

Key Points
The NLRP3 inflammasome in platelets is upregulated in sickle cell disease.
High-mobility group box 1 and Bruton tyrosine kinase regulate the platelet NLRP3 inflammasome in sickle cell disease.

Abstract
A key inflammatory mechanism recently identified in platelets involves the Nod-like receptor nucleotide-binding domain leucine-rich repeat containing protein 3 (NLRP3) and Bruton tyrosine kinase (BTK), which control activation of caspase-1 within inflammasome complexes. We investigated platelet caspase-1 activity in the context of sickle cell disease (SCD) directly in platelets isolated from SCD patients (n = 24) and indirectly by incubating platelets from healthy subjects with plasma obtained from SCD patients (n = 20), both in steady state and during an acute pain crisis (paired samples). The platelet NLRP3 inflammasome was upregulated in SCD patients under steady state conditions compared with healthy controls, and it was further upregulated when patients experienced an acute pain crisis. The results were consistent with indirect platelet assays, in which SCD plasma increased caspase-1 activity of platelets from healthy subjects in an NLRP3-dependent fashion. The damage-associated molecular pattern molecule high-mobility group box 1 (HMGB1) was elevated in plasma of SCD subjects compared with healthy controls and correlated with caspase-1 activity in platelets. Pharmacological or antibody-mediated inhibition of HMGB1, Toll-like receptor 4, and BTK interfered with sickle plasma–induced platelet caspase-1 activation. In Townes SCD mice, caspase-1 activity and aggregation of circulating platelets were elevated, which was suppressed by IV injection of an NLRP3 inhibitor and the BTK inhibitor ibrutinib. Activation of the platelet NLRP3 inflammasome in SCD may have diagnostic and therapeutic implications.
Introduction
Platelets play a critical role in vascular inflammation and thrombosis, key features in sickle cell disease (SCD).1-6 Treatment options for SCD are still limited, which is explained in part by an incomplete understanding of the pathophysiological mechanisms of the disease. Several studies indicate that circulating platelets are activated in SCD patients in steady state and are further activated during acute pain crises.7-11 We and other investigators have shown that platelets express the pattern recognition receptor nucleotide-binding domain leucine-rich repeat containing protein 3 (NLRP3), apoptosis-associated speck-like protein containing a CARD (ASC), and Bruton tyrosine kinase (BTK), which control activation of caspase-1 and cleavage of interleukin-1β (IL-1β) within inflammasome complexes.12,13 Activation of the NLRP3 inflammasome in platelets promotes platelet aggregation, thrombus formation, and vascular leakage and can be targeted by BTK inhibitors.5,12-14 In patients with SCD, platelets secrete increased quantities of IL-1β,15 suggesting possible activation of the platelet NLRP3 inflammasome.
The damage-associated molecular pattern molecule high-mobility group box 1 (HMGB1) is a regulatory trigger of the NLRP3 inflammasome16 ; it induces thrombosis and inflammation when released by stressed or dying cells and activated platelets.17-19 A critical role for HMGB1 in promoting inflammation in SCD has been proposed recently, which was mediated via the pattern recognition receptor Toll-like receptor 4 (TLR4).20 TLR4 signaling is involved in activating the NLRP3 inflammasome, as shown in various disease states.21,22
We hypothesized that the NLRP3 inflammasome in platelets would be upregulated in SCD via HMGB1/TLR4 and that platelet BTK would play a significant role in regulating this process.
Methods
Patients and study design
We evaluated a total of 28 consecutive African American patients with SCD (HbSS) and 13 healthy race-matched controls (HbAA). The direct assays included platelets isolated from 24 patients in their usual state of health (steady state). Ten of the 24 patients also provided platelets during an acute pain crisis (paired samples). For the indirect assays, we used platelet-poor plasma (PPP) from 20 patients, all of whom provided samples in steady state and during crisis (paired samples). A pain crisis was defined as an episode of acute pain that has no evident cause other than SCD, resulting in hospitalization and treatment with parenteral narcotics.23 Steady state was defined as the period from at any time 8 weeks prior to or after a crisis. Sixteen patients were sampled for both direct and indirect assays (supplemental Table 1). Patients were excluded if they were <18 or >80 years of age, pregnant, or had a history of blood transfusion in the previous 8 weeks. The Institutional Review Board approved the studies under National Institutes of Health (NIH) protocol number 17-H-0056 for SCD subjects (clinical trials number NCT03049475) and 03-H-0015 for healthy controls. Written informed consent was obtained from all subjects. The study complied with the Declaration of Helsinki. All authors had access to and approval to access the primary clinical trial data.
Platelets and caspase-1 assay
Venous blood was drawn using a light tourniquet and collected in acid citrate dextrose buffer (Sigma-Aldrich). Direct assays were performed with platelets isolated within 2 hours, as described previously.18 For indirect assays, platelets from healthy controls were incubated for 1 hour with PPP from SCD or control subjects in the presence or absence of a neutralizing anti-HMGB1 antibody (BioLegend), an isotype-control antibody (BioLegend), or the HMGB1 inhibitor glycyrrhizin (Calbiochem) (all 10 µg/mL). For certain experiments, platelets were pretreated for 30 minutes with an inhibitor against NLRP3 (MCC950, 100 nM; Cayman Chemical),24 TLR4 (TAK242, 130 nM; InvivoGen),25 BTK (ibrutinib, 60 µM; Selleckchem),26 or a combination of the 3 inhibitors. Activation of caspase-1 in platelets was measured using a FAM FLICA Caspase-1 Assay Kit (Immunochemistry Technologies), according to the manufacturer’s protocol. Platelets were analyzed in a black 96-well microtiter plate using a plate reader for relative fluorescence units (RFUs).
Immunofluorescence staining of platelets and confocal microscopy
Platelets were fixed with 1% paraformaldehyde, applied to 0.01% poly-l-lysine–coated coverslips, and permeabilized with 0.3% Triton X-100. After blocking with 1% bovine serum albumin–phosphate buffered saline for 1 hour, cells were incubated overnight at 4°C with anti-NLRP3 monoclonal antibody (2 μg/mL, mouse immunoglobulin G2b [IgG2b]; AdipoGen). Platelets were washed with phosphate buffered saline plus 0.3% Triton X-100 plus 0.1% Tween-20 and incubated with Alexa Fluor 488–tagged goat anti-mouse IgG (1:100, Invitrogen) for 2 hours at room temperature. Following another washing step, platelets were incubated with anti-ASC polyclonal antibody (2 μg/mL, rabbit IgG; Biorbyt), washed, and incubated with Alexa Fluor 568–tagged donkey anti-rabbit IgG (1:100; Invitrogen). The corresponding IgG antibodies (AdipoGen and Biorbyt) served as control.
Confocal image stacks were acquired using a Plan-Apo 63× objective (NA 1.4) and a Zeiss 880 confocal laser scanning microscope (Carl Zeiss). Alexa Fluor 488 and Alexa Fluor 568 were excited by 488 and 568 nm excitation, and the emission was collected between 493 and 550 nm and between 568 and 630 nm for the 2 channels, respectively. All images were collected with the pinhole set to produce 1-μm slice thickness and voxel dimensions of 0.08 × 0.8 × 0.3 μm in X, Y, and Z axes, respectively. Image deconvolution was performed using Huygens Essential software (Scientific Volume Imaging), and colocalization analysis and 3-dimensional volumetric visualization were done using Imaris software (Bitplane). Images were exported to Adobe Photoshop for final preparation. Pearson correlation coefficients27 for the colocalization analysis were calculated after masking out noncellular areas of background and automatic thresholding.28 All images shown are representative of 3 independent preparations per group.
HMGB1 enzyme-linked immunosorbent assay
HMGB1 levels in PPP were determined using an HMGB1 ELISA Kit (Shino-Test), following the manufacturer’s protocol.
Platelet aggregometry
Platelet aggregation was evaluated using whole-blood impedance aggregometry (Model 700; Chrono-log), as described previously.13 Collagen (2 μg/mL; Chrono-log) was used as platelet agonist. Aggregation was measured for 6 minutes at 37°C with a stir speed of 1200 rpm. Analysis was performed using Aggrolink-8 software (Chrono-log). Data are reported as area under the curve (AUC), which incorporates the slope and amplitude of the aggregation curves.
SCD mice and inhibitor injections
All animal studies were approved by the NIH Clinical Center Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. We examined humanized SCD mice, the B6;129-Hbatm1(HBA)TowHbbtm2(HBG1,HBB*)Tow/Hbbtm3(HBG1,HBB)Tow/J strain, referred to herein as the Townes strain.29-31 These animals express only human hemoglobin (ie, no mouse hemoglobin) and are known to recapitulate many of the SCD human phenotypes. Control Townes mice express human hemoglobin A, heterozygous Townes mice express ∼30% human sickle hemoglobin and 70% hemoglobin A, and homozygous Townes mice express >94% human sickle hemoglobin.30,31 Mouse breeding and genotyping were performed as previously described.30
The NLRP3 inhibitor MCC 950 (50 mg/kg body weight), the BTK inhibitor ibrutinib (10 mg/kg body weight), or vehicle control (dimethyl sulfoxide) was injected IV (retro-orbitally) in homozygous, heterozygous, and control mice. Four hours later, blood was obtained via cardiac puncture for evaluation of platelet caspase-1 activity and platelet aggregation.
Statistical analysis
Data were analyzed using the Mann-Whitney U test or the Wilcoxon signed-rank test (both 2-tailed), as appropriate. The Friedman test with the Dunn multiple comparison was used for analysis of repeated-measures data with ≥3 conditions. Correlations between HMGB1 and platelet caspase-1 activity were examined using the Spearman correlation coefficient (ρ). For the confocal microscopy colocalization analysis, Pearson correlation coefficients were calculated.27 One-way and 2-way factorial analysis of variance (ANOVA) with the post hoc Bonferroni correction were used to analyze mice data. Data that did not have a normal distribution are shown as median with interquartile range (IQR) and minimum and maximum values. Normally distributed data are shown as mean ± standard deviation (SD).
Data sharing statement
Platelet caspase-1 activity data for all patient samples used in this study can be found in supplemental Table 1. Individual participant data will not be shared.
Results
Demographics and characteristics of the study subjects at the time of blood collection are summarized in Table 1.
Demographics and characteristics of SCD patients and healthy controls for direct and indirect platelet tests
| Variable | Healthy controls | SCD steady state | SCD acute pain crisis |
|---|---|---|---|
| Direct tests | |||
| No. of subjects | 13 | 24 | 10 |
| Age, median (IQR), y | 46 (26-52) | 36 (27-46) | 34 (28-38) |
| Females, n (%) | 7 (53.8) | 16 (66.7) | 8 (80) |
| No. on hydroxyurea (%) | 0 (0) | 20 (83.3) | 6 (60) |
| Hemoglobin, median (IQR), g/dL | 12.6 (11.7-14.8) | 8.8 (8.0-9.3) | 7.8 (7.0-8.9) |
| Reticulocyte count, median (IQR), % | 1.91 (1.36-2.36) | 9.31 (5.81-12.23) | 9.74 (7.96-13.65) |
| White blood cell count, median (IQR), ×109/L | 6.66 (5.01-8.22) | 7.25 (4.61-8.39) | 8.21 (6.64-14.33) |
| Platelet count, median (IQR), ×109/L | 271 (217-371) | 257 (162-355) | 359 (244-483) |
| Total bilirubin, median (IQR), mg/dL | 0.4 (0.3-0.6) | 1.8 (1.1-2.7) | 2.4 (1.7-3.9) |
| Lactate dehydrogenase, median (IQR), U/L | 194 (177-244) | 342 (201-503) | 294 (230-397) |
| High-sensitive C-reactive protein, median (IQR), mg/L | 2.7 (0.7-6.1) | 3.6 (1.3-6.6) | 6.5 (3.1-17.8) |
| Indirect tests | |||
| No. of subjects | 13 | 20 | 20 |
| Age, median (IQR), y | 46 (26-52) | 35 (29-41) | 35 (29-41) |
| Females, n (%) | 7 (53.8) | 14 (70) | 14 (70) |
| No. on hydroxyurea (%) | 0 (0) | 16 (80) | 16 (80) |
| Hemoglobin, median (IQR), g/dL | 12.6 (11.7-14.8) | 9.1 (8.0-9.8) | 8.1 (7.6-9.3) |
| Reticulocyte count, median (IQR), % | 1.91 (1.36-2.36) | 7.39 (5.77-12.09) | 9.24 (6.96-11.72) |
| White blood cell count, median (IQR), ×109/L | 6.66 (5.01-8.22) | 6.69 (4.89-8.80) | 7.10 (6.28-13.79) |
| Platelet count, median (IQR), ×109/L | 271 (217-371) | 310 (216-453) | 319 (224-454) |
| Total bilirubin, median (IQR), mg/dL | 0.4 (0.3-0.6) | 1.8 (1.3-2.3) | 1.7 (1.3-2.5) |
| Lactate dehydrogenase, median (IQR), U/L | 194 (177-244) | 317 (199-542) | 327 (224-508) |
| High-sensitive C-reactive protein, median (IQR), mg/L | 2.7 (0.7-6.1) | 4.0 (1.3-5.8) | 4.2 (1.2-9.9) |
| Variable | Healthy controls | SCD steady state | SCD acute pain crisis |
|---|---|---|---|
| Direct tests | |||
| No. of subjects | 13 | 24 | 10 |
| Age, median (IQR), y | 46 (26-52) | 36 (27-46) | 34 (28-38) |
| Females, n (%) | 7 (53.8) | 16 (66.7) | 8 (80) |
| No. on hydroxyurea (%) | 0 (0) | 20 (83.3) | 6 (60) |
| Hemoglobin, median (IQR), g/dL | 12.6 (11.7-14.8) | 8.8 (8.0-9.3) | 7.8 (7.0-8.9) |
| Reticulocyte count, median (IQR), % | 1.91 (1.36-2.36) | 9.31 (5.81-12.23) | 9.74 (7.96-13.65) |
| White blood cell count, median (IQR), ×109/L | 6.66 (5.01-8.22) | 7.25 (4.61-8.39) | 8.21 (6.64-14.33) |
| Platelet count, median (IQR), ×109/L | 271 (217-371) | 257 (162-355) | 359 (244-483) |
| Total bilirubin, median (IQR), mg/dL | 0.4 (0.3-0.6) | 1.8 (1.1-2.7) | 2.4 (1.7-3.9) |
| Lactate dehydrogenase, median (IQR), U/L | 194 (177-244) | 342 (201-503) | 294 (230-397) |
| High-sensitive C-reactive protein, median (IQR), mg/L | 2.7 (0.7-6.1) | 3.6 (1.3-6.6) | 6.5 (3.1-17.8) |
| Indirect tests | |||
| No. of subjects | 13 | 20 | 20 |
| Age, median (IQR), y | 46 (26-52) | 35 (29-41) | 35 (29-41) |
| Females, n (%) | 7 (53.8) | 14 (70) | 14 (70) |
| No. on hydroxyurea (%) | 0 (0) | 16 (80) | 16 (80) |
| Hemoglobin, median (IQR), g/dL | 12.6 (11.7-14.8) | 9.1 (8.0-9.8) | 8.1 (7.6-9.3) |
| Reticulocyte count, median (IQR), % | 1.91 (1.36-2.36) | 7.39 (5.77-12.09) | 9.24 (6.96-11.72) |
| White blood cell count, median (IQR), ×109/L | 6.66 (5.01-8.22) | 6.69 (4.89-8.80) | 7.10 (6.28-13.79) |
| Platelet count, median (IQR), ×109/L | 271 (217-371) | 310 (216-453) | 319 (224-454) |
| Total bilirubin, median (IQR), mg/dL | 0.4 (0.3-0.6) | 1.8 (1.3-2.3) | 1.7 (1.3-2.5) |
| Lactate dehydrogenase, median (IQR), U/L | 194 (177-244) | 317 (199-542) | 327 (224-508) |
| High-sensitive C-reactive protein, median (IQR), mg/L | 2.7 (0.7-6.1) | 4.0 (1.3-5.8) | 4.2 (1.2-9.9) |
Age is that at the time of blood collection. For the direct tests, assays were performed with platelets isolated within 2 hours of blood collection. For the indirect tests, platelets from healthy subjects were incubated for 1 hour with PPP from SCD patients or healthy controls.
Platelet caspase-1 activity is elevated in sickle cell patients in steady state and further increased during an acute pain crisis
Caspase-1 activity was significantly elevated in circulating platelets isolated from SCD patients in steady state compared with healthy race-matched controls (median RFU, 400.1; IQR, 357.8-467.3 vs median RFU, 291.0; IQR, 252.2-353.9; P < .001) (Figure 1A). In patients from which we had samples from steady state and from acute pain crisis, platelet caspase-1 activity was significantly increased during crisis (median RFU, 482.5; IQR, 392.7-657.5 vs median RFU, 390.4; IQR, 319.6-422.8; P < .01) (Figure 1B).
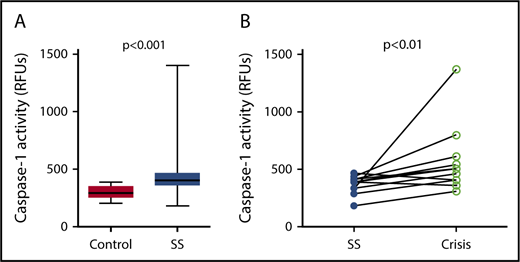
Platelet caspase-1 activity is upregulated in SCD patients in steady state and further increased during an acute pain crisis. Platelet caspase-1 activity is upregulated in SCD patients in steady state (SS) compared with healthy controls (A) and is further elevated when measured in the same SCD patients experiencing an acute pain crisis (paired samples) (B). Data are shown as median with IQR and minimum and maximum values (A) and before-after plot (B). The P values were calculated using the Mann-Whitney U test (A) and the Wilcoxon-signed rank test (B).
Platelet caspase-1 activity is upregulated in SCD patients in steady state and further increased during an acute pain crisis. Platelet caspase-1 activity is upregulated in SCD patients in steady state (SS) compared with healthy controls (A) and is further elevated when measured in the same SCD patients experiencing an acute pain crisis (paired samples) (B). Data are shown as median with IQR and minimum and maximum values (A) and before-after plot (B). The P values were calculated using the Mann-Whitney U test (A) and the Wilcoxon-signed rank test (B).
Activation of the platelet NLRP3 inflammasome in SCD
We sought to define the role of the platelet NLRP3 inflammasome in upregulating platelet caspase-1 activity in SCD. NLRP3 (green) and the adaptor protein ASC (red) were detected in platelets derived from healthy controls and SCD patients, as shown using immunofluorescence staining coupled with confocal laser scanning microscopy (Figure 2A). We determined colocalization of ASC with NLRP3 (quantified by Pearson correlation coefficient, Figure 2B), which was significantly increased in platelets from SCD patients in steady state compared with that in platelets from healthy controls (P < .001), indicating possible platelet NLRP3 inflammasome complex formation in SCD patients.
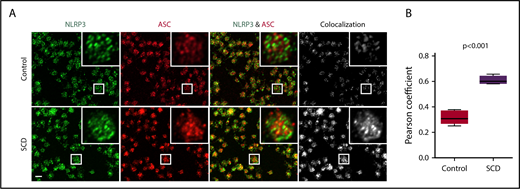
NLRP3 and ASC colocalize in platelets derived from SCD patients. (A) In platelets derived from SCD patients in steady state, intraplatelet ASC (red) colocalizes with NLRP3 (green), as demonstrated with immunofluorescence staining and confocal laser scanning microscopy. Images show maximum intensity projection of confocal images Z-stacks and are representative of 3 independent preparations per group. Scale bar, 2 μm. (B) Quantification of NLRP3/ASC colocalization using the Pearson correlation coefficient. Data are shown as median with IQR and minimum and maximum values from n = 3 subjects per group. The P value was calculated using the Mann-Whitney U test.
NLRP3 and ASC colocalize in platelets derived from SCD patients. (A) In platelets derived from SCD patients in steady state, intraplatelet ASC (red) colocalizes with NLRP3 (green), as demonstrated with immunofluorescence staining and confocal laser scanning microscopy. Images show maximum intensity projection of confocal images Z-stacks and are representative of 3 independent preparations per group. Scale bar, 2 μm. (B) Quantification of NLRP3/ASC colocalization using the Pearson correlation coefficient. Data are shown as median with IQR and minimum and maximum values from n = 3 subjects per group. The P value was calculated using the Mann-Whitney U test.
Next, we examined whether PPP from SCD patients would affect the NLRP3 inflammasome in platelets derived from healthy subjects (indirect platelet tests). PPP from SCD patients in steady state significantly upregulated platelet caspase-1 activity compared with PPP from healthy subjects (median RFU, 389.1; IQR, 350.3-443.1 vs median RFU, 323.1; IQR, 279.9-361.0; P < .01) (Figure 3A). Moreover, activation of platelet caspase-1 was significantly elevated upon incubation with PPP from SCD patients in acute pain crisis compared with PPP from the same subjects under steady state conditions (median RFU, 527.1; IQR, 459.3-612.4 vs median RFU, 389.1; IQR, 350.3-443.1; P < .001) (Figure 3B). The addition of an NLRP3 inhibitor (MCC950) to platelets significantly suppressed platelet caspase-1 activity (control: median percent inhibition, 17.8; IQR, 15.5-23.0; P < .01; steady state: median percent inhibition, 27.1; IQR, 20.8-29.4; P < .001; crisis: median percent inhibition, 30.3; IQR, 27.6-31.6; P < .001) (Figure 3C), indicating that platelet caspase-1 activation depends on NLRP3.
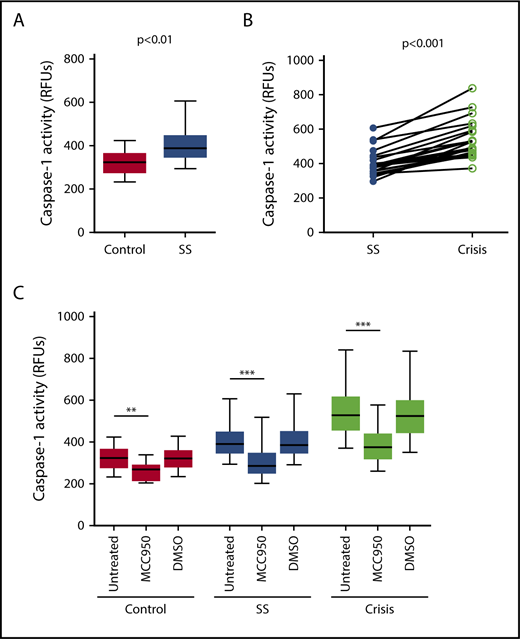
Plasma from SCD patients in steady state and crisis upregulates the NLRP3 inflammasome in platelets from healthy subjects. (A) PPP from SCD patients in steady state (SS) upregulates platelet caspase-1 activity compared with PPP from healthy controls. (B) Platelet caspase-1 activation is further elevated upon incubation with PPP from SCD patients in crisis compared with PPP from the same patients in SS. (C) An NLRP3 inhibitor (MCC950) inhibits caspase-1 activity of platelets in the presence of PPP from healthy subjects and SCD patients in SS and crisis. Data are shown as median with IQR and minimum and maximum values (A,C) and before-after plot (B). The P values were calculated using the Mann-Whitney U test (A), the Wilcoxon signed-rank test (B), and the Friedman test with Dunn multiple comparisons (C). **P < .01, ***P < .001.
Plasma from SCD patients in steady state and crisis upregulates the NLRP3 inflammasome in platelets from healthy subjects. (A) PPP from SCD patients in steady state (SS) upregulates platelet caspase-1 activity compared with PPP from healthy controls. (B) Platelet caspase-1 activation is further elevated upon incubation with PPP from SCD patients in crisis compared with PPP from the same patients in SS. (C) An NLRP3 inhibitor (MCC950) inhibits caspase-1 activity of platelets in the presence of PPP from healthy subjects and SCD patients in SS and crisis. Data are shown as median with IQR and minimum and maximum values (A,C) and before-after plot (B). The P values were calculated using the Mann-Whitney U test (A), the Wilcoxon signed-rank test (B), and the Friedman test with Dunn multiple comparisons (C). **P < .01, ***P < .001.
Upregulation of the platelet NLRP3 inflammasome in SCD depends on HMGB1/TLR4 and BTK
We assessed the underlying mechanism of platelet NLRP3 inflammasome activation in SCD. Plasma HMGB1 levels were significantly (P < .001) elevated in SCD patients in steady state compared with healthy controls (Figure 4A) and further elevated when SCD patients went into an acute crisis (P < .001) (Figure 4B). We found a strong correlation between plasma HMGB1 (healthy subjects and SCD patients) and plasma-induced platelet caspase-1 activity (ρ = 0.70, P < .001) (Figure 4C). A neutralizing anti-HMGB1 antibody significantly suppressed upregulated platelet caspase-1 activity induced by PPP from SCD patients in steady state and during crisis, but it had no significant effect in the presence of PPP from healthy subjects (steady state: median percent inhibition, 18.8; IQR, 16.7-24.1; P < .001; crisis: median percent inhibition, 25.5; IQR, 23.1-28.8; P < .001) (Figure 4D). Similar effects were observed with the HMGB1 inhibitor glycyrrhizin (steady state: median percent inhibition, 21.0; IQR, 18.4-27.9; P < .001; crisis: median percent inhibition, 27.2; IQR, 23.3-32.2; P < .001). Moreover, treatment of platelets with the TLR4 inhibitor TAK242 prior to incubation with PPP significantly inhibited platelet caspase-1 activity induced by PPP from steady state and crisis patients (steady state: median percent inhibition, 21.7; IQR, 18.3-30.0; P < .001; crisis: median percent inhibition, 26.3; IQR, 21.8-30.2; P < .001), a finding that did not occur in the presence of PPP from healthy controls (Figure 4E). The BTK inhibitor ibrutinib (PCI-32765) significantly suppressed PPP-induced caspase-1 activity in platelets (control: median percent inhibition, 13.1; IQR, 10.7-21.6; P < .01; steady state: median percent inhibition, 21.6; IQR, 18.1-24.5; P < .001; crisis: median percent inhibition, 27.1; IQR, 23.4-28.9; P < .001) (Figure 4E). Treatment of platelets with a combination of TAK242, MCC950, and ibrutinib had the strongest inhibitory effect on platelet caspase-1 activity (control: median percent inhibition, 15.4; IQR, 10.3-20.9; P < .001; steady state: median percent inhibition, 31.2; IQR, 25.8-36.3; P < .001; crisis: median percent inhibition, 33.1; IQR, 28.4-37.2; P < .001) (Figure 4E).
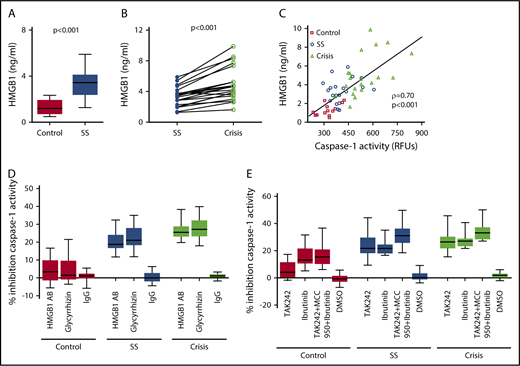
SCD-induced upregulation of the platelet NLRP3 inflammasome depends on HMGB1/TLR4 and BTK. (A) HMGB1 plasma levels are elevated in SCD patients in steady state (SS) compared with healthy controls. (B) HMGB1 plasma levels are elevated in SCD patients in crisis compared with the same patients in SS. (C) HMGB1 plasma levels in healthy subjects and SCD patients in SS and crisis correlate with plasma-induced platelet caspase-1 activity. (D) A neutralizing anti-HMGB1 antibody (HMGB1 AB) or the HMGB1 inhibitor glycyrrhizin inhibits platelet caspase-1 activity induced by PPP from SCD patients in SS and crisis and has no effect in the presence of PPP from healthy controls. (E) Preincubation of platelets with the TLR4 inhibitor TAK242 inhibits platelet caspase-1 activity induced by PPP from SCD patients in SS and crisis. Preincubation of platelets with the BTK inhibitor ibrutinib or a combination of TAK242, MCC950, and ibrutinib inhibits platelet caspase-1 activity in the presence of PPP from healthy and SCD subjects. Data are shown as median with IQR and minimum and maximum values (A,D-E), before-after plot (B), and scatter plot with linear regression (C). Statistical tests used: Mann-Whitney U test (A), Wilcoxon signed-rank test (B), Spearman rank correlation, ρ = correlation coefficient (C), and Friedman test with Dunn multiple comparisons (D-E).
SCD-induced upregulation of the platelet NLRP3 inflammasome depends on HMGB1/TLR4 and BTK. (A) HMGB1 plasma levels are elevated in SCD patients in steady state (SS) compared with healthy controls. (B) HMGB1 plasma levels are elevated in SCD patients in crisis compared with the same patients in SS. (C) HMGB1 plasma levels in healthy subjects and SCD patients in SS and crisis correlate with plasma-induced platelet caspase-1 activity. (D) A neutralizing anti-HMGB1 antibody (HMGB1 AB) or the HMGB1 inhibitor glycyrrhizin inhibits platelet caspase-1 activity induced by PPP from SCD patients in SS and crisis and has no effect in the presence of PPP from healthy controls. (E) Preincubation of platelets with the TLR4 inhibitor TAK242 inhibits platelet caspase-1 activity induced by PPP from SCD patients in SS and crisis. Preincubation of platelets with the BTK inhibitor ibrutinib or a combination of TAK242, MCC950, and ibrutinib inhibits platelet caspase-1 activity in the presence of PPP from healthy and SCD subjects. Data are shown as median with IQR and minimum and maximum values (A,D-E), before-after plot (B), and scatter plot with linear regression (C). Statistical tests used: Mann-Whitney U test (A), Wilcoxon signed-rank test (B), Spearman rank correlation, ρ = correlation coefficient (C), and Friedman test with Dunn multiple comparisons (D-E).
NLRP3 and BTK regulate platelet aggregation in sickle cell mice
We investigated activation of the platelet NLRP3 inflammasome in Townes SCD mice. Platelet caspase-1 activity was significantly increased in homozygous SCD mice compared with heterozygous mice (P < .01) and control mice (P < .001) (homozygous: RFU, 328.8 ± 40.6 [mean ± SD]; heterozygous: RFU, 190.4 ± 53.2 [mean ± SD]; control: RFU, 165.8 ± 21.6 [mean ± SD]) (Figure 5A). IV injection of MCC950 (50 mg/kg body weight) or ibrutinib (10 mg/kg body weight) significantly suppressed upregulated platelet caspase-1 activity in homozygous SCD mice (MCC950: 29.9% ± 12.6% inhibition, P < .01; ibrutinib: 33.3% ± 13.2% inhibition, P < .001), a finding that did not occur in heterozygous and control mice (Figure 5B).
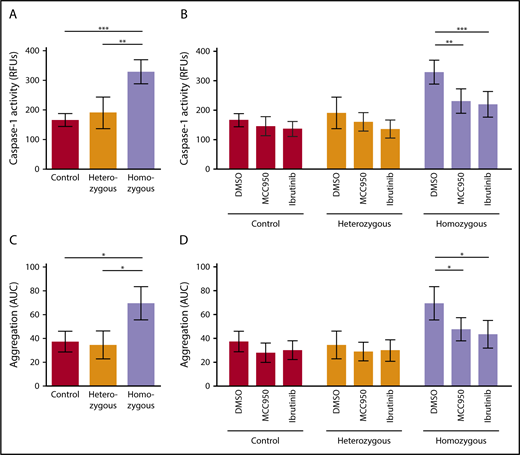
NLRP3 and BTK regulate platelet aggregation in sickle cell mice. (A) Platelet caspase-1 activity is increased in homozygous Townes SCD mice compared with heterozygous and control mice. (B) IV injection of MCC950 (50 mg/kg body weight) or ibrutinib (10 mg/kg body weight) suppresses upregulated platelet caspase-1 activity in homozygous SCD mice. (C) Platelet aggregation is elevated in homozygous Townes SCD mice compared with heterozygous and control mice. (D) IV injection of MCC950 or ibrutinib interferes with upregulated platelet aggregation in homozygous SCD mice. Data are shown as mean ± SD of the results from ≥3 separate experiments (n ≥ 3 mice per group). *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Bonferroni post hoc test (A,C), 2-way ANOVA with Bonferroni post hoc test (B,D).
NLRP3 and BTK regulate platelet aggregation in sickle cell mice. (A) Platelet caspase-1 activity is increased in homozygous Townes SCD mice compared with heterozygous and control mice. (B) IV injection of MCC950 (50 mg/kg body weight) or ibrutinib (10 mg/kg body weight) suppresses upregulated platelet caspase-1 activity in homozygous SCD mice. (C) Platelet aggregation is elevated in homozygous Townes SCD mice compared with heterozygous and control mice. (D) IV injection of MCC950 or ibrutinib interferes with upregulated platelet aggregation in homozygous SCD mice. Data are shown as mean ± SD of the results from ≥3 separate experiments (n ≥ 3 mice per group). *P < .05, **P < .01, ***P < .001, 1-way ANOVA with Bonferroni post hoc test (A,C), 2-way ANOVA with Bonferroni post hoc test (B,D).
We evaluated platelet aggregation in whole blood obtained from these mice. Aggregation of platelets was significantly increased in homozygous SCD mice compared with that in heterozygous mice (P < .05) and control mice (P < .05), as measured by AUC (mean ± SD) (69.5 ± 14.0, 34.6 ± 11.6, and 37.5 ± 8.6, respectively) (Figure 5C). Injection of MCC950 or ibrutinib significantly interfered with upregulated platelet aggregation in homozygous SCD mice (MCC950: 31.2% ± 13.9% inhibition, P < .05; ibrutinib: 37.2% ± 16.5% inhibition, P < .05) (Figure 5D), indicating that NLRP3 and BTK are involved in regulating platelet aggregation in SCD mice.
Discussion
In this study, we show that the NLRP3 inflammasome in platelets is upregulated in patients with SCD and that platelet NLRP3 activation is mediated via HMGB1/TLR4 and BTK. Additionally, we provide evidence that NLRP3 and BTK are involved in regulating platelet aggregation in SCD mice.
The NLRP3 inflammasome recognizes various pathogen-associated molecular patterns, which are predominantly derived from microbes, as well as damage-associated molecular patterns, which are host-derived signals originating from injured or stressed tissues.32 NLRP3 has been described in platelets for the first time in the context of dengue fever.12 In that study, the dengue virus triggered activation of the platelet NLRP3 inflammasome, which resulted in platelet shedding of IL-1β–rich microparticles and increased vascular permeability. In nucleated cells, activation of NLRP3 requires an initial priming signal, such as exposure to lipopolysaccharide, which results in upregulated expression of inflammasome components.33 Zhong et al have recently shown that priming and activation of the NLRP3 inflammasome in macrophages depend on the induction of new mitochondrial DNA synthesis.34 It is unclear whether platelets require such a priming signal due to constitutive expression of functionally active proteins NLRP3, ASC, and BTK.12,13 Interestingly, Brown et al have reported that lipopolysaccharide-induced activation of platelet TLR4 triggers IL-1β signaling in platelets through an autocrine loop.35 Here, we show that activation of the platelet NLRP3 inflammasome in SCD is mediated via HMGB1/TLR4. Autocrine and/or paracrine feed-forward loops on IL-1 receptor–expressing platelets might regulate the HMGB1/TLR4-driven platelet response that we observe in SCD.
Dysregulated activation of the NLRP3 inflammasome has been implicated in various human diseases, including rheumatoid arthritis, type 2 diabetes, and neurodegeneration.36 On the other hand, activation of the NLRP3 inflammasome in immune cells is an important step in host defense against infections, such as Staphylococcus aureus.37 Thus, therapeutically targeting the NLRP3 inflammasome, as with any immunosuppressive therapy, would bear an increased risk for infections and warrants a careful risk–benefit assessment in the context of the respective disease state. Although infections are a significant problem in patients with SCD, uncontrolled “sterile” inflammation plays a major role in the pathophysiology of SCD.2,3,6
Growing evidence on the role of platelets as sentinel innate immune cells demonstrates that platelets play a critical role in disorders associated with abnormal coagulation and inflammation.13,18,38-42 In SCD patients in steady state, circulating platelets are markedly activated, and platelet activation is further increased during acute pain crises and other acute events, such as acute chest syndrome.7-11,43 Activated platelets form aggregates with monocytes and neutrophils, which are significantly elevated in SCD patients44 and cause pulmonary arteriole microembolism in transgenic SCD mice.45 Moreover, platelets have been shown to control the formation of neutrophil–red blood cell aggregates in SCD patients, potentially contributing to vascular inflammation and occlusion.46
The platelet activation marker P-selectin is critically involved in triggering the formation of heterocellular aggregates with platelets. Injections of a neutralizing P-selectin antibody or oral treatment with the ADP receptor antagonist clopidogrel reduced the formation of platelet–neutrophil aggregates and improved lung vascular permeability in SCD mice.47 In a double-blind randomized phase 2 trial, crizanlizumab, a humanized monoclonal antibody against P-selectin, significantly decreased the rate of sickle cell–related pain crises in SCD patients, suggesting that P-selectin expressed by activated platelets and endothelial cells is a therapeutic target.48 On the other hand, the ADP receptor antagonist prasugrel did not significantly affect the rate of vaso-occlusive crises in children and adolescents with SCD in a phase 3 trial.49 The effectiveness of targeting platelets in SCD remains incompletely understood, may depend on the specific platelet target, and requires further studies. The NLRP3 inflammasome in platelets mediates platelet activation/aggregation and thrombus formation13,14 and triggers IL-1β–dependent inflammation and endothelial dysfunction,12,50 key features in SCD.2,4-6,15,51 In this study, IV injection of the NLRP3 inhibitor MCC950 interfered with caspase-1 activation and aggregation of circulating platelets in homozygous SCD mice, but it had no significant effect in heterozygous and control animals. The potential role of platelet NLRP3 inflammasome activation in promoting SCD vasculopathy is currently under investigation.
We and other investigators have recently shown that BTK expressed by immune cells26,52 and platelets13 is a critical regulator of the NLRP3 inflammasome. Ibrutinib is a potent BTK inhibitor that is approved by the US Food and Drug Administration for the treatment of B-cell malignancies. Here, we demonstrate that ibrutinib significantly suppressed upregulation of platelet caspase-1 activity in SCD patients and Townes SCD mice. Moreover, a single ibrutinib injection substantially decreased platelet aggregation in homozygous SCD mice. Despite its risk of exacerbating IL-1β–dependent infections,26 temporary BTK inhibition might have a benefit in breaking a vicious cycle of chronic vascular inflammation and abnormal coagulation in SCD by interfering with NLRP3 signaling. Further studies are needed to investigate NLRP3/BTK in platelets as a potential therapeutic target in SCD and evaluate its potential therapeutic efficacy in comparison with other platelet targets.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Neal Jeffries (National Heart, Lung, and Blood Institute, NIH) for providing valuable statistical advice.
This work was supported by the National Heart, Lung, and Blood Institute, NIH, and by the National Institutes of Health Clinical Center Intramural Research Program.
Authorship
Contribution: S.V., Z.M.N.Q., and S.L.T. designed the study, analyzed data, and wrote the manuscript; S.V. and T.A. performed experiments and analyzed data; X.W. and L.M. performed experiments; and J.N., D.A., A.S.S., and C.A.C. contributed to data acquisition and data analysis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sebastian Vogel, National Heart, Lung, and Blood Institute, National Institutes of Health, 10 Center Dr, Building 10, Room 6N240C, MSC 1589, Bethesda, MD 20814; e-mail: sebastian.vogel@nih.gov.

