined restrictively to exclude individuals with major blood count abnormalities. If unexplained cytopenias and a hematological malignancy-associated mutation are present in the absence of defining criteria for MDS such as extensive dysplasia or excess blasts,25 this is considered “clonal cytopenia of undetermined significance” (CCUS). In 1 multicenter study, CCUS conferred a risk for progression to overt MDS/AML that is 13-fold greater than “idiopathic cytopenias of undetermined significance.”26 CCUS with multiple mutations or spliceosome mutations has a natural history similar to lower-risk MDS. In the future, it seems likely that some forms of CCUS will be considered MDS without dysplasia.
Other forms of clonal hematopoiesis
There are several other clinical settings in which clonal hematopoiesis occurs in the absence of a WHO-diagnosable neoplasm. These may overlap with CHIP but have distinct context-specific features and implications. Clonal hematopoiesis after chemotherapy or radiotherapy for a nonmyeloid neoplasm is often driven by expansion of preexisting TP53 mutant clones during the selective cytotoxic pressure of the treatment.27-29 Clonal hematopoiesis after autologous or allogeneic stem cell transplant (which may be donor derived, especially when using elderly sibling donors) is associated with posttransplant cytopenias and poorer overall survival, and it probably influences the course of graft-versus-host disease in the allogeneic setting.30 In the context of aplastic anemia, clonal mutations in PIGA, BCOR, or BCORL1 are associated with a high rate of response to immunosuppressive therapy and relatively indolent disease, whereas mutations in other genes, such as those commonly associated with CHIP, have more sinister implications (although progression to secondary MDS is by no means inevitable).31 Finally, after induction chemotherapy for AML, detection of clonal mutations by next-generation sequencing or a persistent cell population with the phenotype of the AML cells by sensitive flow cytometry assays (ie, “minimal” or “measurable” residual disease) is a major risk factor for relapse. However, solitary mutations in DNMT3A, ASXL1, or TET2, the 3 most common CHIP-associated mutations, at <30% VAF after induction confer a lower relapse risk than do other mutations, at least after 40 months of follow-up.32,33
How does CHIP result in cardiovascular events?
The idea that a cellular hematological process can promote cardiovascular disease9 is of deep interest to cardiologists.34,35 Murine models have provided insight into the connection between clonal hematopoiesis and accelerated atherogenesis (Figure 1). Using low-density lipoprotein (LDL) receptor–knockout mice fed a fatty diet and transplanted with Tet2-null cells, 2 groups showed that atherogenic plaques progressed more rapidly compared with LDL receptor–knockout mice with an unmanipulated hematopoietic system; heart failure was also increased in 2 models of cardiac injury.9,36,37 The blood counts of the Tet2-null mice were similar to Tet2 wild-type mice, suggesting that the atherosclerosis was not a result of abnormal blood cell numbers. We do not know whether Tet2 missense mutations act similarly to the nonsense/truncating mutations that are modeled in the Tet2-null mice or whether other common CHIP-associated mutations act similarly to TET2. The latter seems likely in view of clinical cardiovascular risk with other mutations (albeit lower with DNMT3A).9
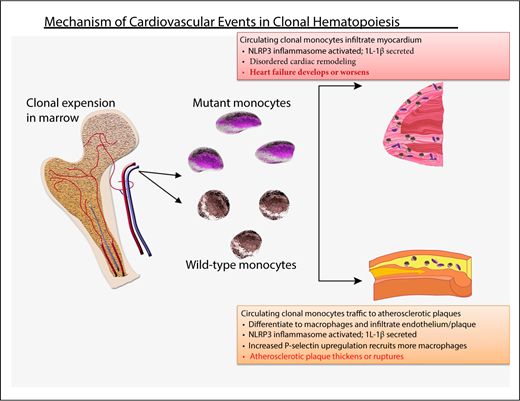
Probable mechanism of cardiovascular injury associated with clonal hematopoiesis. Mutant HSC-derived monocytes circulate and are recruited to plaques in arteries. There, they differentiate into tissue macrophages, which promote inflammation. The inflammasome activates IL-1β, which, in turn, promotes local inflammation, accelerating atherosclerosis, as well as induces expression of the P-selectin glycoprotein, which, in turn, recruits more monocytes (clonally derived and wild-type). Clonally derived platelets may also play an important role. In addition, increased interleukin-1β secretion in endothelial cells within the myocardium promotes disordered remodeling that potentiates heart failure.
Probable mechanism of cardiovascular injury associated with clonal hematopoiesis. Mutant HSC-derived monocytes circulate and are recruited to plaques in arteries. There, they differentiate into tissue macrophages, which promote inflammation. The inflammasome activates IL-1β, which, in turn, promotes local inflammation, accelerating atherosclerosis, as well as induces expression of the P-selectin glycoprotein, which, in turn, recruits more monocytes (clonally derived and wild-type). Clonally derived platelets may also play an important role. In addition, increased interleukin-1β secretion in endothelial cells within the myocardium promotes disordered remodeling that potentiates heart failure.
The atherosclerotic plaques in the Tet2-null LDL receptor–knockout mice contained an increased number of macrophages compared with controls, and the endothelium overexpressed P-selectin, resulting in recruitment of additional proinflammatory cells.37 Strikingly, blocking the NLRP3 inflammasome and, therefore, IL-1β, abrogated the accelerated atherogenesis, indicating a critical role for local inflammation in the animal models.37
The role of inflammation in human cardiovascular events has long been a vibrant area of investigation. Recently, a large prospective randomized clinical trial (CANTOS) of canakinumab, an anti–IL-1β monoclonal antibody, vs placebo in patients with a prior myocardial infarction and persistent elevation of the inflammatory marker C-reactive protein showed lower recurrent events in the intervention group.38 Baseline samples from the patients in that study are being examined to see whether those with CHIP benefitted most from the intervention. If so, this suggests a potential tool for reducing CHIP’s vascular risk, which, given the enormous number of older people with CHIP and the high incidence of stroke and myocardial infarction, is a far greater public health risk than hematological malignancy. Until further information is available, it seems prudent to manage modifiable cardiovascular risk factors in people with CHIP, especially because the risk for a vascular event in patients with CHIP was greatest in those with established risk factors.39 Further studies should also assess the risk for cardiovascular events in patients with established myeloid neoplasms.
More recently, microbial-driven inflammation and dysfunction of the small intestinal barrier associated with elevated IL-6 secretion have been found to be essential for the development of the preleukemic phenotype in Tet2-deficient mice.40 This observation supports the idea that inflammation is important for clonal proliferation and for cardiovascular events, as well as that controlling inflammation may be key in preventing leukemic transformation.
Clonal evolution of CHIP
Investigators have focused on various MDS- and AML-associated phenomena that may aid and abet clonal evolution, which were recently cogently reviewed by Bowman et al.24 The primary driver of progression of CHIP to overt neoplasia has been assumed to be acquisition of new mutation in a clonal cell with self-renewal properties. The observation that CHIP can be stable for years without expansion in clone size indicates that the CHIP-associated mutations, such as DNMT3A, ASXL1, and TET2 are, in general, not as potently leukemogenic as the more common ancestral events in AML that include core binding factor fusion genes or proliferation-inducing mutations in FLT3. In that sense, CHIP mutations collectively are “weak drivers.”
Additionally, although it appears that myeloma virtually always arises from MGUS, there are other pathways to myeloid clonal evolution that bypass CHIP. One large analysis indicated that, even in patients with long-standing CHIP, MDS or AML sometimes resulted from a new somatic event in a cell that was not part of the CHIP clone.41 Drivers of clonal evolution from CHIP to lymphoid neoplasia likely overlap with, but may be distinct from, myeloid malignancies.
Acquisition of secondary mutations is not likely to be completely random. The altered epigenetic programs induced by the 3 most common CHIP-associated mutations may facilitate clonal instability. TP53 loss is also a key driver of clonal instability, but somatic TP53 mutations are relatively uncommon in patients without a history of cytotoxic therapy. Individuals with short telomeres, whether due to well-defined syndromes, such as dyskeratosis congenita, or to less-penetrant polymorphisms in telomere components (and 1 polymorphism in a telomere-associated gene is associated with higher risk for developing clonal hematopoiesis7 ), may develop clonal evolution as a result of gross chromosomal instability consequent to failed DNA damage checkpoint function in critically short telomeres.42
What other factors contribute to CHIP progression?
Even if CHIP mutations are weak drivers, they still increase clonal survival or proliferation, so why do mutant hematopoietic clones so often stop progressing10 and remain at a similar VAF for a long period of time? What holds these mutant clones in check and prevents them from sweeping away all other hematopoietic populations? Endogenous immunity certainly might play a delimiting role, but if clonal cells are recognized by the immune system to the extent that further clonal expansion can be halted, then it must be explained what prevents immunological clearance from continuing to completion.
Given the intricate multifaceted interactions between HSCs and the marrow microenvironment, an unhealthy microenvironment could result in niche-facilitated clonal evolution and leukemogenesis. Conversely, a microenvironment that remains mostly inhospitable to a CHIP population might result in arrest of clonal outgrowth and require acquisition of a stronger driver mutation before further expansion can occur.43,44 The relative importance of microenvironment disease in human myeloid neoplasia is still unclear, but there are numerous murine models of genetic changes introduced into stromal cells that resulted in a hematopoietic defect resembling MDS; some human data support this concept as well, including increased expression of survival pathways in mesenchymal stromal cells obtained from MDS patients.45,46
Since the interaction between CHIP and immunity is poorly studied other than the limited observations in the cardiovascular setting, we can only speculate, based on changes in MDS, what role the immune system has in CHIP progression. MDS cells often express high levels of proinflammatory cytokines, including interferon-γ and tumor necrosis factor, augmenting expression of immunoinhibitory CD274.47 Chronic inflammation resulting from clonal hematopoiesis could increase proliferation pressure on HSCs or progenitor cells, which might then contribute to genomic instability and clonal evolution.48 Early MDS is also characterized by an increase in proinflammatory Th17 cells, and, if true for CHIP, this too might facilitate clonal evolution.49
In parallel, increased reactive oxygen species from iron loading due to ineffective erythropoiesis and repeated transfusions might also contribute to acquisition of new somatic mutations, although evidence of a clinical role for this mechanism of clonal progression is also scant.50 TET2 has a role in inflammation suppression via IL-6, and loss of Tet2 in knockout mice contributed to increased lung and gut injury on toxin exposure, which suggests that specific mutations may augment inflammation.51 The recent observation that vitamin C restores TET2 function suggests a potential mechanism of dampening the proinflammatory effect of TET2 loss, and clinical trials of vitamin C in TET2-mutant myeloid neoplasia are ongoing.52
Regardless of mechanism, prevention of evolution of this relatively benign precursor state to overt neoplasia is critically important; however, prevention of clonal progression is not yet possible in the clinic. The highest-priority population would seem to be those with detectable TP53 mutation following treatment of a solid tumor, given the substantial risk for TP53-mutant clones progressing to therapy-related MDS/AML,27,28,53 but there are no established strategies to eliminate such clones, and currently available treatments, such as hypomethylating agents, only temporary cytoreduce. Future approaches are likely to include a combination of anti-inflammatory treatments and clonally selective immunotherapies.
Conclusion
CHIP represents a vascular risk driven by interactions between clonal monocytes-macrophages and the endothelium and represents a neoplastic progression risk driven by acquisition of additional somatic mutations in the context of many other influences on hematopoiesis and clonal balance. Strategies to reduce the clonal burden associated with CHIP and inhibit key inflammatory pathways driving atherogenesis may improve patient outcomes. Until then, we can only let the CHIPs fall where they may.
Authorship
Contribution: D.P.S. wrote the paper.
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence: David P. Steensma, Dana-Farber Cancer Institute, Division of Hematological Malignancies, Department of Medical Oncology, D2-037, 450 Brookline Ave, Boston, MA 02215; e-mail: david_steensma@dfci.harvard.edu.

