

Abstract
Acute graft-versus-host disease (GVHD) targets the crypts in the gastrointestinal (GI) tract that are responsible for the self-renewal of the intestinal mucosa. Recent advances in the identification and culture of intestinal stem cells have improved our understanding of the interactions between the microbiome and the immune system (both innate and adaptive) that are key to the pathophysiology of GVHD. The identification of serum biomarkers that best predict long-term GVHD outcomes derive from the GI tract and have focused attention on cellular elements that act as shields against GVHD as well as its targets. These biomarkers have illuminated new mechanisms of crypt biology and provided insights that should prove useful both in the design of clinical trials and as guides to GVHD prevention and treatment.
Introduction
Successful allogeneic hematopoietic stem cell transplantation (HCT) relies on a graft-versus-leukemia effect that is mediated primarily by donor T cells. However, the destruction of normal host tissues in the skin, liver, and gastrointestinal (GI) tract by donor T cells results in acute graft-versus-host disease (GVHD), a morbid toxicity that significantly limits this treatment; 1/2 of all allogeneic HCT patients develop clinical GVHD, and <1/2 of patients who receive systemic treatment experience complete resolution of the disease.1 Standard clinical severity scales consider the extent of disease in all 3 organ systems, but the principal target of lethal GVHD is the GI tract, which will serve as the primary focus of this article. The intestinal crypt and its stem cell niche are key cellular targets of GI GVHD, and serum biomarkers released by the intestinal crypts are beginning to illuminate the complex biology of GI GVHD as well as stratify patients into different risk groups.
The intestinal stem cell niche
The epithelial lining of the small intestine is designed to maximize its absorptive surface area. The crypts of Lieberkühn generate all lineages of enterocytes and surround the bases of villi, finger-like protrusions that project into the intestinal lumen. The large intestine does not absorb nutrients, and thus, its surface lacks villi, but its crypts also penetrate the underlying submucosa. Due to the high turnover rate and rapid self-renewal of the intestinal epithelium, the biology of the crypt has emerged as an archetypal adult stem cell model.2 Recent advances in the identification, isolation, and in vitro culture of intestinal stem cells (ISCs) have dramatically improved our understanding of crypt biology and the ISC niche. It should be noted that the vast majority of the mechanistic studies cited here rely on mouse models to establish the roles of specific cell types and proteins, and mouse models do not always illuminate human physiology precisely.
ISCs
For several decades, continuously cycling crypt cells were termed crypt base columnar (CBC) cells based on their location and morphology.3 These cells give rise to progenitors called transit amplifying cells that proliferate vigorously and move upward from the crypt to the villus. As they migrate, the cells differentiate into all lineages of enterocytes that must be renewed every 3 to 5 days. The single exception to this pattern is Paneth cells, which differentiate but do not migrate and are retained at the crypt base, where they survive for 6 to 8 weeks.
A number of studies have shown that CBC cells express leucine-rich repeat containing G protein–coupled receptor 5 (Lgr5), a receptor for the Wnt pathway agonists R-spondins.4 The ability of Lgr5+ cells to both self-renew and differentiate into all types of enterocytes in vitro has firmly established their identity as ISCs.5 A single Lgr5+ stem cell can generate an entire intestinal organoid or “mini-gut” when cultured in vitro with 3 growth factors: R-spondin1, epidermal growth factor (EGF), and Noggin, a bone morphogenetic protein inhibitor. These organoids consist of crypts that join around a central lumen lined by mature epithelial cells of all lineages. Crypts in both the small intestine and the colon contain Lgr5+ ISCs.6
Paneth cells
First recognized over a century ago, Paneth cells are easily identified using light microscopy because of their eosinophilic granules that contain a wide range of antimicrobial peptides (AMPs), including α-defensins, lysozyme, secretory phospholipase A2, and regenerating islet-derived protein 3α (REG3A), earning them the title “guardians of the crypt.”7 AMPs are key elements of the intestinal mucosal barrier that protects the host from enteric pathogens. REG3A (or its mouse homolog Reg3γ) is a bactericidal C-type lectin that preferentially targets gram-positive bacteria.8 REG3A does not alter the overall architecture of the microbiome, but it concentrates in the mucus layer covering the luminal surface of enterocytes and thereby maintains the physical separation between the microbiota and the intestinal epithelial surface.9
The biogeography of the GI microbiome is complex; in the proximal small intestine, higher gradients of antimicrobials (including bile acids), lower pH, and oxygen severely limit the bacterial load compared with the colon, where the numbers of microbes increase exponentially. In addition, tightly adherent mucus layers in both small and large bowels effectively exclude microbiota so that, under normal conditions, the mucosal epithelium remains essentially sterile.10
In addition to proteins that attack microbes, Paneth cells generate essential signals for the proliferation and maintenance of neighboring ISCs,11 which explain their adjacency to ISCs in the crypt and the efficiency of intestinal organoid growth when they are included in culture. One such signal is EGF, a well-characterized mitogen. Another is Wnt3, which maintains expression of receptors on ISCs for the mitogen R-spondin.12 Others are Dll1 and Dll4, ligands for Notch1 and Notch2 receptors that help ISCs maintain their multipotent status along with Noggin.13 In vitro, Lgr5+ ISCs can produce an entire organoid, whereas Paneth cells cannot. However, doublets consisting of 1 stem cell and 1 Paneth cell generate >10-fold as many organoids as a single Lgr5+ stem cell.14 This regenerative synergy is also observed in vivo, where genetic models of Paneth cell ablation result in concomitant loss of Lgr5+ ISCs.11 In the colon, where Paneth cell numbers decrease markedly, the stem cell niche is supported by deep crypt secretory cells that exhibit many functional similarities to Paneth cells.6
Innate lymphoid cells
Clusters of immune cells called cryptopatches reside in close proximity to the crypts and contain innate lymphoid cells (ILCs). ILCs do not express antigen-specific receptors or clonally expand; they include cytotoxic natural killer cells that correspond to CD8+ T cells and helper subsets that may be considered innate counterparts of CD4+ T cells. Helper ILC subtypes are identified by different transcription factors and have different functions. ILC1s (T-bet+) activate macrophages, ILC2s (RORα+) target parasites and produce mucus, and ILC3s (RORγt+) target bacteria and stimulate epithelial proliferation.15 ILC3s have received particular attention, because they secrete interleukin-22 (IL-22) that stimulates the secretion of antimicrobial REG3A by Paneth cells16,17 and expands ISCs in vitro.18 In vivo, ILC3s produce IL-22 in response to methotrexate-mediated crypt damage, and either blockade of IL-22 or elimination of ILC3s impairs ISC regeneration.19
GVHD of the GI Tract
The histologic severity of clinical GVHD in the GI tract is categorized by the degree of crypt damage: isolated apoptotic bodies without crypt loss (grade 1), loss of individual crypts (grade 2), loss of multiple crypts (grade 3), and extensive crypt loss and epithelial denudation (grade 4).20,21 The lack of standards to quantify crypt loss has impeded the ability of this metric to predict long-term clinical outcomes. Standardized quantitation of Paneth cells in clinical samples is more easily achieved, however, and numbers of Paneth cells in duodenal biopsies inversely correlate with clinical severity of GVHD, response to treatment, and transplant-related mortality.22 These observations have highlighted dual roles of Paneth cells as both targets and shields of the GI crypt (Figure 1), illuminating interactions between innate and adaptive immunologic processes in the pathophysiology of GVHD.23,24

Early acute GI GVHD. At steady state, ISCs and Paneth cells are adjacent at the base of the intestinal crypts. Homeostasis is maintained, in part, by large numbers of commensal bacteria that stimulate IL-22 production by ILC3s. During GVHD, activated donor T cells recognize histocompatibility antigens on both hematopoietic (HP) and nonhematopoietic antigen-presenting cells (APCs). Activated APCs and T cells trigger the release of alarmins, such as IL-33, that bind to its receptor ST2; soluble ST2 (both bound to IL-33 and unbound) can enter the villus capillaries. Activated donor T cells destroy ILC3s, Paneth cells, and ISCs, releasing REG3A that was stored in the mucus and Paneth cells into the bloodstream as the epithelial barrier is breached.
Early acute GI GVHD. At steady state, ISCs and Paneth cells are adjacent at the base of the intestinal crypts. Homeostasis is maintained, in part, by large numbers of commensal bacteria that stimulate IL-22 production by ILC3s. During GVHD, activated donor T cells recognize histocompatibility antigens on both hematopoietic (HP) and nonhematopoietic antigen-presenting cells (APCs). Activated APCs and T cells trigger the release of alarmins, such as IL-33, that bind to its receptor ST2; soluble ST2 (both bound to IL-33 and unbound) can enter the villus capillaries. Activated donor T cells destroy ILC3s, Paneth cells, and ISCs, releasing REG3A that was stored in the mucus and Paneth cells into the bloodstream as the epithelial barrier is breached.
The IL-33/ST2 axis serves as a prime example of the complex interactions between the innate and adaptive immune systems that have been carefully analyzed in experimental animal models. IL-33, an alarmin released by damaged stromal, endothelial, and epithelial cells, increases during GVHD, and disease severity is significantly reduced in transplant recipients deficient in IL-33. Expression of membrane-bound ST2, the receptor for IL-33, increases on activated T cells as GVHD progresses and leads to greater secretion of inflammatory proteins, such as interferon-γ. GVHD is reduced if donor T cells lack ST2, and early infusions of soluble ST2 can act as a decoy receptor and scavenge free IL-33, preventing its inflammatory function25 ; it is possible that endogenous soluble ST2 is overwhelmed by IL-33, hampering its ability to regulate tissue inflammation. As discussed below, serum levels of ST2 are powerful predictive markers of severe GVHD in humans.
Changes in the GI microbiome also show the interplay of the innate and adaptive immune systems during GVHD. Numerous studies have highlighted the role of luminal microbial ecology in shaping the mucosal immune system during various diseases of the intestine, including GVHD. GVHD is associated with loss of microbial diversity,26,27 and loss of commensals permits overgrowth of pathogens associated with GI GVHD,28 including the loss of commensals caused by the early use of systemic antibiotics after transplant.29,30
Alteration of microbial metabolites, such as short-chain fatty acids (SCFAs) and tryptophan byproducts, also has profound effects on mucosal immunity. The SCFA butyrate is produced in the large intestine by fermenting anaerobic bacteria, and it induces differentiation of T cells to regulatory T cells through histone acetylation activity31 ; commensal bacteria producing large amounts of butyrate suppress T cell–mediated intestinal damage.32 Butyrate is lost in intestinal tissue during GVHD, and its oral administration decreases GVHD severity.33 Tryptophan metabolites, produced by commensal bacteria such as Lactobacillus, are ligands for the aryl hydrocarbon receptor that stimulates the development of ILC3 cells and drives IL-22 secretion, which promotes ISC growth.34
Administration of the ILC3 cytokine IL-22 reduces crypt damage caused by GVHD, and transplant recipients deficient in IL-22 experience more severe GVHD.18 ILC3s are among the first targets of GI GVHD, and their loss has profound consequences for the rapidly regenerating crypt.35 ILC2s are also lost during GVHD, and infusion of exogenous ILC2s reduces GVHD through the production of IL-13 and the epithelial mitogen amphiregulin.36
As mentioned above, GVHD causes crypt damage and the loss of Paneth cells and their proteins, such as α-defensins, which result in dysbiosis. Oral administration of an α-defensin reduces dysbiosis and attenuates GVHD severity.37 With improved ability to quantitate ISCs through specific markers, such as expression of Lgr5, studies have clearly shown that GVHD targets ISCs as well as Paneth cells.35,38 In several seminal studies, administration of either IL-22 or R-spondin1 stimulates ISC growth, prevents crypt damage, and reduces GVHD mortality.18,38
Our laboratory has recently further illuminated the synergistic relationship between Paneth cells and ISCs during GVHD. The antimicrobial Paneth cell protein REG3A (in humans) or Reg3γ (the mouse homolog) is released into the bloodstream when GVHD damages GI crypts and the integrity of the intestinal mucosa is breached. In both humans and mice, the serum levels of this protein paradoxically increase as the severity of GVHD increases and the levels in the intestine fall.39 Administration of IL-22 protects the crypts from GVHD and prevents leakage of Reg3γ into the bloodstream in normal mice; however, in mice deficient in Reg3γ, IL-22 is unable to reduce apoptosis of either ISCs or Paneth cells and cannot prevent GVHD. This beneficial effect cannot be attributed to changes in the microbiome, which does not differ significantly in mice unable to produce Reg3γ. Mechanistic studies in vitro showed that Reg3γ functions as an antiapoptotic protein for both ISCs and Paneth cells, where addition of Reg3γ to cultured colon epithelial lines enables them to withstand apoptotic stress. Thus, Reg3γ seems to possess 2 distinct roles in the protection of the crypt: (1) a previously established antimicrobial activity that lyses bacteria and (2) a survival signal that prevents inflammatory apoptosis of ISCs and Paneth cells. This insight may generate novel strategies for the prevention and treatment of GVHD by improving the ability of the target tissue itself to withstand immunologically mediated damage, such as has recently been hypothesized.40
Clinical GVHD
As stated earlier, the vast majority of mechanistic insights into acute GVHD pathophysiology derive from animal models. These insights have not yet led to validated therapeutic interventions, however, and no drugs have been approved for either the prevention or treatment of acute GVHD, which is due, in part, to the inability of clinical grading systems to predict the response to therapy. The clinical severity of acute GVHD manifests itself in 3 target organs, and grading systems aggregate the extent of damage in each organ: the body surface area of skin covered by rash, the rise in total bilirubin in the liver, and stool volume for the intestine. All clinical scoring systems have inherent limitations that may lead to inaccurate grading. For example, intestinal symptoms and hyperbilirubinemia can be caused by other concurrent conditions, stool volume measurement is unreliable, and skin rash measurement is subject to wide interobserver variability. Traditional scoring systems have 4 grades (Glucksberg grades 1-4 or International Bone Marrow Transplant Registry grades A-D), and transplant-related mortality significantly correlates with the 2 highest grades, which are often combined in analysis.41,42 These systems are most accurate in predicting mortality when maximum GVHD grade is considered, which can only be determined retrospectively. Outcomes for lower GVHD grades are heterogeneous and inconsistent between studies, because their demarcation is often not clear and they may progress after treatment. A recent development in this regard is the Minnesota score that assigns only 2 grades: standard risk (single-organ or limited 2-organ involvement) and high risk (all others).43 Standard-risk patients experience the same mortality as the overall group (standard and high risk combined), and low-risk patients are not identifiable in this system.
Acute GVHD biomarkers
The need for reliable, actionable information with better predictive power than clinical symptom severity has stimulated a vigorous effort to identify and validate serum biomarkers that can estimate risk for individual patients and help guide treatment decisions. Several categories of biomarkers have been evaluated, including microRNAs, proteins, and cellular subsets. The most promising candidates have been identified through proteomic screens using either array-based or mass spectrometry analyses.44
Two biomarkers with the strongest predictive power, ST2 and REG3A, derive from the GI tract and have complementary roles in the pathophysiology of GVHD as described above.45-47 Using samples from hundreds of patients enrolled in the multicenter Mount Sinai Acute GVHD International Consortium (MAGIC), we generated an algorithm that weights the concentration of each of these 2 biomarkers as measured by enzyme-linked immunosorbent assay (ELISA), which produces a predictive probability of nonrelapse mortality (NRM) for that individual patient. Thresholds have been tested and validated that divide patients into either 2 risk categories (high and low) or 3 risk categories (high, intermediate, and low). At the onset of GVHD, the algorithm divides patients into 3 groups (Ann Arbor Scores of 1-3) with distinct outcomes for response to systemic treatment and NRM (Figure 2).48 With use of more sensitive capture antibodies for ST2 in the ELISA, the 2-biomarker algorithm improves prediction, identifying 1/2 of the patients as low risk with an NRM at 6 months of 8%. These probabilities do not correlate with relapse, and thus, overall survival is also distinctly different in all 3 risk groups.

Risk stratification by application of a 2-biomarker MAGIC algorithm at the onset of GVHD. Six-month cumulative incidence of NRM in Ann Arbor risk groups AA1, AA2, and AA3 as defined by the MAGIC algorithm and applied to serum samples obtained at the onset of GVHD symptoms (n = 212): AA3: 46% (95% confidence interval, 32-58); AA2: 24% (95% confidence interval, 14-36); and AA1: 8% (95% confidence interval, 4-15). The proportions of patients in each risk group, as represented by the bar graph, were AA3: 27% (n = 57); AA2: 28% (n = 59); and AA1: 45% (n = 96). Adapted from Hartwell et al48 with permission.
Risk stratification by application of a 2-biomarker MAGIC algorithm at the onset of GVHD. Six-month cumulative incidence of NRM in Ann Arbor risk groups AA1, AA2, and AA3 as defined by the MAGIC algorithm and applied to serum samples obtained at the onset of GVHD symptoms (n = 212): AA3: 46% (95% confidence interval, 32-58); AA2: 24% (95% confidence interval, 14-36); and AA1: 8% (95% confidence interval, 4-15). The proportions of patients in each risk group, as represented by the bar graph, were AA3: 27% (n = 57); AA2: 28% (n = 59); and AA1: 45% (n = 96). Adapted from Hartwell et al48 with permission.
The same algorithm predicts outcomes for patients after 1 week of systemic treatment of GVHD, and it is particularly useful for patients who have not responded to therapy and are likely to have a poor outcome. A single threshold separates patients into 2 groups with markedly different mortality in the separate cohorts (Figure 3).49 The survival of the group with low NRM probabilities is equivalent to that of patients who had responded to treatment. Univariate analysis of relevant clinical variables shows that only biomarker probability, response to treatment, and Minnesota risk score at 1 week of therapy predict response after 4 weeks of systemic treatment and 1-year NRM. When these variables are compared through the creation of receiver operating characteristic curves, the area under the curve for biomarker probabilities (0.82) is significantly greater than that for clinical response (0.68) or Minnesota risk (0.72), providing sensitivity, specificity, positive predictive value, and negative predictive value of 74%, 83%, 58%, and 91%, respectively. The addition of clinical variables to biomarker probabilities do not significantly increase the predictive accuracy of the algorithm.

Long-term outcomes by biomarker probabilities in patients with GVHD that is resistant to early treatment. Patients with GVHD in a validation cohort (n = 68) who did not have a complete response to systemic treatment were subdivided into low (N = 49; blue) and high (N = 19; red) groups based on biomarker probabilities. Differences in 12-month cumulative incidence of NRM (low 14% vs high 75%, P < .001) and overall survival (low 78% vs high 14%, P < .001) were highly significant. Adapted from Major-Monfried et al49 with permission.
Long-term outcomes by biomarker probabilities in patients with GVHD that is resistant to early treatment. Patients with GVHD in a validation cohort (n = 68) who did not have a complete response to systemic treatment were subdivided into low (N = 49; blue) and high (N = 19; red) groups based on biomarker probabilities. Differences in 12-month cumulative incidence of NRM (low 14% vs high 75%, P < .001) and overall survival (low 78% vs high 14%, P < .001) were highly significant. Adapted from Major-Monfried et al49 with permission.
The 2-biomarker algorithm also provides useful information when used 1 week after HCT and before the onset of GVHD symptoms in any patients (Figure 4).48 In a large dataset (n = 1287), the algorithm identifies between 16% and 20% of all HCT patients with a 3 to 4 times higher risk for NRM. The algorithm identifies both high- and low-risk patients independent of the degree of HLA match between donor and recipient, graft source, or intensity of the conditioning regimen. For patients with an adverse clinical characteristic, the algorithm assigns a greater percentage of patients to the high-risk group (for example, 21% of unrelated donor grafts vs 12% of related donor grafts). As a result, the algorithm performs consistently across pretransplant risk factors, which may be attributable in part to the rigorous crossvalidation strategy in 75 different combinations of training sets used in its development.
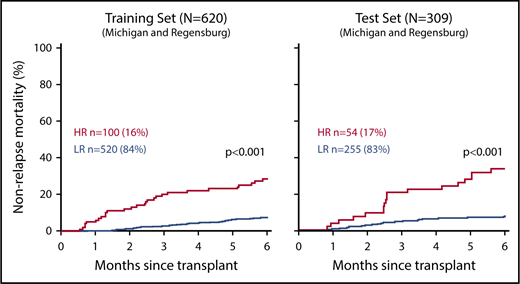
Cumulative incidence of 6-month NRM according to MAGIC risk stratification of samples from day +7 after HCT. High risk (HR; red) and low risk (LR; blue) were defined by the MAGIC algorithm using concentrations of 2 serum biomarkers (ST2 and REG3A) measured before the onset of any GVHD symptoms on day +7 of HCT and compared using Gray’s test. Training set (n = 620): HR: 28% (95% confidence interval, 20-37); LR: 7% (95% confidence interval, 5-10); test set (n = 309): HR: 33% (95% confidence interval, 21-46); LR: 7% (95% confidence interval, 5-11). In total, 16% and 17% of patients were identified as HR in the training and test sets, respectively. Adapted from Hartwell et al48 with permission.
Cumulative incidence of 6-month NRM according to MAGIC risk stratification of samples from day +7 after HCT. High risk (HR; red) and low risk (LR; blue) were defined by the MAGIC algorithm using concentrations of 2 serum biomarkers (ST2 and REG3A) measured before the onset of any GVHD symptoms on day +7 of HCT and compared using Gray’s test. Training set (n = 620): HR: 28% (95% confidence interval, 20-37); LR: 7% (95% confidence interval, 5-10); test set (n = 309): HR: 33% (95% confidence interval, 21-46); LR: 7% (95% confidence interval, 5-11). In total, 16% and 17% of patients were identified as HR in the training and test sets, respectively. Adapted from Hartwell et al48 with permission.
The ability of the algorithm to predict long-term outcomes before, during, and after the onset of GVHD symptoms likely relates to the fact that it quantitates damage to GI crypts in advance of lower GI symptoms. For example, in patients who had received systemic treatment for 1 week but were not experiencing diarrhea, biomarkers identified a high-risk group with at least 30% greater 1-year NRM, and most of them developed GI symptoms later in their course. Among patients with lower GI symptoms, those with unfavorable biomarkers experienced 4-fold greater mortality than those with favorable biomarkers.49 It is thus reasonable to consider the biomarker probability as a liquid biopsy of GI crypt damage, particularly relevant to the distal ileum with its preponderance of Paneth cells.
It is possible that additional biomarkers may improve the current 2-biomarker algorithm, and such possibilities will be easily tested in the future. The current 2-biomarker algorithm has sufficient sensitivity and specificity to be clinically useful in the setting of steroid-resistant GVHD. It should be emphasized that specific interventions based on biomarker probabilities have not yet been shown, but several clinical trials are currently in progress testing novel therapeutic strategies as either preemptive or primary treatment of acute GVHD in high-risk patients.
Patients with low-risk biomarkers will also be candidates for novel strategies. For example, patients presenting with skin disease and low probability biomarkers will be candidates for novel monotherapy strategies that avoid the toxicities of systemic steroids, such as increased risks of infection or relapse of malignancy. Such a trial has been conducted by the Bone Marrow Transplant Clinical Trials Network (Clinical Trials Identifier NCT02806947), and the results should be available in the near future. Serial biomarker measurements in asymptomatic patients early after HCT that show repeated low-risk status could act as an indication for a safe early taper of prophylactic immunosuppression to accelerate immunologic reconstitution. Such a taper could be stopped or modified if additional biomarker measurements showed evidence of subclinical disease. Such trials are currently in the design phase and eventually will provide the evidence needed to determine best practices in biomarker monitoring for the prevention and treatment of acute GVHD.
Acknowledgments
This work was supported by National Cancer Institute grants R21CA173459, P01CA039542, and P30CA196521 and American Cancer Society Clinical Research Professorship grant CRP-13-306-06-COUN (J.L.M.F.)
This article was selected by the Blood Advances and Hematology 2018 American Society of Hematology Education Program editors for concurrent submission to Blood Advances and Hematology 2018. It is reprinted in Hematology Am Soc Hematol Educ Program. 2018;2018:221-227.
Authorship
Contribution: M.S.C. drafted the manuscript and J.L.M.F. edited the manuscript and created the figures.
Conflict-of-interest disclosure: J.L.M.F. is a coinventor on a graft-versus-host disease biomarker patent. M.S.C. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence: James L. M. Ferrara, Hematologic Malignancies Translational Research Center, The Tisch Cancer Institute and Division of Hematology/Medical Oncology, Icahn School of Medicine at Mount Sinai, Hess Center for Science and Medicine, 1470 Madison Ave, 6th Floor, New York, NY 10029; e-mail: james.ferrara@mssm.edu.

