

Key Points
About half of the mutations other than KIT D816V identified in SM patients were germline in nature.
Somatic EZH2 gene mutations provide prognostic information in addition to that of the well-established S/A/R gene panel.

Abstract
Systemic mastocytosis (SM) is a highly heterogeneous disease with indolent and aggressive forms, with the mechanisms leading to malignant transformation still remaining to be elucidated. Here, we investigated the presence and frequency of genetic variants in 34 SM patients with multilineal KIT D816V mutations. Initial screening was performed by targeted sequencing of 410 genes in DNA extracted from purified bone marrow cells and hair from 12 patients with nonadvanced SM and 8 patients with advanced SM, followed by whole-genome sequencing (WGS) in 4 cases. Somatic mutations were further investigated in another 14 patients with advanced SM. Despite the fact that no common mutation other than KIT D816V was found in WGS analyses, targeted next-generation sequencing identified 67 nonsynonymous genetic variants involving 39 genes. Half of the mutations were somatic (mostly multilineal), whereas the other half were germline variants. The presence of ≥1 multilineal somatic mutation involving genes other than KIT D816V, ≥3 germline variants, and ≥1 multilineal mutation in the SRSF2, ASXL1, RUNX1, and/or EZH2 genes (S/A/R/E genes), in addition to skin lesions, splenomegaly, thrombocytopenia, low hemoglobin levels, and increased alkaline phosphatase and β2-microglobulin serum levels, were associated with a poorer patient outcome. However, the presence of ≥1 multilineal mutation, particularly involving S/A/R/E genes, was the only independent predictor for progression-free survival and overall survival in our cohort.
Introduction
Systemic mastocytosis (SM) comprises a heterogeneous group of hematological disorders that is characterized by the accumulation of abnormal mast cells (MCs) in multiple tissues that usually include the skin and bone marrow (BM).1 According to the World Health Organization (WHO) criteria,2,3 most SM patients (>90%) have indolent SM (ISM) and a normal life expectancy4-6 ; however, a fraction of the patients might present with (or progress to) advanced forms of the disease, such as aggressive SM (ASM), SM associated with another hematological neoplasm (SM-AHN), and, less frequently, MC leukemia (MCL).3-5,7 The mechanisms leading to malignant transformation of SM remain to be fully elucidated.
The KIT D816V somatic mutation is present in the majority of adult SM patients,8,9 particularly among ISM and ASM cases.10 Thus, although this KIT mutation might represent the genetic driver of SM, on its own it cannot explain malignant transformation of the disease. However, multilineal involvement of BM hematopoiesis by the KIT D816V mutation, found in approximately one third of ISM cases and the great majority of advanced forms of SM,6,10,11 particularly when this mutation is already present in an early pluripotent precursor cell also involving mesenchymal stem cells (MSCs), significantly enhances the probability of progression from ISM to advanced forms of SM.12 Altogether, these findings suggest that acquisition of additional genetic alterations along with the KIT mutation and/or the existence of a specific genetic background might be required for progression of ISM to more severe forms of the disease.13-15 Hence, recent studies based on relatively limited gene panels have shown that advanced forms of SM, including 177 of 284 SM-AHN cases, 28 of 284 ASM cases, and 8 of 284 MCL cases,13,15-18 often carry mutations in genes previously reported to be altered in other myeloid neoplasms,19-21 in addition to the KIT mutation. However, relatively limited information exists about the frequency of mutations in those genes in diagnostic subtypes of SM other than SM-AHN (eg, ISM and smoldering SM [SSM] in addition to ASM and MCL). Also, these mutations have been found in the other hematological neoplasm component of the disease but not in the MC compartment.22,23 In addition, it remains unknown whether the occurrence of such mutations in an early hematopoietic precursor would also confer a worse prognosis to SM patients, as demonstrated by Jawhar et al for SM-AHN cases17 and previously reported for KIT D816V.12
Here, we first investigated the presence and frequency of genetic variants, for a total of 410 genes, on purified BM MCs, maturing neutrophils, and T cells (plus hair in cases with multilineal gene involvement) from 20 SM patients presenting with a multilineal KIT D816V mutation, followed by whole genome sequencing (WGS) in 4 cases. In a second step, the somatic mutations identified were investigated in whole BM samples from another 14 advanced SM patients. To define the clonal hierarchy of the genetic variants identified, patients’ genomic (g)DNA obtained from different (purified) BM cell populations and hair was sequenced in parallel. Then the number and type of nonsynonymous (coding) genetic variants identified were compared among the distinct diagnostic subtypes of SM and related to patient outcome. Our results show, for the first time, a high frequency of germline mutations in advanced SM, in addition to the previously described somatic mutations, most of which were also found to involve multiple hematopoietic cell lineages. Somatic EZH2 gene mutations provide prognostic information, in addition to that of the well-established SRSF2, ASXL1, and/or RUNX1 (S/A/R) gene panel, in our series of SM patients.
Materials and methods
Patients
Overall, 34 patients (15 females and 19 males), 12 with nonadvanced SM but high-risk features (ie, ISM with multilineal involvement by the KIT D816V mutation and high serum baseline tryptase levels) and 22 with advanced forms of SM (ie, 4 SSM, 11 ASM, and 7 SM-AHN), diagnosed at the reference center (Virgen del Valle Hospital) of the Spanish Network on Mastocytosis (REMA) were studied (median age at diagnosis, 52 years; range, 0-76 years) based on (1) the presence of multilineal KIT D816V mutation, including involvement of MSCs in every ISM case tested, (2) high (>100 µg/L) tryptase serum levels, and (3) follow-up time from diagnosis ≥ 1 year (median, 6.5 years; range, 1-48). In an initial series (test series) of 20 patients (12 nonadvanced and 8 advanced SM cases; patients 1-20 in Table 1) sufficient (≥5 × 104) highly purified BM MCs, maturing neutrophils, and T cells were also required to perform further molecular analyses. In all 20 patients, hair was collected in parallel with the purified BM cell populations. In the additional 14 advanced SM cases (cases 21-34 in Table 1), availability of a whole BM sample was required to enter the study. Diagnosis and classification of SM were reviewed based on 2016 WHO criteria.2 All patients showed BM MC aggregates in histology with CD25+KIT D816V mutated and cytologically altered BM MCs; in addition, median serum baseline tryptase levels at diagnosis in nonadvanced and advanced SM cases were 268.5 µg/L (range, 115-1298) and 224 µg/L (range, 112-1469), respectively. To detect SM-AHN, conventional WHO cytomorphology and immunophenotypic criteria,2 based on the EuroFlow ALOT and acute myeloid leukemia (AML)/myelodysplastic syndrome (MDS) antibody panels,24 were used, respectively; at diagnosis, no patient showed BM infiltration by blast cells or other cells (in the absence of pathological MCs) compatible with a non-MC myeloid malignancy. Information about patient treatment is available in supplemental Table 7. Prior to entering the study, each individual gave his/her written informed consent to participate according to the Declaration of Helsinki, and the study was approved by the local institutional Ethics Committees. During the study period, 20 of 34 patients showed disease progression or died from SM-related causes, whereas the remaining 14 cases had stable disease (Table 1), after a median follow-up from initial diagnosis of 6.5 years. Briefly, 9 ISM cases progressed to SSM (n = 3) or ASM (n = 6), 8 ASM patients showed progression to ASM-AHN (n = 6) or died from SM-related causes (n = 2), and 3 SM-AHN patients died from SM-related causes. None of the patients showed progression to MCL, and none of the 34 SM patients had a history of familial mastocytosis.
Clinical and biological features of the 34 SM patients carrying the multilineal KIT mutation analyzed in this study
| Patient ID | Sex | Age, y | Diagnosis | Progression | PFS, y | Follow-up (at disease progression or last visit) | Alive | OS, y | Cause of progression | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WHO subtype | BM MC aggregates in histology | sBT, ng/mL | Age, y | WHO subtype | sBT, ng/mL | ||||||||
| 1 | M | 46 | ISM | + | 115 | No | 17 | 63 | ISM | 105 | Yes | 17 | – |
| 2 | M | 48 | ISM | + | 210 | No | 19 | 67 | ISM | 258 | Yes | 19 | – |
| 3 | M | 67 | ISM | + | 167 | No | 3 | 70 | ISM | 143 | Yes | 3 | – |
| 4 | F | 71 | ISM | + | 208 | Yes | 5 | 76 | SSM | 289 | Yes | 10 | sBT > 200; SPLEN |
| 5 | F | 39 | ISM | + | 175 | Yes | 15 | 54 | SSM | 240 | Yes | 17 | sBT > 200; SPLEN |
| 6 | M | 45 | ISM | + | 267 | Yes | 2 | 46 | SSM | 310 | Yes | 9 | HEP-SPLEN; DBS |
| 7 | M | 66 | ISM | + | 270 | Yes | 2 | 68 | ASM | 305 | No | 6 | HEP-SPLEN; NEUP |
| 8 | F | 11 | ISM | + | 1077 | Yes | 14 | 25 | ASM | 1970 | Yes | 30 | HEP-SPLEN*; DBS |
| 9 | M | 0 | ISM | + | 332 | Yes | 32 | 31 | ASM | 430 | Yes | 48 | HEP-SPLEN*; DBS |
| 10 | M | 32 | ISM | + | 1298 | Yes | 30 | 62 | ASM | 2036 | Yes | 39 | HEP*; DBS |
| 11 | M | 57 | ISM | + | 362 | Yes | 6 | 63 | ASM | 1507 | No | 7 | HEP*; IDA |
| 12 | F | 47 | ISM | + | 290 | Yes | 2 | 49 | ASM | 312 | No | 6 | SPLEN*; DBS; TRP |
| Subtotal | 66% M, 34% F | 46.5 (0-71) | 270 (167-1298) | 9/12 (75%) | 10 (2-32) | 62.5 (25-76) | 307.5 (105-2036) | 9/12 (75%) | 13.5 (3-48) | ||||
| 13 | M | 51 | ASM | + | 238 | No | 6 | 57 | ASM | 174 | Yes | 6 | – |
| 14 | F | 60 | ASM | + | 260 | No | 10 | 70 | ASM | 288 | Yes | 10 | – |
| 15 | M | 72 | ASM | + | 1469 | No | 4 | 76 | ASM | 1469 | Yes | 4 | – |
| 16 | M | 66 | ASM | + | 123 | Yes | 4 | 70 | SM-AHN | 53 | No | 4 | MPN |
| 17 | F | 37 | ASM | + | 201 | Yes | 1 | 38 | SM-AHN | NA | Yes | 3 | AML |
| 18 | M | 65 | ASM | + | 150 | Yes | 3 | 68 | SM-AHN | 516 | No | 4 | MDS |
| 19 | M | 45 | ASM | + | 548 | Yes | 4 | 49 | SM-AHN | 477 | No | 10 | AML |
| 20 | M | 58 | ASM | + | 178 | Yes | 1 | 59 | SM-AHN | 235 | No | 2 | MDS |
| 21 | F | 76 | ASM | + | 279 | Yes | 5 | 81 | ASM | 400 | No | 5 | SM-related death |
| 22 | F | 49 | SM-AHN | + | 180 | No | 10 | 50 | SM-AHN | 173 | Yes | 10 | – |
| 23 | F | 41 | SM-AHN | + | 184 | No | 3 | 44 | SM-AHN | 123 | Yes | 3 | – |
| 24 | F | 53 | SM-AHN | + | 159 | No | 3 | 56 | SM-AHN | 85 | Yes | 3 | – |
| 25 | F | 51 | SM-AHN | + | 112 | Yes | 11 | 62 | SM-AHN | 147 | No | 11 | SM-related death |
| 26 | F | 56 | SM-AHN | + | 1376 | Yes | 1 | 57 | SM-AHN | 1354 | No | 1 | SM-related death |
| 27 | M | 64 | ASM | + | 180 | Yes | 12 | 76 | SM-AHN | 107 | Yes | 12 | AML |
| 28 | M | 70 | ASM | + | 308 | Yes | 1 | 71 | ASM | 308 | No | 1 | SM-related death |
| 29 | M | 59 | SM-AHN | + | 160 | Yes | 1 | 60 | SM-AHN | 257 | No | 1 | SM-related death |
| 30 | M | 15 | SM-AHN | + | 210 | No | 13 | 28 | SM-AHN | 107 | Yes | 13 | – |
| 31 | F | 66 | SSM | + | 892 | No | 4 | 70 | SSM | 386 | Yes | 4 | – |
| 32 | M | 41 | SSM | + | 304 | No | 18 | 59 | SSM | 226 | Yes | 18 | – |
| 33 | F | 52 | SSM | + | 316 | No | 5 | 57 | SSM | 282 | Yes | 5 | – |
| 34 | F | 33 | SSM | + | 187 | No | 10 | 43 | SSM | 166 | Yes | 10 | – |
| Subtotal | 50% M 50% F | 54.5 (15-76) | 224 (112-1469) | 11/22 (50%) | 4 (1-18) | 59 (28-81) | 235 (53-1469) | 13/22 (59%) | 4.5 (1-18) | ||||
| Total | 56% M 44% F | 51.5 (0-76) | 249 (112-1469) | 20/34 (59%) | 4 (1-32) | 62 (25-81) | 282 (53-2036) | 22/34 (65%) | 6.5 (1-48) | ||||
| Patient ID | Sex | Age, y | Diagnosis | Progression | PFS, y | Follow-up (at disease progression or last visit) | Alive | OS, y | Cause of progression | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WHO subtype | BM MC aggregates in histology | sBT, ng/mL | Age, y | WHO subtype | sBT, ng/mL | ||||||||
| 1 | M | 46 | ISM | + | 115 | No | 17 | 63 | ISM | 105 | Yes | 17 | – |
| 2 | M | 48 | ISM | + | 210 | No | 19 | 67 | ISM | 258 | Yes | 19 | – |
| 3 | M | 67 | ISM | + | 167 | No | 3 | 70 | ISM | 143 | Yes | 3 | – |
| 4 | F | 71 | ISM | + | 208 | Yes | 5 | 76 | SSM | 289 | Yes | 10 | sBT > 200; SPLEN |
| 5 | F | 39 | ISM | + | 175 | Yes | 15 | 54 | SSM | 240 | Yes | 17 | sBT > 200; SPLEN |
| 6 | M | 45 | ISM | + | 267 | Yes | 2 | 46 | SSM | 310 | Yes | 9 | HEP-SPLEN; DBS |
| 7 | M | 66 | ISM | + | 270 | Yes | 2 | 68 | ASM | 305 | No | 6 | HEP-SPLEN; NEUP |
| 8 | F | 11 | ISM | + | 1077 | Yes | 14 | 25 | ASM | 1970 | Yes | 30 | HEP-SPLEN*; DBS |
| 9 | M | 0 | ISM | + | 332 | Yes | 32 | 31 | ASM | 430 | Yes | 48 | HEP-SPLEN*; DBS |
| 10 | M | 32 | ISM | + | 1298 | Yes | 30 | 62 | ASM | 2036 | Yes | 39 | HEP*; DBS |
| 11 | M | 57 | ISM | + | 362 | Yes | 6 | 63 | ASM | 1507 | No | 7 | HEP*; IDA |
| 12 | F | 47 | ISM | + | 290 | Yes | 2 | 49 | ASM | 312 | No | 6 | SPLEN*; DBS; TRP |
| Subtotal | 66% M, 34% F | 46.5 (0-71) | 270 (167-1298) | 9/12 (75%) | 10 (2-32) | 62.5 (25-76) | 307.5 (105-2036) | 9/12 (75%) | 13.5 (3-48) | ||||
| 13 | M | 51 | ASM | + | 238 | No | 6 | 57 | ASM | 174 | Yes | 6 | – |
| 14 | F | 60 | ASM | + | 260 | No | 10 | 70 | ASM | 288 | Yes | 10 | – |
| 15 | M | 72 | ASM | + | 1469 | No | 4 | 76 | ASM | 1469 | Yes | 4 | – |
| 16 | M | 66 | ASM | + | 123 | Yes | 4 | 70 | SM-AHN | 53 | No | 4 | MPN |
| 17 | F | 37 | ASM | + | 201 | Yes | 1 | 38 | SM-AHN | NA | Yes | 3 | AML |
| 18 | M | 65 | ASM | + | 150 | Yes | 3 | 68 | SM-AHN | 516 | No | 4 | MDS |
| 19 | M | 45 | ASM | + | 548 | Yes | 4 | 49 | SM-AHN | 477 | No | 10 | AML |
| 20 | M | 58 | ASM | + | 178 | Yes | 1 | 59 | SM-AHN | 235 | No | 2 | MDS |
| 21 | F | 76 | ASM | + | 279 | Yes | 5 | 81 | ASM | 400 | No | 5 | SM-related death |
| 22 | F | 49 | SM-AHN | + | 180 | No | 10 | 50 | SM-AHN | 173 | Yes | 10 | – |
| 23 | F | 41 | SM-AHN | + | 184 | No | 3 | 44 | SM-AHN | 123 | Yes | 3 | – |
| 24 | F | 53 | SM-AHN | + | 159 | No | 3 | 56 | SM-AHN | 85 | Yes | 3 | – |
| 25 | F | 51 | SM-AHN | + | 112 | Yes | 11 | 62 | SM-AHN | 147 | No | 11 | SM-related death |
| 26 | F | 56 | SM-AHN | + | 1376 | Yes | 1 | 57 | SM-AHN | 1354 | No | 1 | SM-related death |
| 27 | M | 64 | ASM | + | 180 | Yes | 12 | 76 | SM-AHN | 107 | Yes | 12 | AML |
| 28 | M | 70 | ASM | + | 308 | Yes | 1 | 71 | ASM | 308 | No | 1 | SM-related death |
| 29 | M | 59 | SM-AHN | + | 160 | Yes | 1 | 60 | SM-AHN | 257 | No | 1 | SM-related death |
| 30 | M | 15 | SM-AHN | + | 210 | No | 13 | 28 | SM-AHN | 107 | Yes | 13 | – |
| 31 | F | 66 | SSM | + | 892 | No | 4 | 70 | SSM | 386 | Yes | 4 | – |
| 32 | M | 41 | SSM | + | 304 | No | 18 | 59 | SSM | 226 | Yes | 18 | – |
| 33 | F | 52 | SSM | + | 316 | No | 5 | 57 | SSM | 282 | Yes | 5 | – |
| 34 | F | 33 | SSM | + | 187 | No | 10 | 43 | SSM | 166 | Yes | 10 | – |
| Subtotal | 50% M 50% F | 54.5 (15-76) | 224 (112-1469) | 11/22 (50%) | 4 (1-18) | 59 (28-81) | 235 (53-1469) | 13/22 (59%) | 4.5 (1-18) | ||||
| Total | 56% M 44% F | 51.5 (0-76) | 249 (112-1469) | 20/34 (59%) | 4 (1-32) | 62 (25-81) | 282 (53-2036) | 22/34 (65%) | 6.5 (1-48) | ||||
Subtotal and total results are expressed as percentage of cases (and range) for sex (male [M]/female [F]); as median (and range) for age, serum baseline tryptase (sBT), PFS, and OS; and as number of cases (and percentage) for progression and survival.
DBS, diffuse bone sclerosis; HEP, hepatomegaly; IDA, iron-deficiency anemia; MPN, myeloproliferative neoplasm; NEUP, neutropenia; SPLEN, splenomegaly; TRP, thrombocytopenia; +, positive; –, no progression.
With organ failure.
Purification of BM cell populations
Identification and isolation of antibody-stained (supplemental Table 1) BM MCs, maturing neutrophils, T cells, and MSC populations were performed in the initial cohort of 20 patients, as described in supplemental Methods, using well-established stain-and-then-lyse-and-wash procedures10 and a 4-way fluorescence-activated cell sorter (FACSAria III) equipped with FACSDiva software (both from BD, San Jose, CA), as described elsewhere.10,25 The purity of the FACS-sorted cells was systematically >98%, in the absence of cross-contamination by MCs (<0.001%) or any other KIT D816V+ BM cell population.
Analysis of the KIT D816V mutation
Positivity for the KIT D816V mutation was assessed in gDNA of FACS-purified BM MCs, maturing neutrophils, T cells, and MSCs using a quantitative real-time allele-specific oligonucleotide PCR method, as described.26,27
Targeted gDNA sequencing
Overall, 40 ng of gDNA from purified BM MCs and maturing neutrophils was used for targeted sequencing of all exons of 409 genes in a first cohort of 20 cases (patients 1-20 in Table 1). For this purpose, the AmpliSeq Comprehensive Cancer Panel was analyzed on an Ion Proton platform (both from Life Technologies, Carlsbad, CA), according to the manufacturer’s instructions. Only those nonsynonymous coding genetic variants identified based on the GRCh37 reference genome28 with ≥100× allele coverage were selected.
Subsequently, the following additional filters were applied to discriminate between acquired genetic variants (ie, somatic mutations) and germline mutations vs genetic variants present in the Spanish population (ie, single nucleotide polymorphisms). First, the above results were compared with the 5000 exomes,29 the IBS 1000 Genome Project,30 and the ExAC31 population databases. Then, only those genetic variants that had not been previously reported in Spanish and/or in European (non-Finnish) populations and/or those with a minor allele frequency <0.001 were considered in this study. Subsequently, the exome of 36 control gDNA samples representative of the Spanish healthy population (Spanish National DNA Bank Carlos III, University of Salamanca, Salamanca, Spain; http://www.bancoadn.org), pooled into 3 groups of 12 individuals per group, was sequenced to discriminate between the actual genetic variants found in SM patients and technical sequencing artifacts. Then, potentially deleterious mutations, as defined by the SIFT32 and PolyPhen33 algorithms, were selected and confirmed by Sanger sequencing on gDNA extracted from purified BM MCs, neutrophils, and T lymphocytes. Finally, the somatic vs germline nature of the genetic variants was evaluated by Sanger sequencing of gDNA obtained from paired patient’s hair. Germline mutations were defined as those gene variants detected in gDNA from hair with an allele frequency ∼50%. In 1 patient (case 9 in Table 1), the germline nature (vs early acquisition of the mutation during embryonic development) could be tested (and confirmed) in gDNA from his mother’s hair. Additionally, the SRSF2-p.P9534 mutational hotspot was investigated through Sanger sequencing of BM-derived gDNA from all 20 patients.
The new mutations identified were classified according to their pattern of distribution in different cell compartments, as follows: (1) hematopoietic (acquired) somatic mutations either restricted to MCs or (2) shared by MCs and other myeloid and/or lymphoid cells (ie, multilineal mutations) and (3) inherited rare germline variants or mutations acquired early during embryonic development, when the genetic variant was also present in gDNA from hair (supplemental Table 2).
Based on the somatic mutations found, a customized library for target sequencing of 15 genes (ie, ASXL1, CDH11, DNMT3A, EPHA7, EZH2, ICK, IKZF1, ITGA10, KAT6B, PIK3CD, ROS1, RUNX1, SF3B1, SRSF2, and TET2) was designed and analyzed in an additional cohort of 14 advanced SM patients (4 SSM, 3 ASM and 7 SM-AHN; patients 21-34 in Table 1). A total of 200 ng of BM gDNA per patient was used to prepare DNA libraries with a TruSeq Custom Amplicon Low Input kit (Illumina, San Diego, CA) and sequenced at 2 × 150-bp read length on an Illumina HiSeq 2500 genome sequencer, following the manufacturer’s instructions.
WGS
gDNA from purified BM MCs and T lymphocytes from patients 9, 10, 13, and 14 was used to generate a single short-insert library. Libraries were sequenced on an Illumina HiSeq 2500 instrument to generate paired-end 2 × 100-bp reads at a minimum of 110 G of data per sample (average coverage of 48.4 total reads; range, 41.9-53.9). The raw reads from both cell population libraries were processed and aligned to the GRCh37/hg19 reference sequence28 according to GATK best practices.35 Bulk read statistics were calculated using samtools36 (version 1.3.1, stats subcommand). Exon coverage was calculated using bedtools37 (version 2.26.0, coverage subcommand) with exon ranges from UCSC Known Genes38 downloaded with the UCSC Table Browser.39 Only those genetic variants with ≥20× allele coverage were considered. Sequencing metrics for each specimen are provided in supplemental Table 3.
Statistical analyses
The Kruskal-Wallis and Mann-Whitney U tests were used to assess the statistical significance (set at P < .05) of differences observed among groups. Overall survival (OS; calculated from the time of diagnosis to death or the last follow-up visit) and progression-free survival (PFS; calculated from the time of diagnosis to disease progression/death or the last follow-up visit in case of stable disease) curves were plotted according to the Kaplan-Meier method and compared using the Breslow (ie, generalized Wilcoxon) test, as suggested by Bouliotis and Billingham40 for data with a nonproportional hazard pattern. Receiver operating characteristic curve analysis was used to identify the most sensitive cutoff for the number of genetic variants that discriminated between patients with distinct OS and PFS. For multivariate analyses, the covariate adjustment model41 was applied to those variables found to be statistically different in the univariate analysis, and the Cox proportional hazard regression model was then used. SPSS software (SPSS version 20.0; IBM Corporation, Armonk, NY) was used for all statistical analyses.
Results
Nonsynonymous coding genetic variants identified in SM patients
In addition to KIT D816V, targeted next-generation sequencing (NGS) analyses of purified BM cell populations obtained from the first cohort of 20 SM patients showed 37 583 genetic variants (median: 1347 per case; range: 662-2431) that resulted in 52 nonsynonymous coding genetic variants involving 39 genes, after excluding population allele filters and technical artifacts (supplemental Table 2). Only 10 of 39 genes (ASXL1, DCC, DNMT3A, EZH2, IKZF1, LRP1B, RUNX1, SF3B1, SRSF2, and TET2) were recurrently mutated (ie, present in ≥2 patients) (supplemental Table 2). Interestingly, 46% (24/52) of the nonsynonymous coding genetic variants found are reported here for the first time (supplemental Table 2).
Fifteen of the 39 mutated genes harbored half of the genetic variants detected (26/52), which were confirmed to be somatic mutations present in hematopoietic cells but absent in hair gDNA; in contrast, the remaining 24 mutated genes carried germline genetic variants (or early acquired mutations), because they were present at ∼50% allele burden in all hematopoietic cells tested and in the patients’ hair (supplemental Table 2). Of note, the germline nature of the IGF2R mutation in patient 9 was confirmed in gDNA extracted from his mother’s hair. Also, most somatic mutations (20/26, 77%) corresponded to multilineal mutations that involved myeloid (17/26, 65%) or myeloid plus lymphoid (3/26, 12%) hematopoietic BM cells, in addition to MCs, and only a few mutations (6/26, 23%) were restricted to the MC compartment in BM (supplemental Table 2).
Subsequent WGS analysis of BM MCs and T cells from 4 ASM cases showed a genome-wide mutation frequency of between 0.008 and 0.19 mutations per megabase (Mb; average of 0.12 mutations per Mb) (Table 2). After applying filters to discriminate for MC-specific genetic variants (supplemental Methods), only 10 nonsynonymous coding or canonical splice site MC-specific variants (range: 0-4 per case) were found (supplemental Table 5). Only 1 of 10 MC-specific genetic variants identified in the PIK3CD gene by WGS analysis was also screened by targeted NGS. Interestingly, similar allele burdens were found, despite the distinct sequencing methods used (ie, 33% for WGS and 37% for targeted NGS).
Number and type of genetic variants identified in purified BM MCs from SM patients by WGS (n = 4)
| Patient ID | SNVs | Indels | Total variants (SNVs + indels) | Mutations per Mb |
|---|---|---|---|---|
| 9 | 15 | 7 | 22 | 0.008 |
| 10 | 486 | 15 | 501 | 0.18 |
| 13 | 534 | 21 | 555 | 0.19 |
| 14 | 267 | 9 | 276 | 0.10 |
| Average | 326 | 13 | 339 | 0.12 |
| Patient ID | SNVs | Indels | Total variants (SNVs + indels) | Mutations per Mb |
|---|---|---|---|---|
| 9 | 15 | 7 | 22 | 0.008 |
| 10 | 486 | 15 | 501 | 0.18 |
| 13 | 534 | 21 | 555 | 0.19 |
| 14 | 267 | 9 | 276 | 0.10 |
| Average | 326 | 13 | 339 | 0.12 |
Indels, insertions and deletions; SNV, single nucleotide variant.
Ten of the 14 advanced SM patients included in the second patient cohort (71%) harbored a total of 15 mutations, other than KIT D816V, involving 9 genes, of which 2 (RUNX1, SRSF2) were recurrently mutated. Interestingly, 5 of these 15 mutations (33%) were reported here for the first time (supplemental Table 3).
Clonal hierarchy of somatic mutations involving hematopoietic cells
All 20 patients screened for mutations within purified BM cell populations showed multilineal involvement of BM hematopoietic cells by KIT D816V, and KIT D816V–mutated MSCs were detected in 11 of 13 cases tested, including 9 of 9 ISM cases (patients 2, 3, 5, 6, 7, 9, 10, 11, and 12) and 2 of 4 ASM cases (patients 13 and 14). Also, targeted NGS analyses showed that 11 of 20 patients carried additional mutations.
Detailed analysis of the mutated allele burden within the MC population and the different hematopoietic cell lineages involved (supplemental Table 2) revealed heterogeneous patterns of distribution of somatic mutations in hematopoietic cells. Briefly, KIT D816V apparently emerged as the first somatic mutation acquired by hematopoietic cells in 4 of 11 cases: patients 5 (ISM), 14 (ASM), 17 (ASM), and 18 (ASM) (supplemental Figure 1A). In 1 ISM patient (patient 7) who had KIT and RUNX1 mutations, the KIT mutation was associated with a greater degree of involvement of hematopoiesis (including KIT D816V–mutated MSCs), pointing out its potential emergence at an earlier stage than the RUNX1 mutation; however, lower percentages of mutated MCs for KIT vs RUNX1 (24% vs 50% allele burden, respectively) were observed, which may indicate that the 2 mutations might had been acquired in different clones/subclones, in the absence of chromosomal/genetic gains and/or losses (supplemental Figure 1B). Likewise, a pattern consistent with the coexistence of 2 different clones/subclones was found in 2 other ASM cases (patients 19 and 20) that progressed to ASM-AHN (supplemental Figure 1B). Of note, in patient 19, AML blasts identified at progression tested positive for the EZH2 mutation but lacked KIT D816V. In turn, in another ASM case (patient 16) that progressed to ASM-AHN, KIT D816V was a secondary event to the TET2 mutation (supplemental Figure 1C). Finally, in 2 ISM cases (patients 11 and 12) and 1 ASM case (patient 15), the precise clonal hierarchy could not be defined with the available data.
Mutational profiles in ISM vs ASM
A higher median number of nonsynonymous coding genetic variants per patient was detected in ASM (median, 3 mutations per case; range, 1-11 mutations per case) compared with ISM (median, 1 mutation per case; range, 0-4 mutations per case; P = .02) (Table 3). Likewise, similar differences were also found when we restricted the analysis to the number of somatic mutations that involved multiple BM hematopoietic cell lineages (Table 3): 3 of 12 ISM cases (25%) vs 7 of 8 ASM cases (88%) (P = .008). In contrast, similar numbers of somatic mutations restricted to the MC compartment in the BM and of germline mutations/variants were observed in ISM vs ASM patients (Table 3).
Number of genes carrying nonsynonymous coding genetic variants (somatic vs germline mutations) per SM patient grouped according to the distinct diagnostic subtypes of the disease
| Patient ID | WHO diagnostic subtype | Hematopoietic acquired somatic mutations | Germline genetic variants or early acquired mutations | Mutations, median (range), n | Patients with ≥2 mutations (%) | Total genetic variants | Mutations, median (range), n | Patients with ≥3 mutations (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MC restricted mutations | Mutations, median (range), n | Mutated patients (%) | Multilineal mutations | Mutations, median (range), n | Mutated patients (%) | Total somatic mutations | Mutations, median (range), n | Mutated patients (%) | ||||||||
| 1 | ISM | – | 0 (0-2) | 3/12 (25) | – | 0 (0-2) | 3/12 (25) | – | 0 (0-3) | 4/12 (33) | – | 1 (0-2) | 3/12 (25) | – | 1 (0-4) | 3/12 (25) |
| 2 | ISM | – | – | – | 1 (EP400) | 1 | ||||||||||
| 3 | ISM | – | – | – | 2 (RECQL4, NSD2) | 2 | ||||||||||
| 4 | ISM | – | – | – | 1 (DCC) | 1 | ||||||||||
| 5 | ISM | 1 (ITGA10) | – | 1 | – | 1 | ||||||||||
| 6 | ISM | – | – | – | – | – | ||||||||||
| 7 | ISM | – | 1 (RUNX1) | 1 | 2 (CSF1R, MARK4) | 3 | ||||||||||
| 8 | ISM | – | – | – | 1 (SYNE1) | 1 | ||||||||||
| 9 | ISM | – | – | – | 2 (IGF2R, ITPKA) | 2 | ||||||||||
| 10 | ISM | – | – | – | 1 (HSP90AA1) | 1 | ||||||||||
| 11 | ISM | 2 (EZH2, SF3B1) | 1 (DNMT3A) | 3 | – | 3 | ||||||||||
| 12 | ISM | 1 (IKZF1) | 2 (ASXL1, DNMT3A) | 3 | 1 (DCC) | 4 | ||||||||||
| 13 | ASM | – | 0 (0-1) NS | 2/8 (25) NS | – | 2 (0-5) P = .004 | 7/8 (88) P = .008 | – | 2 (0-5) P = .02 | 7/8 (88) P = .02 | 1 (SDHC) | 1 (0-6) NS | 3/8 (38) NS | 1 | 3 (1-11) P = .02 | 6/8 (75) P = .03 |
| 14 | ASM | 1 (PIK3CD) | 1 (EPHA7) | 2 | – | 2 | ||||||||||
| 15 | ASM | – | 2 (EZH2, IKZF1) | 2 | 1 (DST) | 3 | ||||||||||
| 16 | ASM | – | 2 (SRSF2, TET2) | 2 | 1 (CREBBP) | 3 | ||||||||||
| 17 | ASM | 1 (KAT6B) | 2 (ASXL1, RUNX1) | 3 | – | 3 | ||||||||||
| 18 | ASM | – | 3 (EZH2, ROS1, SF3B1) | 3 | 3 (EPHB6, LRP1B, RPS6KA2) | 6 | ||||||||||
| 19 | ASM | – | 1 (EZH2) | 1 | 3 (CYP2C19, LRP1B, TCF3) | 4 | ||||||||||
| 20 | ASM | – | 5 (CDH11, ICK, SRSF2, RUNX1, TET2) | 5 | 6 (ADGRB3, MBD1, MUC1, NFKB2, NOTCH4, SOCS1) | 11 | ||||||||||
| Patient ID | WHO diagnostic subtype | Hematopoietic acquired somatic mutations | Germline genetic variants or early acquired mutations | Mutations, median (range), n | Patients with ≥2 mutations (%) | Total genetic variants | Mutations, median (range), n | Patients with ≥3 mutations (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MC restricted mutations | Mutations, median (range), n | Mutated patients (%) | Multilineal mutations | Mutations, median (range), n | Mutated patients (%) | Total somatic mutations | Mutations, median (range), n | Mutated patients (%) | ||||||||
| 1 | ISM | – | 0 (0-2) | 3/12 (25) | – | 0 (0-2) | 3/12 (25) | – | 0 (0-3) | 4/12 (33) | – | 1 (0-2) | 3/12 (25) | – | 1 (0-4) | 3/12 (25) |
| 2 | ISM | – | – | – | 1 (EP400) | 1 | ||||||||||
| 3 | ISM | – | – | – | 2 (RECQL4, NSD2) | 2 | ||||||||||
| 4 | ISM | – | – | – | 1 (DCC) | 1 | ||||||||||
| 5 | ISM | 1 (ITGA10) | – | 1 | – | 1 | ||||||||||
| 6 | ISM | – | – | – | – | – | ||||||||||
| 7 | ISM | – | 1 (RUNX1) | 1 | 2 (CSF1R, MARK4) | 3 | ||||||||||
| 8 | ISM | – | – | – | 1 (SYNE1) | 1 | ||||||||||
| 9 | ISM | – | – | – | 2 (IGF2R, ITPKA) | 2 | ||||||||||
| 10 | ISM | – | – | – | 1 (HSP90AA1) | 1 | ||||||||||
| 11 | ISM | 2 (EZH2, SF3B1) | 1 (DNMT3A) | 3 | – | 3 | ||||||||||
| 12 | ISM | 1 (IKZF1) | 2 (ASXL1, DNMT3A) | 3 | 1 (DCC) | 4 | ||||||||||
| 13 | ASM | – | 0 (0-1) NS | 2/8 (25) NS | – | 2 (0-5) P = .004 | 7/8 (88) P = .008 | – | 2 (0-5) P = .02 | 7/8 (88) P = .02 | 1 (SDHC) | 1 (0-6) NS | 3/8 (38) NS | 1 | 3 (1-11) P = .02 | 6/8 (75) P = .03 |
| 14 | ASM | 1 (PIK3CD) | 1 (EPHA7) | 2 | – | 2 | ||||||||||
| 15 | ASM | – | 2 (EZH2, IKZF1) | 2 | 1 (DST) | 3 | ||||||||||
| 16 | ASM | – | 2 (SRSF2, TET2) | 2 | 1 (CREBBP) | 3 | ||||||||||
| 17 | ASM | 1 (KAT6B) | 2 (ASXL1, RUNX1) | 3 | – | 3 | ||||||||||
| 18 | ASM | – | 3 (EZH2, ROS1, SF3B1) | 3 | 3 (EPHB6, LRP1B, RPS6KA2) | 6 | ||||||||||
| 19 | ASM | – | 1 (EZH2) | 1 | 3 (CYP2C19, LRP1B, TCF3) | 4 | ||||||||||
| 20 | ASM | – | 5 (CDH11, ICK, SRSF2, RUNX1, TET2) | 5 | 6 (ADGRB3, MBD1, MUC1, NFKB2, NOTCH4, SOCS1) | 11 | ||||||||||
Results are expressed as number of genetic variants per case after classifying the genetic variants into hematopoietic restricted mutations and germline genetic variants or early acquired mutations (ie, during embryonic development).
NS, not statistically significantly different.
Impact of the mutational profile on the outcome of SM
Receiver operating characteristic curve analysis showed that the presence of ≥3 genetic variants in genes other than KIT was the most sensitive cutoff for discriminating between patients with distinct OS and PFS. Thus, SM patients who carried ≥3 nonsynonymous coding genetic variants other than KIT D816V showed a significantly shortened PFS (median: 3 vs 30 years) and OS (median: 6 years vs not reached) compared with patients with <3 mutations (P = .002 and P < .001, respectively) (Figure 1A). Similarly, the presence of ≥3 germline variants was also associated with a shorter PFS (median, 3 vs 14 years) and OS (median, 4 years vs not reached) compared with cases with <3 germline variants (P = .016 and P = .004, respectively) (Figure 1B), whereas the presence of ≥1 somatic mutation (other than KIT D816V) involving multiple hematopoietic cell lineages showed an adverse impact on PFS (median, 3 vs 30 years) and OS (median: 6 years vs not reached) compared with the absence of somatic mutations other than KIT D816V (P = .009 and P = .001, respectively) (Figure 1C). In contrast, the presence of somatic mutations (other than KIT D816V) restricted to the MC compartment showed no prognostic impact on PFS or on OS of SM patients (Figure 1D).
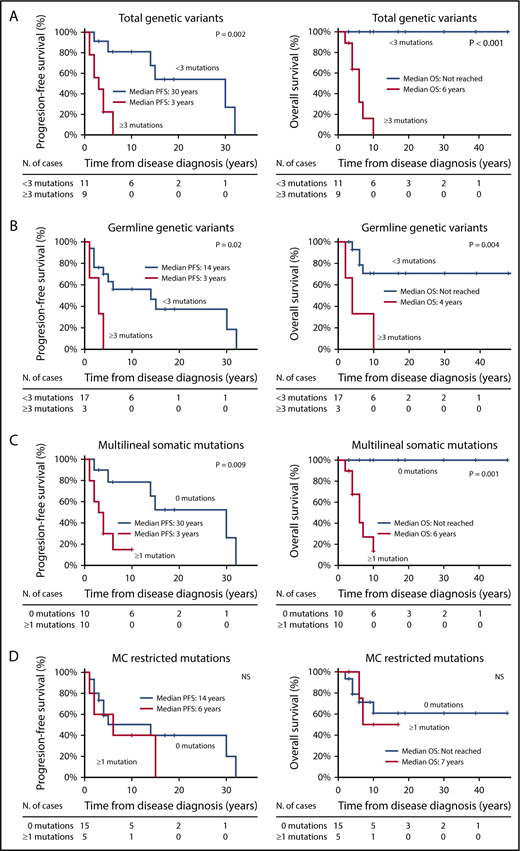
Kaplan-Meier estimates of PFS and OS of SM patients (n = 20) grouped according to the number and type of mutations detected. Panels describe PFS (left panels) and OS (right panels) of patients grouped according to the number of total genetic variants (A), germline genetic variants (B), multilineal somatic mutations (C), and MC restricted (somatic) mutations (D). Values are expressed in years from diagnosis to disease progression (PFS) and death (OS) or the last follow-up visit. Median PFS and/or OS indicates the time point at which half of the patients have progressed to more aggressive forms of the disease or died, respectively.
Kaplan-Meier estimates of PFS and OS of SM patients (n = 20) grouped according to the number and type of mutations detected. Panels describe PFS (left panels) and OS (right panels) of patients grouped according to the number of total genetic variants (A), germline genetic variants (B), multilineal somatic mutations (C), and MC restricted (somatic) mutations (D). Values are expressed in years from diagnosis to disease progression (PFS) and death (OS) or the last follow-up visit. Median PFS and/or OS indicates the time point at which half of the patients have progressed to more aggressive forms of the disease or died, respectively.
Interestingly, when we restricted the analysis to those mutations involving the SRSF2, ASXL1 and/or RUNX1 (S/A/R) gene panel2,17 among all 34 SM patients, a significant impact on PFS and OS (P < .001 and P = .002, respectively) was also observed (Table 4; Figure 2A). Interestingly, the status of the EZH2 gene (ie, the most frequently mutated gene among our patients) in S/A/R-negative patients further identified, among S/A/R-nonmutated cases, 2 patient groups with distinct outcomes (Figure 2B). Hence, ≥1 mutation in the new SRSF2, ASXL1, RUNX1, and/or EZH2 (S/A/R/E) gene panel showed an increased significant impact on PFS (P < .001) and OS (P < .001) vs S/A/R genes alone (Table 4; Figure 2C). Moreover, increased alkaline phosphatase serum levels (P = .01), thrombocytopenia (P = .03), and splenomegaly (P = .04) showed an impact on patients’ PFS (Table 4). In turn, the presence of skin lesions (P = .02), thrombocytopenia (P = .04), decreased hemoglobin levels (P = .001), and increased alkaline phosphatase (P = .01) and β2-microglobulin serum levels (P = .04) at diagnosis were all significantly associated with a shorter OS in the univariate analysis (Table 4).
SM patients: prognostic factors for PFS and OS (n = 34)
| Disease features | n | Univariate analysis | |||
|---|---|---|---|---|---|
| PFS, y | OS, y | ||||
| Median (range) | P | Median (range) | P | ||
| Clinical and laboratory features | |||||
| Diagnosis | |||||
| Nonadvanced SM | 12 | 14 (2-32) | NS | NR (3-48) | NS |
| Advanced SM | 22 | 11 (1-18) | 11 (1-18) | ||
| Age at diagnosis, y | |||||
| <60 | 23 | 14 (1-32) | NS | NR (1-48) | NS |
| ≥60 | 11 | 5 (1-10) | 6 (1-12) | ||
| Skin lesions | |||||
| No | 10 | 4 (1-6) | NS | 6 (1-13) | 0.02 |
| Yes | 24 | 14 (1-32) | NR (3-48) | ||
| BM MCs, % | |||||
| <1 | 14 | 5 (1-19) | NS | 11 (1-19) | NS |
| ≥1 | 18 | 14 (1-32) | NR (1-48) | ||
| sBT, µg/L | |||||
| <200 | 10 | 15 (1-17) | NS | NR (1-17) | NS |
| ≥200 | 22 | 6 (1-32) | NR (1-48) | ||
| Hemoglobin, g/L | |||||
| <100 | 5 | 3 (1-6) | NS | 4 (1-7) | .001 |
| ≥100 | 29 | 14 (1-32) | NR (1-48) | ||
| Platelets, ×109/L | |||||
| <100 | 11 | 4 (1-15) | .03 | 4 (1-17) | .04 |
| ≥100 | 23 | 14 (1-32) | NR (1-48) | ||
| β2-microglobulin, µg/mL | |||||
| <2.5 | 7 | NR (2-18) | NS | NR (3-17) | .04 |
| ≥2.5 | 20 | 5 (1-32) | 10 (1-48) | ||
| SAP, U/L | |||||
| <150 | 16 | 32 (2-32) | .01 | NR (3-48) | .01 |
| ≥150 | 14 | 4 (1-30) | 10 (1-39) | ||
| Splenomegaly | |||||
| No | 11 | 30 (1-30) | .04 | NR (1-39) | NS |
| Yes | 23 | 5 (1-32) | 11 (1-48) | ||
| Hepatomegaly | |||||
| No | 16 | 15 (1-19) | NS | NR (1-19) | NS |
| Yes | 18 | 5 (1-32) | 11 (1-48) | ||
| Number of nonsynonymous coding genetic variants | |||||
| Total somatic mutations | |||||
| 0 | 9 | 30 (2-32) | .02 | NR (3-48) | .005 |
| ≥1 | 11 | 4 (1-15) | 6 (2-17) | ||
| MC restricted | |||||
| 0 | 15 | 14 (1-32) | NS | NR (2-48) | NS |
| ≥1 | 5 | 6 (1-15) | 7 (6-17) | ||
| Multilineal | |||||
| 0 | 10 | 30 (2-32) | .009 | NR (3-48) | .001 |
| ≥1 | 10 | 3 (1-10) | 6 (2-10) | ||
| Germline genetic variants | |||||
| <3 | 17 | 14 (1-32) | .016 | NR (4-48) | .004 |
| ≥3 | 3 | 3 (1-4) | 4 (2-10) | ||
| Total genetic variants | |||||
| <3 | 11 | 30 (2-32) | .002 | NR (3-48) | <.001 |
| ≥3 | 9 | 3 (1-6) | 6 (2-10) | ||
| Gene panel mutational status | |||||
| S/A/R | |||||
| WT | 23 | 15 (2-32) | <.001 | NR (3-48) | .002 |
| Mutated | 11 | 2 (1-4) | 6 (1-13) | ||
| S/A/R/E | |||||
| WT | 19 | 30 (2-32) | <.001 | NR (3-48) | <.001 |
| Mutated | 15 | 3 (1-6) | 6 (1-13) | ||
| Disease features | n | Univariate analysis | |||
|---|---|---|---|---|---|
| PFS, y | OS, y | ||||
| Median (range) | P | Median (range) | P | ||
| Clinical and laboratory features | |||||
| Diagnosis | |||||
| Nonadvanced SM | 12 | 14 (2-32) | NS | NR (3-48) | NS |
| Advanced SM | 22 | 11 (1-18) | 11 (1-18) | ||
| Age at diagnosis, y | |||||
| <60 | 23 | 14 (1-32) | NS | NR (1-48) | NS |
| ≥60 | 11 | 5 (1-10) | 6 (1-12) | ||
| Skin lesions | |||||
| No | 10 | 4 (1-6) | NS | 6 (1-13) | 0.02 |
| Yes | 24 | 14 (1-32) | NR (3-48) | ||
| BM MCs, % | |||||
| <1 | 14 | 5 (1-19) | NS | 11 (1-19) | NS |
| ≥1 | 18 | 14 (1-32) | NR (1-48) | ||
| sBT, µg/L | |||||
| <200 | 10 | 15 (1-17) | NS | NR (1-17) | NS |
| ≥200 | 22 | 6 (1-32) | NR (1-48) | ||
| Hemoglobin, g/L | |||||
| <100 | 5 | 3 (1-6) | NS | 4 (1-7) | .001 |
| ≥100 | 29 | 14 (1-32) | NR (1-48) | ||
| Platelets, ×109/L | |||||
| <100 | 11 | 4 (1-15) | .03 | 4 (1-17) | .04 |
| ≥100 | 23 | 14 (1-32) | NR (1-48) | ||
| β2-microglobulin, µg/mL | |||||
| <2.5 | 7 | NR (2-18) | NS | NR (3-17) | .04 |
| ≥2.5 | 20 | 5 (1-32) | 10 (1-48) | ||
| SAP, U/L | |||||
| <150 | 16 | 32 (2-32) | .01 | NR (3-48) | .01 |
| ≥150 | 14 | 4 (1-30) | 10 (1-39) | ||
| Splenomegaly | |||||
| No | 11 | 30 (1-30) | .04 | NR (1-39) | NS |
| Yes | 23 | 5 (1-32) | 11 (1-48) | ||
| Hepatomegaly | |||||
| No | 16 | 15 (1-19) | NS | NR (1-19) | NS |
| Yes | 18 | 5 (1-32) | 11 (1-48) | ||
| Number of nonsynonymous coding genetic variants | |||||
| Total somatic mutations | |||||
| 0 | 9 | 30 (2-32) | .02 | NR (3-48) | .005 |
| ≥1 | 11 | 4 (1-15) | 6 (2-17) | ||
| MC restricted | |||||
| 0 | 15 | 14 (1-32) | NS | NR (2-48) | NS |
| ≥1 | 5 | 6 (1-15) | 7 (6-17) | ||
| Multilineal | |||||
| 0 | 10 | 30 (2-32) | .009 | NR (3-48) | .001 |
| ≥1 | 10 | 3 (1-10) | 6 (2-10) | ||
| Germline genetic variants | |||||
| <3 | 17 | 14 (1-32) | .016 | NR (4-48) | .004 |
| ≥3 | 3 | 3 (1-4) | 4 (2-10) | ||
| Total genetic variants | |||||
| <3 | 11 | 30 (2-32) | .002 | NR (3-48) | <.001 |
| ≥3 | 9 | 3 (1-6) | 6 (2-10) | ||
| Gene panel mutational status | |||||
| S/A/R | |||||
| WT | 23 | 15 (2-32) | <.001 | NR (3-48) | .002 |
| Mutated | 11 | 2 (1-4) | 6 (1-13) | ||
| S/A/R/E | |||||
| WT | 19 | 30 (2-32) | <.001 | NR (3-48) | <.001 |
| Mutated | 15 | 3 (1-6) | 6 (1-13) | ||
NR, not reached; NS, not statistically significant (P > .05); SAP, serum alkaline phosphatase; WT, wild-type.
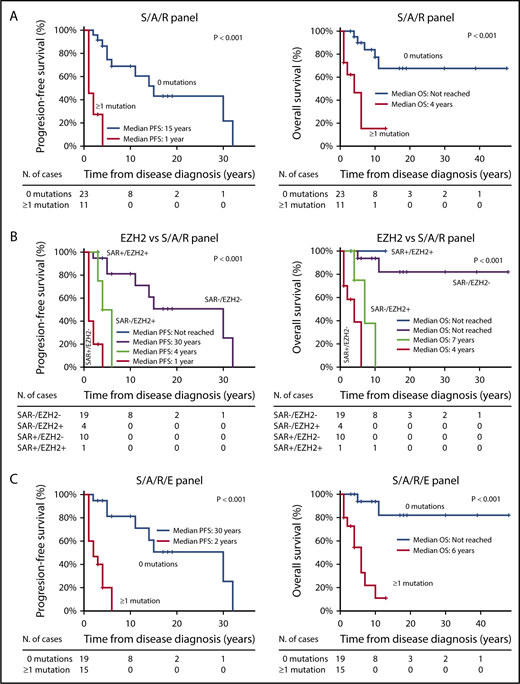
Kaplan-Meier estimates of PFS and OS of SM patients (n = 34) grouped according to the mutational status ofSRSF2,ASXL1,RUNX1 andEZH2 genes. PFS (left panels) and OS (right panels) of SM patients grouped according to the presence vs absence of mutated S/A/R genes (A), the EZH2 mutational status within the S/A/R gene panel (B), and the presence vs absence of mutated S/A/R/E genes (C). Values are expressed in years from diagnosis to disease progression (PFS) and death (OS) or the last follow-up visit. Median PFS and/or OS indicates the time point at which half of the patients have progressed to more aggressive forms of the disease or died, respectively.
Kaplan-Meier estimates of PFS and OS of SM patients (n = 34) grouped according to the mutational status ofSRSF2,ASXL1,RUNX1 andEZH2 genes. PFS (left panels) and OS (right panels) of SM patients grouped according to the presence vs absence of mutated S/A/R genes (A), the EZH2 mutational status within the S/A/R gene panel (B), and the presence vs absence of mutated S/A/R/E genes (C). Values are expressed in years from diagnosis to disease progression (PFS) and death (OS) or the last follow-up visit. Median PFS and/or OS indicates the time point at which half of the patients have progressed to more aggressive forms of the disease or died, respectively.
Multivariate analysis of prognostic factors, for the 20 SM patients in whom the somatic vs germline nature of the mutations identified was assessed, including those variables that had a significant impact on patient outcome in the univariate analysis (alkaline phosphatase serum levels and the number of multilineal, somatic, and total [somatic plus germline] mutations, for PFS, and the former variables plus platelet counts and the number of germline genetic variants, for OS), showed that the presence of ≥1 somatic mutation (other than KIT D816V) with multilineal hematopoietic involvement was the only independent predictor for PFS (P = .003; hazard ratio [HR], 2.8; 95% confidence interval [CI], 1.4-5.5) and OS (P = .002; HR, 9.3; 95% CI, 2.2-39.2). In turn, multivariate analysis of prognostic factors based on those variables with a significant impact in the univariate analysis that were available in all 34 SM patients studied (ie, alkaline phosphatase serum levels, platelet count, splenomegaly, and presence of S/A/R or S/AR/E gene mutations, for PFS, and these variables plus skin lesions and hemoglobin and β2-microglobulin serum levels, for OS) showed that the presence of S/A/R/E gene mutations (HR, 7.6; 95% CI, 2.2-26; P = .001 for PFS and HR, 13.1; 95% CI, 2.7-64; P = .001 for OS) was the only independent predictor for a worse patient outcome (Table 5).
SM: multivariate analyses of prognostic factors for PFS and OS for the entire patient series (n = 34)
| Disease features | n | PFS | OS | ||
|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | ||
| Clinical and laboratory features | |||||
| Hemoglobin, g/L | |||||
| <100 | 5 | NT | NS | ||
| ≥100 | 29 | ||||
| Skin lesions | |||||
| No | 10 | NT | NS | ||
| Yes | 24 | ||||
| Platelets, ×109/L | |||||
| <100 | 11 | NS | NS | ||
| ≥100 | 23 | ||||
| β2-microglobulin, µg/mL | |||||
| <2.5 | 7 | NT | NS | ||
| ≥2.5 | 20 | ||||
| SAP, U/L | |||||
| <150 | 16 | NS | NS | ||
| ≥150 | 14 | ||||
| Splenomegaly | |||||
| No | 11 | NS | NS | ||
| Yes | 23 | ||||
| Gene panel mutational status | |||||
| S/A/R | |||||
| WT | 23 | NS | NS | ||
| Mutated | 11 | ||||
| S/A/R/E | |||||
| WT | 19 | 7.6 (2.2-2.6) | .001 | 13.1 (2.7-64) | .001 |
| Mutated | 15 | ||||
| Disease features | n | PFS | OS | ||
|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | ||
| Clinical and laboratory features | |||||
| Hemoglobin, g/L | |||||
| <100 | 5 | NT | NS | ||
| ≥100 | 29 | ||||
| Skin lesions | |||||
| No | 10 | NT | NS | ||
| Yes | 24 | ||||
| Platelets, ×109/L | |||||
| <100 | 11 | NS | NS | ||
| ≥100 | 23 | ||||
| β2-microglobulin, µg/mL | |||||
| <2.5 | 7 | NT | NS | ||
| ≥2.5 | 20 | ||||
| SAP, U/L | |||||
| <150 | 16 | NS | NS | ||
| ≥150 | 14 | ||||
| Splenomegaly | |||||
| No | 11 | NS | NS | ||
| Yes | 23 | ||||
| Gene panel mutational status | |||||
| S/A/R | |||||
| WT | 23 | NS | NS | ||
| Mutated | 11 | ||||
| S/A/R/E | |||||
| WT | 19 | 7.6 (2.2-2.6) | .001 | 13.1 (2.7-64) | .001 |
| Mutated | 15 | ||||
NS, not statistically significant (P > 0.05); NT, not tested.
Discussion
Despite the (potentially) early acquisition of the KIT D816V mutation during ontogeny,12 the onset of clinical symptoms associated with advanced forms of SM often occurs at middle or late adulthood.5,42 Such observations suggest that continuous activation of the defective (ie, D816V-mutated) SCF/KIT pathway in an early progenitor cell does not confer a malignant phenotype per se, whereas it might facilitate malignant transformation of the disease, either because of an increased susceptibility to acquire secondary “oncogenic” mutations and/or by cooperating with a particular (preexisting) genetic background. Thus, results derived from murine models43,44 and SM patients13,42,43 indicate that coexistence of different mutations along with KIT D816V is required for the development of advanced SM. Hence, recent studies have found higher numbers of additional (somatic) mutations in advanced SM vs ISM patients.13 Despite these observations, neither additional somatic mutations (apart from KIT D816V) nor a specific genetic background have been reported in common among advanced SM cases.13,16-18,34,43,45-47
This is the first study in which a large number of genes that might be involved in malignant transformation of nonadvanced to advanced forms of SM have been investigated in a group of SM patients presenting with multilineal KIT D816V mutation and high serum tryptase levels. For this purpose, the complete coding regions of 410 genes (including the SRSF2 p.P95 hotspot) were first analyzed in gDNA from purified BM cell populations from 20 SM patients.
Despite the fact that progression of ISM to more aggressive forms of the disease is a relatively rare event,2 a high rate of progression was observed within our ISM cases; this high rate of progression could be explained by the patient-selection criteria (advanced SM patients plus nonadvanced ISM cases with high-risk features, such as high serum tryptase levels and multilineal involvement of the hematopoiesis by the KIT D816V mutation,6 including, in the great majority of cases, KIT D816V–mutated BM MSCs12 ) and the long follow-up of our cases.
Overall, no common mutation shared among ASM and ISM cases that showed disease progression was found. Hence, except for the IKZF1 p.N159S, RUNX1 p.R162K, and SRSF2 p.P95R mutations found in 2 patients each, all other (specific) genetic variants identified were detected in our series in a single case. In line with this as well, only 2 of 67 variants (SF3B1 p.K666T and SRSF2 p.P95R mutations) identified in this study had been reported in SM patients.16 Furthermore, WGS performed in a subset of 4 ASM patients (ie, ASM without an another hematological neoplasm [AHN]), confirmed the absence of common nonsynonymous (coding) genetic variants, other than KIT D816V. Interestingly, the mutation rate observed within purified BM clonal MCs in these 4 cases (0.12 per Mb) was significantly lower than that reported for other myeloid neoplasms48 and other types of cancer.49 Such a reduced overall mutational rate of ASM cases could be due to the lower genetic/genomic instability of advanced SM compared with other myeloid malignancies; however, despite the fact that the number of BM MCs is typically increased in ASM vs ISM, the most relevant “tumor” cell in advanced SM patients might be hematopoietic precursor cells10,50 and not pathological BM MCs, because all of these cases had multilineal involvement of the hematopoiesis by KIT D816V. If this holds true, neutrophils and, to a lesser extent, T cells would probably share most of the genetic variants found in pathological MCs. In line with this hypothesis, our results confirmed that most (77%) somatic mutations identified in BM MCs were also present in the myeloid or the myeloid plus lymphoid BM cells analyzed.
Surprisingly, half of all genetic variants found in BM MCs were also detected in gDNA from paired hair samples at ∼50% allele burden, suggesting that they would correspond to germline genetic events or to mutations acquired early during ontogeny. Interestingly, most patients (70%) carried ≥1 (potentially deleterious) germline genetic event for the 410 genes investigated in this study, which reveals an extremely high frequency compared with the 4% to 30% cases with germline mutations identified in pediatric and adult populations with other types of cancers.51-53 Despite this, the presence of germline mutations only showed a prognostic impact for PFS and OS when ≥3 germline variants were present in the same patient. However, all 3 cases displaying ≥3 germline variants (patients 18, 19, and 20) also carried a greater number of additional (multilineal) somatic mutations, identifying a potential greater genomic instability among these patients. This might also explain the greater and independent prognostic impact of the number of somatic vs germline mutations among our patients. Despite all of the above, the small number of cases analyzed here and the lack of uniform criteria across different laboratories to label a germline mutation as a deleterious variant,54 including the use of different filtering criteria for their identification, patients with germline mutations in cancer-related genes are being identified more frequently and, due to their distinct clinical behavior, they now represent a unique subtype of myeloid malignancies in the current WHO classification.3
Recently, age-related clonal hematopoiesis (ARCH) has been associated with somatic mutations in genes also found to be altered frequently in myeloid neoplasms (eg, the ASXL1, DNMT3A, EZH2, RUNX1, SF3B1, SRSF2, and TET2 genes) involving ∼10% of otherwise healthy individuals by the age of 70 years.55,56 Of note, these individuals would have a 12-fold higher relative risk for developing a hematological cancer; therefore, ARCH is considered an age-related preleukemic condition.57,58 In line with this finding, half of our SM patients who harbored ≥1 ARCH mutation (n = 10/18 cases; 56%) already exhibited an AHN at diagnosis or subsequently developed an AHN in addition to the SM, whereas these mutations affected a smaller percentage of SSM (11%) and ASM (33%) cases. These findings might suggest that acquisition of ARCH-associated gene mutations in ISM patients could be a trigger for a (myeloid) AHN, but not progression to SSM or ASM.
Analysis of the potential sequence of acquisition of somatic mutations in multimutated patients (coexistence of ≥2 somatic mutations) showed a potentially early acquisition of the KIT mutation in approximately one third of cases. In another third of our cases, the KIT D816V mutation emerged in a distinct clone or later than other multilineal somatic mutations involving, for example, the TET2, RUNX1, and EZH2 genes, with most of these patients having or developing SM-AHN. Altogether, these findings support previous observations in (mostly) SM-AHN patients that showed a predominance of gene mutations, other than KIT D816V alone or in combination with KIT mutations, in (in vitro) expanded colony-forming cells from SM patients.16 Moreover, in the only case in which we could confirm that the KIT mutation was a secondary genetic event, the development of a myeloid proliferative neoplasm occurred just prior to death. The apparent discrepancy between our data and previous data indicating that the KIT mutation might frequently occur as a distinct and late event16 in advanced mastocytosis might be due to the fact that all multimutated patients analyzed in such series (12/12) corresponded to SM-AHN patients16 with preferential growth of KIT wild-type vs KIT-mutated tumor cell precursors, whereas our series was selectively enriched for multilineal KIT-mutated ISM and ASM cases. However, clonogenic assays and/or single-cell analyses have not been performed in this study; therefore, our conclusions should be viewed with caution.
Interestingly, among our cases, most genes targeted by acquired somatic mutations, other than KIT D816V (ie, ASLXL1, DNMT3A, EZH2, IKZF1, RUNX1, SF3B1, SRSF2, TET2), had previously been found to be mutated in advanced SM13,16,17,34,46,47 and/or in other myeloid neoplasias,19,21,45,55,59-62 and their multilineal nature was shown in this study for the first time. In addition, the number of mutated genes (apart from KIT) with multilineal involvement of hematopoiesis increased significantly from ISM to ASM cases, in line with previous observations.17 Altogether, these findings further support the role of the KIT D816V mutation as a trigger for advanced SM under a “dangerous” genetic background, defined not only by the (number of) preexisting germline variants, as well as by the presence of (previously) acquired multilineal mutations in genes other than KIT. Actually, the presence of ≥1 somatic mutation with multilineal hematopoietic involvement, in addition to KIT, or the presence of ≥3 nonsynonymous coding genetic variants (including somatic and germline events) in the genes investigated in this study significantly increased the probability of progression of SM and of shortening the OS of SM patients.19,20,60
To explore the potential clinical utility of NGS screening based on a reduced number of genes, we investigated the prognostic impact of the previously reported (adverse) S/A/R gene panel17 in our patients. Our results confirmed that patients carrying ≥1 mutation in S/A/R genes had significantly poorer PFS and OS.17 However, inclusion of EZH2 into a S/A/R/E gene panel revealed that the combination of mutations in these 4 genes had greater predictive value for PFS and OS than did the well-established S/A/R gene panel, because S/A/R/E gene mutations were shown to be the only independent predictor for PFS and OS in our SM patients. Of note, EZH2 was 1 of the most frequently mutated genes in our series and 1 of the most frequently mutated genes in other cohorts of patients with SM13,16 and other myeloid neoplasms,19,20,63 where its mutations emerged as never being shared with SRSF2 mutations64,65 ; this might contribute to explain its additional prognostic information over the S/A/R gene mutations. This observation differs from data reported by other investigators17,62 who also tested the influence of mutations in the EZH2 gene; however, both studies17,62 included a significantly shorter median patient follow-up than ours (eg, 3.6 and 2 years vs 6.5 years, respectively), a period during which less than half of our ISM patients who progressed would have transformed to SSM and other forms of advanced SM. Thus, further investigations in larger series of SM patients, with a long follow-up, are required to confirm these findings.
In summary, our results show that no individual genetic lesion apart from the (multilineal) KIT mutation appears to be an independent predictor for malignant transformation of SM; however, progression of ISM to more advanced forms of the disease most likely requires an altered multimutated genetic background of both (KIT-mutated) tumor MCs and other hematopoietic BM cells. Germline and multilineal somatic mutations (particularly those involving the SRSF2, ASXL1, and RUNX1 genes in addition to the EZH2 gene) emerge as critical genetic markers associated with disease progression and a shortened OS for ISM patients at higher risk of disease progression.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Cell Sorting Service and Spanish National DNA Bank Carlos III (NUCLEUS, University of Salamanca) for technical assistance.
This work was supported by grants from the Instituto de Salud Carlos III (ISCIII) and fondos FEDER (PI16/00642 and CIBERONC [CB16/12/00400]), Madrid, Spain; Consejería de Educación, Junta de Castilla y León (SA013U16, FEDER), Valladolid, Spain; and Fundación Ramón Areces (CIVP16A1806), Madrid, Spain. M.J.-A. was supported by Ministerio de Economia y Competitividad (PTA-2016)–Universidad de Salamanca. A. Mayado was supported by CIBERONC (CB16/12/00400). J.I.S.-G. was supported by Consejería de Educación, Junta de Castilla y León (SA013U16), and FEDER. Spanish National DNA Bank Carlos III was supported by ISCIII and fondos FEDER (PT13/0001/0037 and PT17/0015/0044).
Authorship
Contribution: J.I.M.-G. performed experiments, analyzed data, and wrote the manuscript; M.J.-A., I.A.-T., and C.T. helped with the statistical analysis, the data analysis, and the review of the manuscript; C.C. and A. Mayado helped with data analysis and reviewed the manuscript; Y.H., K.M.R., and A.G.T. analyzed data and reviewed the manuscript; A.H. provided patient clinical data and reviewed the manuscript; L.S.-M., A. Matito, J.I.S.-G., and N.D.-F. reviewed the manuscript; and J.D.M., J.R.G., L.E., A.O., and A.C.G.-M. designed the experiments, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alberto Orfao, Centro de Investigación del Cáncer, Campus Miguel de Unamuno, 37007 Salamanca, Spain; e-mail: orfao@usal.es; and Andrés C. García-Montero, Centro de Investigación del Cáncer, Campus Miguel de Unamuno, 37007 Salamanca, Spain; e-mail: angarmon@usal.es.

