TO THE EDITOR:
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma in the western world.1 It is a biologically heterogenous disease, with 8% to 14% of the cases demonstrating MYC rearrangement.2 High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (HGBL double-hit [DH]/triple-hit [TH]) is a newly created lymphoma subtype in the 2016 World Health Organization (WHO) classification of lymphoid neoplasms, characterized by concurrent chromosomal rearrangements involving MYC and BCL2 and/or BCL6.2,3 HGBL-DH/TH constitutes ∼4% to 7% of newly diagnosed DLBCL and is frequently associated with early treatment failure.4-6 Intensive frontline immunochemotherapy regimens appear to be associated with better outcomes in retrospective and single-arm studies6-9 ; however, emerging data have challenged the role of intensive inductions.10 Patients with relapsed or refractory (rel/ref) HGBL-DH/TH have a poor rate of survival.5 Thus, novel treatment strategies that will improve response rates and duration of remission remain an unmet need.
Loncastuximab tesirine is an antibody-drug conjugate comprising a humanized anti-CD19 monoclonal antibody stochastically conjugated to a pyrrolobenzodiazepine (PBD) dimer cytotoxin, SG3199.11,12 After rapid internalization by CD19-expressing cells, loncastuximab tesirine is transported to the lysosomes, where the PBD dimer is released after cleavage of the protease-sensitive linker. The PBD dimer binds to the minor groove of DNA, where it causes interstrand DNA crosslinks with minimal DNA distortion. The pivotal phase 2 single-arm LOTIS-2 study (registered on https://clinicaltrials.gov as #NCT03589469) enrolled 145 patients with rel/ref DLBCL (including HGBL-DH/TH) from August 2018 through September 2019, after they had received ≥2 multiagent systemic therapies, and led to US Food and Drug Administration approval of loncastuximab tesirine in this setting.13 Eligibility criteria have been reported but included measurable disease defined by the 2014 Lugano Classification, Eastern Cooperative Oncology Group performance status of 0 to 2, and adequate organ function.13 Patients with bulky disease, defined as a tumor ≥10 cm in the longest dimension, autologous stem cell transplant within 30 days, allogenic stem cell transplant within 60 days, and active central nervous system lymphoma, represented the major exclusion criteria. The study was performed in accordance with regulatory requirements, and the protocol was approved by institutional review boards and ethics committees at individual sites. All patients provided written informed consent. Treatment consisted of single-agent loncastuximab tesirine 0.150 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles for up to 1 year or until relapse or progression of disease, unacceptable toxicity, or death. The primary study end point was overall response rate, defined as the proportion of patients with best overall complete response (CR) or partial response assessed by independent review. Investigator assessment of histopathology according to the 2016 WHO classification was used to define HGBL-DH/TH.2 We also included in the present analysis patients who received a diagnosis of DLBCL-DH/TH before the current WHO classification. Median progression-free survival (PFS) and overall survival (OS) measured the time from first dose to the point when the probability of the events reach 50%. In this report, we sought to evaluate the outcomes of patients with HGBL-DH/TH enrolled in the LOTIS-2 trial with a data cutoff of 1 March 2021.
Fifteen (10%) patients with HGBL-DH/TH were enrolled in the LOTIS-2 clinical trial. Twelve of those patients (80%) exhibited MYC and BCL2 or BCL6 rearrangements (DH), and 3 (20%) showed MYC, BCL2, and BCL6 rearrangements (TH). Ten patients were women (66.7%), and 5 (33.3%) were men. The median age was 74 (range, 27-85) years, with 6 patients (40%) ≥75 years of age. The majority of the patients (n = 11; 73%) had received ≥3 lines of therapy, 3 patients (20%) had undergone autologous stem cell transplant, and 4 patients (26.7%) had received chimeric antigen receptor (CAR)–modified T-cell therapy with a current biopsy specimen demonstrating persistent CD19 expression by immunohistochemistry (Table 1). Only 1 patient received a polatuzumab vedotin–containing regimen before enrollment. The median time from diagnosis to first dose of loncastuximab tesirine was 22.2 (range, 5.4-86.6) months, and the median time from completion of immediate last therapy to initiation of the study treatment was 1.7 (range, 0.6-62.4) months.
The overall response rate of loncastuximab tesirine in HGBL-DH/TH was 33.3% (95% confidence interval [CI], 11.8% to 61.6%) with all 5 patients achieving CR (Figure 1A). The median time to first CR was 43 (38-148) days. Among the patients with HGBL-DH/TH treated with loncastuximab tesirine after CAR T-cell progression (n = 4), 1 patient achieved CR. With a median follow-up of 5.8 (range, 0.7-28.1) months, the median PFS and OS were 3.7 (95% CI, 1.28-not reached) and 9.2 (95% CI, 1.84-not reached), respectively. All responding patients with HGBL-DH/TH had a duration of response of more than 12 months, with a median duration of response not reached at the time of data cutoff (Figure 1B). Treatment-emergent adverse events were similar to those of patients with DLBCL not otherwise specified. The most common included grade ≥3 neutropenia (26%), thrombocytopenia (18%), and increased γ-glutamyl transferase (17%).
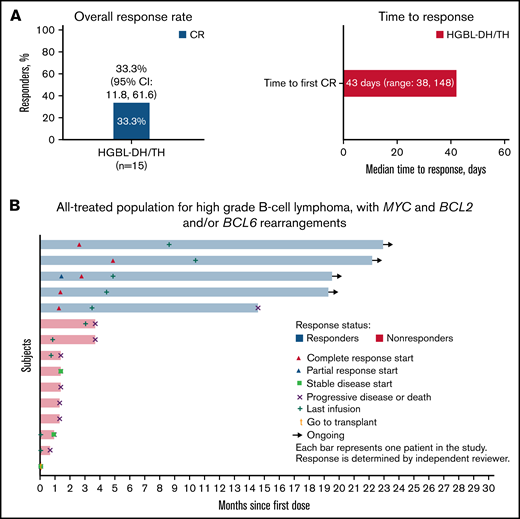
Efficacy of loncastuximab tesirine in patients with high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (HGBL-DH/TH). Overall response rate, time to response (A), and time to events (B) by patient in 15 patients enrolled in the LOTIS-2 trial.
Efficacy of loncastuximab tesirine in patients with high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (HGBL-DH/TH). Overall response rate, time to response (A), and time to events (B) by patient in 15 patients enrolled in the LOTIS-2 trial.
Patients with rel/ref HGBL-DH/TH have a poor prognosis. In a retrospective multicenter study, patients with HGBL-DH/TH (n = 12) were more likely to present primary refractory disease or relapse within 12 months from frontline therapy (83%), achieved only partial response (75%) to salvage chemotherapy, and demonstrated a 4-year OS of 25%.5 CD19-directed CAR T-cell therapy has changed the landscape of treatment options in patients with aggressive lymphomas; however, efficacy in patients with HGBL-DH/TH appears different by cellular product. Nastoupil et al reported similar 12-month PFS (39% vs 48%; P = .43) and OS (69% vs 68%; P = .99) in patients with HGBL-DH/TH compared with other rel/ref DLBCL subtypes treated with axicabtagene ciloleucel.14 Similar results were achieved in this population with lisocabtagene maraleucel, with an objective response rate of 75.8% and not-reached median OS.15 In contrast, a recent update of the JULIET study reported that patients with HGBL-DH/TH treated with tisagenlecleucel exhibited lower overall response rates (20% vs 42.1%).16 CD19-directed CAR T-cell therapy denotes a significant advance in the treatment of patients with rel/ref HGBL-DH/TH, but requires extensive health care coordination, is limited by manufacturing time lines, and carries the risk of substantial treatment-related toxicity. Loncastuximab tesirine is a valuable off-the-shelf treatment option for patients with rel/ref HGBL-DH/TH associated with an acceptable safety profile. Current US Food and Drug Administration–approved regimens in patients with rel/ref aggressive lymphoma include polatuzumab vedotin in combination with bendamustine and rituximab and tafasitamab with lenalidomide. However, the efficacy of these agents in HGBL-DH/TH remains unknown, as patients with this disease subtype were not included in pivotal clinical trials of these therapeutic combinations.17,18 Acknowledging the small number of patients included in our analysis and the need for future larger confirmatory studies, our data support the selection of loncastuximab tesirine in rel/ref HGBL-DH/TH.
In summary, response rates in this small subgroup of patients with heavily pretreated HGBL-DH/TH (33.3%) are consistent with those observed in the DLBCL-not otherwise specified population previously reported in the LOTIS-2 study. Initial responses were achieved within a median of 6.1 weeks, with all responding patients achieving CR. Moreover, the patients achieved long-term disease control, underscoring the efficacy of loncastuximab tesirine in overcoming poor disease prognoses in rel/ref HGBL-DH/TH.
Acknowledgments: The authors thank the patients who participated in the study and their families and the study research nurses, research coordinators, and site staff for their support of the study.
Funding for the original study was provided by ADC Therapeutics, SA. J.P.A. is supported by the Peykoff Initiative of the Lymphoma Research Foundation.
Contribution: J.P.A. and P.F.C. designed and performed the research, analyzed the data, were involved in data acquisition, and wrote the manuscript with input and approval of the final version from all coauthors; W.Z.A., J.R., M.S., K.M.A., M.A.L., B.T.H., P.L.Z., A.S., C.C.-S., M.H., and B.S.K., designed and performed the research, analyzed the data, were involved in data acquisition, and critically reviewed and approved the manuscript; E.Y. contributed to the statistical analysis; and D.U., T.K., E.Y., and Y.Q. were involved in designing the original study and/or analyzing the data on which this work was based and provided critical review and approval of the manuscript for publication.
Conflict-of-interest disclosure: J.P.A. is a consultant to and has obtained research funding for ADC Therapeutics. An immediate family member has served on the advisory boards of Puma Biotechnology, Inovio Pharmaceuticals, Agios Pharmaceuticals, Forma Therapeutics, and Foundation Medicine. W.Z.A. has been a consultant to ADC Therapeutics, Nurix, Kite Pharma, and Kymera Therapeutics. J.R. has been a consultant to ADC Therapeutics, Bristol Myers Squibb, Kite Pharma, and Takeda; has ownership interests in AstraZeneca (spouse) and GlaxoSmithKline (self); has received research funding from Takeda and honoraria from Takeda, Bristol Myers Squibb, and ADC Therapeutics; and is on the speakers’ bureaus of Takeda and ADC Therapeutics. M.S. is a consultant to Amgen and Bristol Myers Squibb; has received research funding from ADC Therapeutics and Partner Therapeutics; and is on the speakers’ bureaus of Bristol Myers Squibb, GSK, AbbVie, and Celgene. K.M.A. has received research funding from Novartis, Bristol Myers Squibb, Autolus Therapeutics, ADC Therapeutics, Pharmacyclics, and Janssen and honoraria from BeiGene, Celgene, Novartis, and Roche; is on the Board of Directors or advisory boards of Gilead, BeiGene, Celgene, Novartis, and Roche. M.A.L. served as a consultant and has financial relationships with BeiGene, Karyopharm, Gilead/Kite Pharma, Daiichi Sankyo, Novartis, Kyowa Kirin, AbbVie, Celgene, Verastem, Janssen, Myeloid Therapeutics, AstraZeneca, Acrotech, ADC Therapeutics, Legend, Spectrum, MorphoSys, and TG Therapeutics. B.T.H. is on the speakers’ bureaus of Bristol Myers Squibb and AstraZeneca and is an advisory board member of ADC Therapeutics. P.L.Z. is consultant to Verastem, MSD, EUSA Pharma, and Sanofi; is a member of the Board of Directors, speakers’ bureaus, or advisory committees of ADC Therapeutics (advisory board agreement), Verastem, Celltrion, Gilead, Janssen-Cilag, Bristol Myers Squibb, Servier, Sandoz, MSD, Immune Design, Celgene, Portola, Roche, EUSA Pharma, and Kyowa Kirin. A.S. served as consultant to Bayer and Eli Lilly and advisor to Roche and Janssen, received institutional research funding from Roche, AbbVie, Pfizer, Bayer, Merck, Novartis, ADC Therapeutics, and MEI Therapeutics, and received travel grants from AbbVie and PharmaMar. C.C.-S. has been a consultant to Sanofi; has received research funding from ADC Therapeutics, Roche, and Sanofi and honoraria from AstraZeneca, Bristol Myers Squibb, Incyte, Janssen Oncology, Takeda, and ADC Therapeutics; is a member of the Board of Directors, speakers’ bureaus, or advisory committees of Sanofi, ADC Therapeutics, Bristol Myers Squibb, Celgene, Karyopharm Therapeutics, and Roche. M.H. has received research funding from Takeda, Spectrum Pharmaceuticals, and Astellas Pharma; has been a consultant to Janssen, Incyte Corporation, ADC Therapeutics, Celgene Corporation, Omeros, Verastem, and MorphoSys; and has been on the speaker’s bureaus of Sanofi Genzyme, AstraZeneca, and BeiGene. B.S.K. has been a consultant to AbbVie, ADC Therapeutics, AstraZeneca, BeiGene, Celgene, Teva, Janssen, MTEM, Bayer, Incyte, Adaptive, Genentech, Roche, MEI, KITE, TG Therapeutics, Epizyme, and Takeda. D.U., T.K., and Y.Q. are employees of ADC Therapeutics with ownership interests. E.Y. is an employee of ADC Therapeutics and has ownership interests in Zentalis Pharma and Merck. P.F.C. has received research funding from ADC Therapeutics and grants from Genentech; has been a consultant to ADC Therapeutics, Kite Pharmaceuticals, Verastem, Seattle Genetrics, Amgen, and TG Therapeutics and is on the speakers’ bureau of Celgene.
Correspondence: Juan Pablo Alderuccio, Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, 1475 NW 12th Ave, Miami, Florida, 33136; e-mail: jalderuccio@med.miami.edu.