

Key Points
Apparent ETV6-PDGFRA fusions identified by FISH analysis in t(4;12)(q12;p13) AML should be confirmed by sequencing.
Sequence-confirmed ETV6-PDGFRA fusions have not been identified in patients with t(4;12)(q12;p13) AML without eosinophilia.

Abstract
Acute myeloid leukemia (AML) with t(4;12)(q12;p13) translocation is rare and often associated with an aggressive clinical course and poor prognosis. Previous reports based on fluorescence in situ hybridization (FISH) analysis have suggested that ETV6::PDGFRA fusions are present in these patients, despite the absence of eosinophilia, which is typically found in other hematopoietic malignancies with PDGFRA-containing fusions. We first detected an ETV6-SCFD2 fusion by targeted RNA sequencing in a patient with t(4;12)(q12;p13) who had been diagnosed with an ETV6-PDGFRA fusion by FISH analysis but failed to respond to imatinib. We then retrospectively identified 4 additional patients with AML and t(4;12)(q12;p13) with apparent ETV6-PDGFRA fusions using chromosome and FISH analysis and applied targeted RNA sequencing to archival material. We again detected rearrangements between ETV6 and non-PDGFRA 4q12 genes, including SCFD2, CHIC2, and GSX2. None of the 3 patients who received imatinib based on the incorrect assumption of an ETV6-PDGFRA fusion responded. Our findings highlight the importance of using a sequencing-based assay to confirm the presence of targetable gene fusions, particularly in genomic regions, such as 4q12, with many clinically relevant genes that are too close to resolve by chromosome or FISH analysis. Finally, combining our data and review of the literature, we show that sequence-confirmed ETV6-PDGFRA fusions are typically found in eosinophilic disorders (3/3 cases), and patients with t(4;12)(q12;p13) without eosinophilia are found to have other 4q12 partners on sequencing (17/17 cases).
Introduction
Chromosomal rearrangements resulting in gene fusions play a significant role in tumorigenesis and are predicted to be a key driver in 20% of human cancers.1,2 The importance of fusion genes is particularly well recognized in hematologic malignancies and soft tissue tumors, where disease entities are often defined by the presence of a specific fusion.3,4 The identification of recurrent gene fusions in cancer has also led to the development of highly effective targeted therapies, such as imatinib for BCR-ABL1+ chronic myeloid leukemia and acute lymphoblastic lymphoma.5 Methods of identifying chromosomal rearrangements in common clinical use include chromosome banding analysis, fluorescence in situ hybridization (FISH) analysis, polymerase chain reaction (PCR)-based techniques, and various next-generation sequencing (NGS) platforms. These methods vary in availability, cost, turnaround time, and resolution. For example, chromosome banding analysis can identify chromosomal rearrangements, but the resolution of 5 to 10 Mb is insufficient to identify the exact genes involved. However, even with the higher resolution provided by FISH analysis (∼100 kb to 1 Mb), identification of fusions is highly dependent on the choice of probe, which requires preexisting knowledge of the gene targets and expected breakpoints.6 Similarly, PCR-based methods are rapid and cost effective but are only useful with well-defined targets. Although many sequencing platforms are not truly agnostic because they depend on primer selection, sequencing is the only modality that can efficiently identify novel breakpoints with base pair precision. In this article, we describe 5 patients who were initially diagnosed with ETV6-PDGFRA fusions based on chromosome and FISH analysis but were later found, using targeted RNA sequencing, to have ETV6 rearrangements that did not involve PDGFRA.
ETV6 is a ubiquitously expressed transcriptional repressor, in the ETS family of transcription factors, that is encoded by the ETV6 gene (previously known as TEL, for translocation-ETS-leukemia virus) located on chromosomal band 12p13. Band 12p13 is frequently involved in chromosomal translocations in hematologic malignancies, and ETV6 is the most commonly rearranged gene in this region in that context.7 ETV6 has 8 exons with the PNT (pointed) homodimerization domain encoded by exons 3 and 4 and the ETS DNA-binding domain encoded by exons 6 through 8.8 More than 40 ETV6-containing fusion genes have been reported, with the majority identified in hematologic malignancies and soft tissue tumors (see De Braekeleer et al9 and Biswas et al10 for additional details). Breakpoints are highly variable and can result in fusion genes missing either domain. The functional consequences of these rearrangements depend on the domain(s) involved, but most can be broadly grouped into (1) constitutive activation of the partner kinase, (2) modification of transcription factor function, (3) loss of function of ETV6 or its fusion partner, or (4) deregulation of nearby genes.9,11 Fusions that fall into mechanism group 1 are of particular clinical interest when the kinase is targetable with US Food and Drug Administration–approved medications, as is the case with PDGFRA, which is highly sensitive to imatinib.12
PDGFRA is a receptor tyrosine kinase subunit that is expressed in cells of mesenchymal origin and encoded by the PDGFRA gene located on chromosomal band 4q12. Like ETV6, numerous PDGFR fusion proteins have been identified in hematologic malignancies, although the majority involve PDGFRB.13 PDGFRA has a canonical receptor tyrosine kinase structure with N-terminal extracellular immunoglobulin-like domains, a juxtamembrane WW-like domain that is critical for autoregulation (exon 12), and 2 C-terminal intracellular tyrosine kinase domains (exons 14 and 18).8 Breakpoints in PDGFRA rearrangements typically involve exon 12, disrupting the autoinhibitory domain and resulting in chimeric proteins with an unregulated constitutively active kinase domain.14,15 Eight PDGFRA fusion partners have been described in the literature, with FIP1L1 being the most commonly identified (see Appiah-Kubi et al13 and Cools et al16 for additional details). These fusions are implicated primarily in clonal eosinophilias ranging from hypereosinophilic syndrome to leukemia and less commonly in noneosinophilic disorders, such as atypical chronic myeloid leukemia and B- and T-cell acute lymphoblastic leukemias (ALLs). Most of these cases respond well to the tyrosine kinase inhibitor imatinib, which was designed to target the ABL kinases but also inhibits the PDGF receptors.12
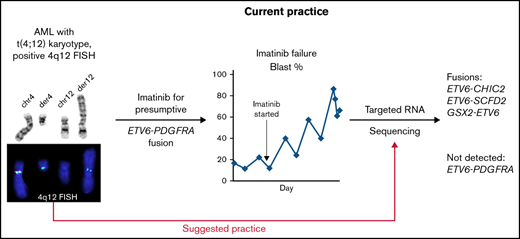
This series was prompted after a patient with t(4;12)(q12;p13) acute myeloid leukemia (AML) failed to respond to imatinib. Subsequent RNA sequencing identified an in-frame ETV6-SCFD2 fusion and no evidence to support the presumed ETV6-PDGFRA fusion based on FISH analysis. We then retrospectively identified 4 additional patients with similar findings and sequenced archival material, confirming ETV6-4q12 rearrangements that did not include PDGFRA. Three of the 5 patients had received imatinib based on the incorrect assumption of an ETV6-PDGFRA fusion, and none responded. None of these cases had associated eosinophilia. These findings highlight the importance of using a clinically validated sequencing assay to confirm the presence of a PDGFRA-containing fusion in patients with cytogenetic and/or FISH evidence of t(4;12)(q12;p13) rearrangements.
Methods
Chromosome analysis and FISH
GTG-banded metaphases were obtained from unstimulated 24-hour bone marrow (BM) cultures according to standard cytogenetic protocols. Interphase FISH analysis was performed on remaining fixed pellet BM cultures according to standard genetic protocols and the manufacturer’s recommended hybridization conditions. FISH probes were purchased from Abbott Molecular (Des Plaines, IL) and were used as follows: Vysis LSI ETV6 (Cen) and Vysis LSI ETV6 (Tel) probes at 12p13 to identify 12p13 rearrangements and Vysis LSI 4q12 Tri-color Rearrangement FISH Probe kit to detect 4q12 rearrangements. In all samples, a positive result was based on the cutoff value used by the Brigham and Women's Hospital cytogenetics laboratory for each probe.
Fusion detection
Fusion detection for all patients was performed at Massachusetts General Hospital. Using NGS and a clinically validated anchored multiplex PCR-based targeted assay (Heme Fusion Assay v3), fusion transcripts involving genes more commonly rearranged in hematological malignancies were identified.17,18 Briefly, total nucleic acid was isolated from BM aspirate or fixed pellets. Double-stranded complementary DNA was created and then end repaired, adenylated, and ligated with a half-functional adapter. Using ArcherDx Heme Fusion kit primers, 2 hemi-nested PCR reactions were performed to create a fully functional sequencing library that targets 86 genes (supplemental Figure 1). Illumina NextSeq 2 × 151 bp paired-end sequencing results were aligned to the hg19 human genome reference with BWA-MEM10, and a laboratory-developed algorithm was used for fusion transcript detection and annotation through split-read analysis of primary and secondary alignments. At least 5 reads per potential fusion transcript were manually checked for alignment to the hg19 human genome reference using the UCSC genome browser, and the reading frame was confirmed using ExPASy Translate.19,20
Detection of single nucleotide variants
Identification of single nucleotide variants and small insertions/deletions (indels) for patients 1 and 2 was performed at Massachusetts General Hospital. A clinically validated anchored multiplex PCR-based assay (Heme SNaPshot v3) targeting 103 genes was used for the detection of single nucleotide variants and indels in genes recurrently mutated in hematological malignancies using the ArcherDx platform and Illumina NextSeq NGS.17,21 Results were aligned to the hg19 human genome reference, and an ensemble-based variant calling approach and a laboratory-developed hotspot caller were applied for single nucleotide variant and indel variant detection. Analogous studies for patients 4 and 5 were performed at Brigham and Women’s Hospital using a clinically validated assay targeting 95 genes for the detection of single nucleotide variants and indels in genes recurrently mutated in hematological malignancies using the Illumina TruSeq Custom Amplicon platform.22 The commercially available MiSeq Reporter was used for alignment and single nucleotide and small indel variant calling. The custom FLT3 internal tandem duplication caller was developed using DotNetBio 3.0.
Literature review
We searched PubMed and Google Scholar for published English language studies with no constraint on publication year using the following terms (independent searches): t(4;12)(q12;p13); t(4;12) AND hematologic malignancy; t(4;12) AND AML; ETV6-PDGFRA; ETV6 AND AML; TEL AND AML; and PDGFRA AND AML. Seventy-six publications were reviewed in detail. Of those, publications in which the following conditions were met were included: the cases were of hematologic origin and there was ≥1 case with t(4;12)(q12;p13) karyotype or 3-way rearrangement involving 4q12 and 12p13, ETV6 was found to be rearranged by sequencing or FISH analysis, and the putative ETV6 partner on 4q12 was identified by sequencing or FISH analysis.
Results
In total, we retrospectively identified 5 patients with AML and t(4;12)(q12;p13) rearrangements identified by chromosome banding analysis with or without FISH analysis (Tables 1 and 2). As in prior studies of t(4;12)(q12;p13) AML,23-25 the majority of our patients had only a small subset of blasts that were CD7+ and MPO− or MPO+. Patients 1 and 3 progressed while on imatinib. Patient 4 had stable disease while on imatinib and hydroxyurea with a white blood cell count >70 000 per microliter and >90% blasts for 5 months before dying. Patient 2 was not given imatinib on diagnosis; however, long-term clinical follow-up was unavailable. The clinical team was alerted to the absence of a ETV6-PDGFRA fusion in patient 5 prior to initiating cytotoxic chemotherapy, precluding the initiation of imatinib.
FISH analysis was performed on abnormal metaphases and/or nuclei in patients 1 through 3 using a Vysis LSI 4q12 Tri-Color Rearrangement FISH Probe Kit (Abbott), which consists of 3 probes targeting 4q12 and spanning a region ∼274 kb upstream of SCFD2 to 117 kb downstream of KIT (supplemental Figure 2). Patient 2 showed 1 tricolor overlap signal on chromosome 4, indicating an intact 4q12 region (Figure 1A). The second group of signals was split, with the SpectrumAqua “PDGFRA” probe on derivative chromosome 12, the SpectrumGreen “SCFD2” probe on derivative chromosome 4, and loss of the SpectrumOrange “LNX1” probe. Patient 1 had identical 4q12 FISH findings (data not shown). Interphase FISH in patient 3 showed similar results without loss of the SpectrumOrange “LNX1” signal (Figure 1B). ETV6 rearrangements were also detected by break-apart FISH analysis in patients 2 and 3 (Figure 1). Patient 4 reportedly had similar results of interphase FISH analysis performed at an outside hospital using the same commercial ETV6 and 4q12 probes (Table 2).
![Selected cytogenetic and FISH results for patients 2 and 3. (A) Patient 2 partial karyotype showing t(4;12) (top panel). 4q12 metaphase FISH analysis shows 1 normal chromosome 4 [chr(4)] with overlap of all 3 probes, the SpectrumAqua (PDGFRA) probe on the derivative chromosome 12 [der(12)], and the SpectrumGreen (SCFD2) probe on the derivative chromosome 4 [der(4)] (middle panel). There is loss of 1 SpectrumOrange signal, indicating loss of LNX1 or adjacent material. ETV6 break-apart metaphase FISH analysis shows 1 normal chromosome 12 [chr(12)] with overlap of the 5′ ETV6 SpectrumOrange and 3′ ETV6 SpectrumGreen probes, the derivative chromosome 12 [der(12)t(4;12)] with 3′ ETV6 SpectrumGreen and dim 5′ ETV6 SpectrumOrange signals, and the derivative chromosome 4 [der(4)t(4;12)] with the 5′ ETV6 SpectrumOrange probe only (bottom panel). (B) Patient 3 partial karyotype showing t(4;12) (top panel). 4q12 interphase FISH showing 1 normal tricolor signal, 1 separate SpectrumGreen (SCFD2)/SpectrumOrange (LNX1) overlap signal, and 1 isolated SpectrumAqua (PDGFRA) signal (middle panel). ETV6 interphase break-apart FISH showing 1 isolated ETV6 5′ signal in addition to 1 normal unsplit pair and 1 unsplit pair with dim 5′ ETV6 signal (bottom panel).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/3/10.1182_bloodadvances.2021005280/4/m_advancesadv2021005280f1.png?Expires=1765057234&Signature=sr2~MWLsTPHZksNXt1Wjx2Lk2vh89AfgW36PIFsDoeyOhiHtU9O2RCx56H-qHuaKPBqeu9vE8EDHkkD7At55Xu6OMrqywMscQxNqEl1XeDkHwPbQ1ZLfA0yWbzRIJ6OncV7Bfsu2go0G4ydIIv5945Tn0I8UyQo~p~W-NfqUrhTmULk6552vi~Bd--w5fM~hBgjQFDboIUboRp2wV-GDhTYN9OKhxzl3-II-tCBetwNZTV-yu~ZdI~~7TViI2~6q0drWWEZuWdxNpCOwQ4C8Z2zUZy0fbEBkcF8uLxVDfBbJfSpKwXtn4LLnsfk3Z4byU2D9MR5RYLAp41qcE8d90Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Selected cytogenetic and FISH results for patients 2 and 3. (A) Patient 2 partial karyotype showing t(4;12) (top panel). 4q12 metaphase FISH analysis shows 1 normal chromosome 4 [chr(4)] with overlap of all 3 probes, the SpectrumAqua (PDGFRA) probe on the derivative chromosome 12 [der(12)], and the SpectrumGreen (SCFD2) probe on the derivative chromosome 4 [der(4)] (middle panel). There is loss of 1 SpectrumOrange signal, indicating loss of LNX1 or adjacent material. ETV6 break-apart metaphase FISH analysis shows 1 normal chromosome 12 [chr(12)] with overlap of the 5′ ETV6 SpectrumOrange and 3′ ETV6 SpectrumGreen probes, the derivative chromosome 12 [der(12)t(4;12)] with 3′ ETV6 SpectrumGreen and dim 5′ ETV6 SpectrumOrange signals, and the derivative chromosome 4 [der(4)t(4;12)] with the 5′ ETV6 SpectrumOrange probe only (bottom panel). (B) Patient 3 partial karyotype showing t(4;12) (top panel). 4q12 interphase FISH showing 1 normal tricolor signal, 1 separate SpectrumGreen (SCFD2)/SpectrumOrange (LNX1) overlap signal, and 1 isolated SpectrumAqua (PDGFRA) signal (middle panel). ETV6 interphase break-apart FISH showing 1 isolated ETV6 5′ signal in addition to 1 normal unsplit pair and 1 unsplit pair with dim 5′ ETV6 signal (bottom panel).
Selected cytogenetic and FISH results for patients 2 and 3. (A) Patient 2 partial karyotype showing t(4;12) (top panel). 4q12 metaphase FISH analysis shows 1 normal chromosome 4 [chr(4)] with overlap of all 3 probes, the SpectrumAqua (PDGFRA) probe on the derivative chromosome 12 [der(12)], and the SpectrumGreen (SCFD2) probe on the derivative chromosome 4 [der(4)] (middle panel). There is loss of 1 SpectrumOrange signal, indicating loss of LNX1 or adjacent material. ETV6 break-apart metaphase FISH analysis shows 1 normal chromosome 12 [chr(12)] with overlap of the 5′ ETV6 SpectrumOrange and 3′ ETV6 SpectrumGreen probes, the derivative chromosome 12 [der(12)t(4;12)] with 3′ ETV6 SpectrumGreen and dim 5′ ETV6 SpectrumOrange signals, and the derivative chromosome 4 [der(4)t(4;12)] with the 5′ ETV6 SpectrumOrange probe only (bottom panel). (B) Patient 3 partial karyotype showing t(4;12) (top panel). 4q12 interphase FISH showing 1 normal tricolor signal, 1 separate SpectrumGreen (SCFD2)/SpectrumOrange (LNX1) overlap signal, and 1 isolated SpectrumAqua (PDGFRA) signal (middle panel). ETV6 interphase break-apart FISH showing 1 isolated ETV6 5′ signal in addition to 1 normal unsplit pair and 1 unsplit pair with dim 5′ ETV6 signal (bottom panel).
Chimeric sequences involving ETV6 and 4q12 genes were identified in all 5 patients using a targeted RNA sequencing assay relying on anchored multiplex PCR technology (Heme Fusion Assay) (Table 2). Patients 1 and 2 had in-frame fusions of ETV6 exon 1 and SCFD2 exon 5 (Figure 2A-C). The reciprocal SCFD2-ETV6 transcript was not identified. Patient 3 had an in-frame fusion of ETV6 exon 1 and CHIC2 exon 4 (Figure 2D), also with no reciprocal transcript detected. The proximal ETV6 breakpoints explain the dim 5′ ETV6 SpectrumOrange signals seen in patients 2 and 3 (Figure 1); the probe overlaps with intron 1 and exon 2 as well resulting in some binding to the derivative chromosome 12 (supplemental Figure 2). Patient 4 had an out-of-frame rearrangement involving ETV6 exon 2 and CHIC2, and patients 1, 4, and 5 also had out-of-frame rearrangements involving ETV6 exon 2 or 3 and GSX2. Although this assay has been used successfully at Massachusetts General Hospital to identify FIP1L1-PDGFRA fusions in patients with eosinophilia (data not shown), no rearrangement between PDGFRA and other chromosome 12 genes was identified in any of the 5 patients.
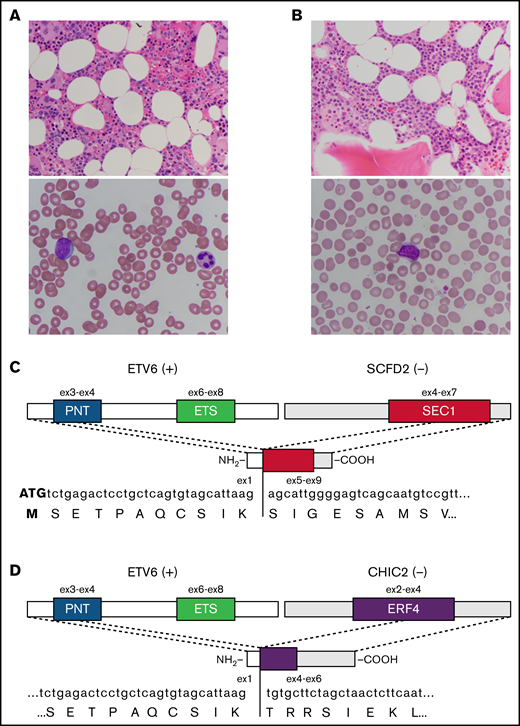
Representative pathology and schematics of in-frame fusion transcripts. (A) Representative BM histology (original magnification ×400; hematoxylin and eosin stain) (upper panel) and blast morphology on peripheral blood smear (original magnification ×1000; Wright-Giemsa stain) (lower panel) from patient 1. (B) Representative BM histology (original magnification ×400; hematoxylin and eosin stain) (upper panel) and blast morphology on peripheral blood smear (original magnification ×1000; Wright-Giemsa stain) (lower panel) from patient 2. (C) Schematic diagram of the in-frame ETV6-SCFD2 fusion identified in patients 1 and 2. Exon 1 of ETV6 is upstream of exons 5 through 9 of SCFD2, which includes part of the SEC1 domain sequence (exons 4-7). The fusion transcript does not contain the coding sequence for the PNT or ETS domains. (D) Schematic diagram of the in-frame ETV6-CHIC2 fusion identified in patient 3. Exon 1 of ETV6 is upstream of exons 4 through 6 of CHIC2, which includes the distal portion of the ERF4 domain sequence (exons 2-4). As above, the PNT and ETS domains are not included in the fusion transcript. +, positive strand gene; −, negative strand gene.
Representative pathology and schematics of in-frame fusion transcripts. (A) Representative BM histology (original magnification ×400; hematoxylin and eosin stain) (upper panel) and blast morphology on peripheral blood smear (original magnification ×1000; Wright-Giemsa stain) (lower panel) from patient 1. (B) Representative BM histology (original magnification ×400; hematoxylin and eosin stain) (upper panel) and blast morphology on peripheral blood smear (original magnification ×1000; Wright-Giemsa stain) (lower panel) from patient 2. (C) Schematic diagram of the in-frame ETV6-SCFD2 fusion identified in patients 1 and 2. Exon 1 of ETV6 is upstream of exons 5 through 9 of SCFD2, which includes part of the SEC1 domain sequence (exons 4-7). The fusion transcript does not contain the coding sequence for the PNT or ETS domains. (D) Schematic diagram of the in-frame ETV6-CHIC2 fusion identified in patient 3. Exon 1 of ETV6 is upstream of exons 4 through 6 of CHIC2, which includes the distal portion of the ERF4 domain sequence (exons 2-4). As above, the PNT and ETS domains are not included in the fusion transcript. +, positive strand gene; −, negative strand gene.
None of the patients for whom data were available had elevated eosinophil counts peripherally or on BM differential (when available) (Table 1). Review of the literature showed 17 publications describing 38 cases of hematologic neoplasms with t(4;12)(q12;p13), the majority of which are cases of undifferentiated AML (AML-M0) (Table 3). Of these, there were only 3 published cases of sequence-proven ETV6-PDGFRA rearrangement, and all occurred in the setting of hypereosinophilia, chronic eosinophilic leukemia (CEL), or AML transformed from CEL. As in our 5 cases, the remaining 12 published cases for which sequencing was performed did not feature eosinophilia and have non-PDGFRA fusion partners.
Discussion
Hematologic neoplasms with t(4;12)(q12;p13) are uncommon, with only 38 cases reported in the literature (Table 3). Most cases with this translocation are AML-M0; however, it has also been described in AML of other types; myeloid/natural killer cell leukemia; myeloproliferative neoplasms, unclassifiable (MPN-U); CEL; and hypereosinophilia. The first report of this translocation used FISH analysis with laboratory-developed P1-derived artificial chromosomes (PACs) and reverse transcription PCR, followed by sequencing to identify a fusion transcript containing exons 1 through 3 of CHIC2 (previously named BTL) and exons 2 through 8 of ETV6.26 Subsequent publications used similar techniques to show that these rearrangements essentially always included ETV6 juxtaposed with a variety of in-frame and out-of-frame partners on 4q12. Of the 11 cases with sequence-defined partners, 5 involved CHIC2, 3 involved PDGFRA, 2 involved GSX2, and 1 involved LINC02260 (Table 3).
The first ETV6-PDGFRA fusion gene was reported in a patient with CEL and t(4;12)(q2?3;p1?2) karyotype.27 Sequencing identified an in-frame whole exon fusion between ETV6 exon 6 and PDGFRA exon 12. Although this fusion gene had an intact WW-like domain, in vitro studies have shown that dimerization of the chimeric proteins enforced by the ETV6 portion overcomes its inhibitory function, increasing kinase activity sufficiently to induce transformation.15 Unsurprisingly, this patient’s disease was sensitive to imatinib, with complete cytogenetic response at 9 months. A subsequent report identified another patient with CEL and t(4;12)(q12;p13) corresponding to a fusion between ETV6 exon 7 and PDGFRA exon 23.28 This patient was not responsive to 5 months of imatinib therapy, likely as a result of the fusion protein containing only the C-terminal exon of PDGFRA, which is downstream of the tyrosine kinase domains. The functional consequences of the reciprocal fusion, if any, are unclear.
Another recent study used phospho-flow cytometry targeting anti–phospho-PDGFRA Y720 to identify a patient with hypereosinophilia who was subsequently found to have a (4;12)(q12;p13) karyotype with an ETV6-PDGFRA fusion confirmed by sequencing.29 Response to treatment was not described. The remaining published reports used chromosome and FISH analysis alone to identify cases of potential ETV6-PDGFRA rearrangements. These include a myeloid neoplasm with eosinophilia that later transformed to AML,30 a case of imatinib-resistant AML that evolved from chronic myelomonocytic leukemia with t(4;12;6)(q12;p13;p21),31 and 7 cases of aggressive AML with t(4;12) rearrangements.24 None of the 7 patients in the larger study were treated with imatinib; however, 1 patient was treated unsuccessfully with dasatinib.24
To our knowledge, all of the published ETV6-PDGFRA fusions that were associated with noneosinophilic disorders were diagnosed using chromosome analysis and the Abbott Vysis LSI 4q12 Tri-Color Rearrangement FISH Probes, without confirmation by sequencing (Table 3). Given their respective locations on the positive strand of 12p13 and the positive strand of 4q12, ETV6-PDGFRA fusions can only occur in the setting of insertions or in translocations that involve an inversion. Although ETV6-PDGFRA fusion cannot occur as the result of a simple reciprocal translocation alone, small inversions are not detectable by conventional chromosome banding analysis; therefore, a t(4;12)(q12;p13) karyotype does not rule out an ETV6-PDGFRA fusion. The Abbott 4q12 kit was originally validated for the detection of del(4)(q12q12), which is associated with FIP1L1-PDGFRA fusion in diverse eosinophilia-associated hematologic disorders.32 However, based on the probe locations, the resolution of the Abbott 4q12 kit is insufficient to definitively identify 4q12 genes involved in other rearrangements, including t(4;12)(q12;p13) (supplemental Figure 2). Based on our findings of false-positive ETV6-PDGFRA fusions using these methods in imatinib-insensitive patients without eosinophilia, we hypothesize that the rearrangements in published cases lacking eosinophilia did not produce an intact ETV6-PDGFRA fusion protein, or perhaps did not involve PDGFRA at all, and should be confirmed by sequencing.
However, all of our patients did have rearrangements between ETV6 and other 4q12 genes that explain the cytogenetic/FISH findings of t(4;12)(q12;p13) (Table 2). Based on patient 1’s cytogenetic/FISH data and lack of reciprocal fusion sequence, we hypothesize that the rearrangement also involves a deletion up to ∼956 kb in length between SCFD2 exon 5 and the 5′ end of GSX2. Although patient 2 had only 1 fusion detected, the loss of the SpectrumOrange (LNX1) signal by FISH analysis suggests a similar structure. Patient 3 retained two copies of the SpectrumOrange signal and, therefore, may have a translocation without loss of material. FISH analysis was not performed for patients 4 and 5, but the presence of the GSX2 exon 2–ETV6 exon 3 reads, in addition to the ETV6 exon 1–CHIC2 exon 2 fusion, in patient 4 is reminiscent of patient 1 (Table 2).
The wide variety of breakpoints in 4q12 identified in the literature and in our study is unsurprising because of its location in FRA4B, one of many common fragile sites in the human genome. As the name implies, these domains are common and were originally described as areas of recurrent double-stranded breaks in cultured lymphocytes.33 These regions are enriched in long AT repeat sequences and tend to form secondary structures that interfere with replication fork progression, leading to double-strand breaks in the setting of replication stress, whether induced chemically or by oncogenic mutations (see Lukusa et al34 for additional details).
Given that the ETV6 breakpoints identified in our study are primarily in exons 1 and 2 (upstream of its functional domains), these rearrangements are likely phenotypically similar to ETV6 loss, which has been implicated as pathogenic in a variety of hematologic malignancies, including AML, ALL, and myelodysplastic syndrome.35-37 Additionally, ETV6 rearrangements that disrupt the coding region but do not generate functional fusion proteins have been identified in AML, ALL, and MPN-U.38-41 These rearrangements are generally thought to be pathogenic as a result of the deregulation of nearby genes in addition to ETV6 loss of function (see Rasighaemi et al11 for additional details). As in our patient cohort (Table 1), none of the ETV6 deletions, truncations, or nonfunctional rearrangements cited above were associated with eosinophilia.
Although the ETV6 partner genes identified in our study are not well understood, it is possible that disruption of these genes contributed to AML pathogenesis in our patients. SCFD2 (Sec1 family domain containing 2) is a reverse-strand gene located ∼863 kb upstream of PDGFRA on chromosome 4q12. SCDF2 is ubiquitously expressed in human tissues and was detectable in every subtype of peripheral blood mononuclear cell in the Human Protein Atlas.42 Several in-frame SCFD2 fusions with non-ETV6 partners have been identified, primarily in epithelial tumors and astrocytomas. However, their mechanisms of action were undetermined or due to kinase activity of the partner gene.43-48 SCFD2 function is not well studied, but there is some evidence suggesting a role in tumorigenesis; 1 study showed p53 binding to the SCFD2 promoter after hypoxia and DNA damage, and another showed that SCFD2 knockdown suppressed proliferation of breast cancer cells in vitro.49,50
CHIC2 (cysteine-rich hydrophobic domain 2) is another reverse-strand gene located ∼146 kb upstream of PDGFRA on chromosome 4q12. It was originally named BTL (Brx-like translocated in leukemia) and was first described fused to ETV6 in a report of 4 patients with AML with t(4;12)(q11-q12;p13).26 Subsequent case reports identified several other in-frame and out-of-frame ETV6-CHIC2 fusions in AML.36,51,52 The pathogenic effects of these ETV6-CHIC2 fusions seem to be due to the deregulated expression of the nearby gene GSX2, potentially as a result of the proximity to regulatory elements of the partner gene. For example, 1 study showed that GSX2 expression was elevated in 4 patients with AML with both in-frame and out-of-frame ETV6-CHIC2 fusions and that overexpression of GSX2 in vitro was sufficient to transform NIH3T3 cells.53 These findings were duplicated in a subsequent study of a similar out-of-frame fusion in a patient with AML.52 This study also noted elevated PDGFRA expression in 1 patient.
GSX2 (genetic-screened homeobox 2, formerly GSH2) is a forward strand gene adjacent to PDGFRA (∼127 kb upstream). GSX2 contains a homeobox domain encoded by exon 2 and is expressed during early specification of lateral ganglionic eminence progenitors in the telencephalon.8,54 Although GSX2 overexpression has been shown to transform NIH3T3 cells as mentioned above,53 its role in the pathogenesis of solid tumors is unclear. Promoter hypermethylation is common in pancreatic cancer55 and astrocytomas,56 suggesting a possible tumor suppressor function. Conversely, increased GSX2 expression is associated with higher-risk disease in low-grade gliomas.57 However, the data are more consistent in AML, for which elevated GSX2 expression has been observed in multiple case series of patients with aggressive t(4;12)(q12;p13) AML.25,53 GSX2-containing fusions do not appear to be common; however, there is 1 case report of an in-frame NUP98-GSX2 fusion transcript in a patient with acute myelomonocytic leukemia and t(4;11)(q12;p15) translocation.58 All of the sequences involving GSX2 in our study are out-of-frame (Table 2).
It is important to note that other oncogenic drivers were identified in our patients. Each of the patients for whom there are data available have ≥1 pathogenic single nucleotide variant (Table 2).59 Additionally, the 4q12 SpectrumOrange signal, which covers a 448-kb region including the LNX1 gene, was lost in patients 1 and 2 (Figure 1; Table 2). LNX1 was originally thought to be a tumor suppressor because of its downregulation in gliomas, but recent evidence points to a potential oncogenic role in shortening the half-life of p53 via destabilization of Numb.60,61
In summary, we identified 5 patients with AML without eosinophilia with cytogenetic and FISH findings suggestive of ETV6-PDGFRA rearrangement who were found to have rearrangements involving ETV6 and other 4q12 genes on NGS. Two in-frame fusions were identified (ETV6-SCFD2 and ETV6-CHIC2); however, they are likely functionally analogous to ETV6 loss given the proximal breakpoints. Most importantly, 3 of the 5 patients were initially treated with imatinib to target the putative PDGFRA fusion, delaying the initiation of more appropriate chemotherapy. Based on these findings and review of the literature, we propose that NGS-based testing should be performed in cases of t(4;12)(q12;p13) AML instead of using the Abbott Vysis LSI 4q12 Tri-Color Rearrangement FISH Probe Kit. We further suggest that true ETV6-PDGFRA fusions may be rare in the absence of eosinophilia; analysis of a larger cohort is required to better define this relationship.
Finally, although sequencing is the gold standard in identifying breakpoints with single nucleotide resolution, the turnaround time is often relatively slow due to batching and the need for specialized review of the data (as well as delays for specimen shipment if the technology is not available in-house). Depending on the assay design, targeted NGS may miss fusions between uncovered exons/genes and will not detect changes in the expression of wild-type genes adjacent to rearrangements, which is known to occur in the 4q12 region, as described above. Whole-transcriptome sequencing overcomes the coverage limitations of targeted sequencing62 ; however, it still suffers from the same relatively slow turnaround time and lack of wide accessibility that affect sequencing-based assays in general. More rapid functional readouts of PDGFRA activity (such as phosphor-flow cytometry) may be useful to expedite the selection of patients who could benefit from tyrosine kinase inhibition while NGS results are pending.29
Acknowledgment
The authors thank Alexander Farahani for technical assistance.
Authorship
Contribution: V.N., P.D.C., S.B.M., H.D.M., D.D.-S., L.P.L., J.K.L., and A.J.I. designed the study and interpreted data; A.M.B., M.J.W., M.R.L., D.J.D.A., and R.M.S. were responsible for patient care and provided clinical data; S.B.M., P.D.C., and V.N. wrote the manuscript; and all authors reviewed, edited, and approved the final version of the manuscript.
Conflict-of-interest disclosure: A.J.I. and L.P.L. have partial ownership in and have acted as advisors for ArcherDx. The remaining authors declare no competing financial interests.
Correspondence: Valentina Nardi, Massachusetts General Hospital, MGH Pathology Service, Warren 219, 55 Fruit St, Boston, MA 02114; e-mail: vnardi@partners.org.

![Selected cytogenetic and FISH results for patients 2 and 3. (A) Patient 2 partial karyotype showing t(4;12) (top panel). 4q12 metaphase FISH analysis shows 1 normal chromosome 4 [chr(4)] with overlap of all 3 probes, the SpectrumAqua (PDGFRA) probe on the derivative chromosome 12 [der(12)], and the SpectrumGreen (SCFD2) probe on the derivative chromosome 4 [der(4)] (middle panel). There is loss of 1 SpectrumOrange signal, indicating loss of LNX1 or adjacent material. ETV6 break-apart metaphase FISH analysis shows 1 normal chromosome 12 [chr(12)] with overlap of the 5′ ETV6 SpectrumOrange and 3′ ETV6 SpectrumGreen probes, the derivative chromosome 12 [der(12)t(4;12)] with 3′ ETV6 SpectrumGreen and dim 5′ ETV6 SpectrumOrange signals, and the derivative chromosome 4 [der(4)t(4;12)] with the 5′ ETV6 SpectrumOrange probe only (bottom panel). (B) Patient 3 partial karyotype showing t(4;12) (top panel). 4q12 interphase FISH showing 1 normal tricolor signal, 1 separate SpectrumGreen (SCFD2)/SpectrumOrange (LNX1) overlap signal, and 1 isolated SpectrumAqua (PDGFRA) signal (middle panel). ETV6 interphase break-apart FISH showing 1 isolated ETV6 5′ signal in addition to 1 normal unsplit pair and 1 unsplit pair with dim 5′ ETV6 signal (bottom panel).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/3/10.1182_bloodadvances.2021005280/4/m_advancesadv2021005280f1.png?Expires=1765057235&Signature=WwWHfpTxod5e-ySm3AZiR86Meym8lWxCS4ri4vm6DIqDSpGuttE5RnR6vV5pNT671f5OXaWNzxw8dEo9C-v11hMKbWYJfvBCU1JcwGdedxvofM5FiV4SrE-4nP2R69CHqWwo~Z5NtZ8gjswAReLKeQC1KNnaI~tKWXds4~XZlWxZD4k9IA1oBOvFv2oBUpMG5PAJViplOaZaepTtko~fxzdz8FTI6xG0O3KRDpX~hQNzlu3GxNQhpQpb3ZEMXG3htCgcJrbbdxnwioiBv~sK0apFutlXNOK8ETH3xUuxAy68Uj9G9tUX2QyxHJDID5Jx6yXJW~XLPvAMguXAyIcN3w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)


