TO THE EDITOR:
The advent of chimeric antigen receptor T-cell therapy (CAR-T) has substantially improved clinical outcomes for refractory B-cell malignancies.1-4 Real-world evidence has underlined the role of hematological toxicity, which is common and substantially contributes to infectious complications and non-relapse mortality (NRM) after CD19 CAR-T.5-8 Hematotoxicity can be delayed and prolonged in nature, and neutrophil recovery is typically biphasic with intermittent recovery followed by a second dip.9,10 Clinically challenging cases of profound granulocyte colony-stimulating factor (G-CSF) refractory aplasia have been reported.8,11,12 While the pathomechanism remains incompletely understood, impaired pre–CAR-T hematopoietic reserve and inflammation10 and cytokine release syndrome (CRS)-related cytokine patterns13 have been discussed. For refractory cytopenia, stem cell transplantation represents the last resort, either from an allogeneic donor or an autologous product from previous collection. However, its role in the context of CAR-T–related hematotoxicity remains ill defined, and experience is limited.11,12 In this retrospective observational study, we therefore describe clinical characteristics, bone marrow (BM) findings, engraftment and hematopoietic reconstitution, and survival outcomes in 12 patients receiving stem cell boost for severe CAR-T–related hematological toxicity.
We surveyed institutional databases across 6 European CAR-T centers (supplemental Figure 1). Exclusion criteria included active infection and poor performance status. Clinical metadata were collected with institutional review board approval and in accordance with the Declaration of Helsinki. The indication was severe pancytopenia or long-lasting G-CSF or transfusion dependency (persistent cytopenia group). Immunotoxicity was graded as previously described (supplemental Methods).7,10,14 Neutrophil engraftment was defined as the first of 3 consecutive days achieving a sustained absolute neutrophil count (ANC) >500/µL without growth factor support.15,16 Platelet engraftment was defined as a platelet count >20 G/L and transfusion independence ≥7 days.17 Cumulative incidence curves were calculated as the time to engraftment from Kaplan-Meier estimates, censoring the observation time on the date of progression, or death. Patients already fulfilling the criteria for lineage-specific engraftment on the day of transplant were excluded from the respective analysis. Efficacy outcomes were assessed according to Lugano criteria for B-cell Non-Hodgkin Lymphoma (B-NHL) and minimal residual disease (MRD) status for B-cell precursor acute lymphoblastic leukemia (BCP-ALL).3 NRM was defined as death after CAR-T without prior relapse or progression. Kaplan-Meier estimates were used to assess progression-free survival (PFS) and overall survival (OS).
We encountered 13 cases between April 2019 and March 2022. One patient was excluded due to active infection and poor performance status (supplemental Figure 1). Nine patients received axicabtagene ciloleucel, 2 received tisagenlecleucel, and 1 received brexucabtagene autoleucel (Table 1). At lymphodepletion, the median Eastern Cooperative Oncology Group Performance Status was 1 (range, 0-2). Six patients presented with a prior history of stem cell transplantation, and an additional 6 patients had collected but not received autologous CD34+ peripheral stem cells due to progressive disease. Baseline laboratory findings were notable for an elevated lactate dehydrogenase (median 332 U/L) and pronounced baseline cytopenia and inflammation (supplemental Table 1), as evidenced by high CAR-HEMATOTOX scores (median, 4; range, 2-7).10 Hypocellularity and underlying infiltration were frequent in the 8 patients with pre–CAR-T BM studies (supplemental Table 2). The toxicity profile between CAR-T and stem cell boost was dominated by hematotoxicity and severe infections (supplemental Table 3). Median duration of severe neutropenia and thrombocytopenia was 42 days, respectively (Figure 1A). Severe infections were noted in 58% of patients, whereas grade ≥3 CRS was not observed. One patient died of an invasive fungal infection on day 58 after CAR T in the setting of burgeoning hematopoietic recovery after boost (patient #3; supplemental Figure 2). Peak inflammatory markers were particularly notable for high serum ferritin levels (median, 6542; supplemental Table 1). Post–CAR-T BM studies revealed hypocellular marrow in 9/9 patients and T-cell lymphocytosis in 3/9 patients (supplemental Table 2). To overcome neutropenia, all patients received G-CSF, and half received a TPO-agonist (Figure 1B), initiated after a median of 17 and 42.5 days, respectively (supplemental Table 4). Anticytokine therapy was infrequently applied; however, transfusion dependency was common.
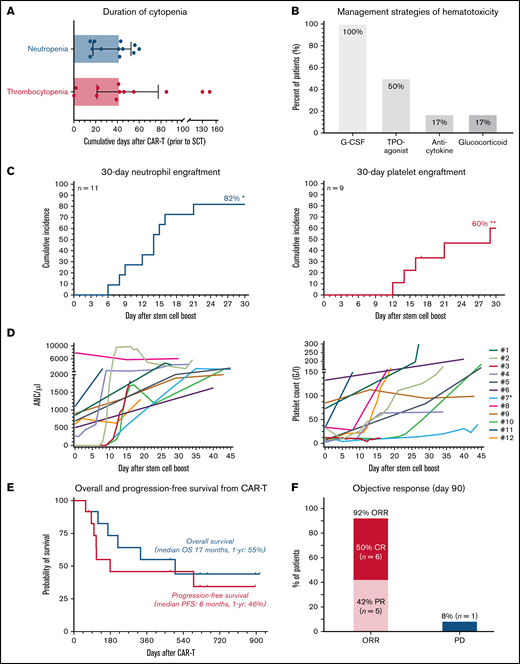
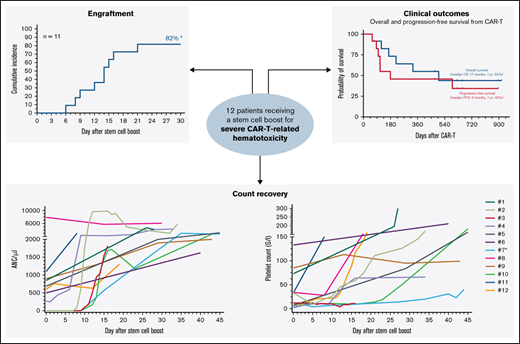
Stem cell boost for severe CAR T-related hematotoxicity is safe and clinically feasible. (A) Median cumulative duration of severe thrombocytopenia (platelet count < 50 G/L) and severe neutropenia (ANC/µ) in days between CAR infusion and stem cell rescue. Box and whiskers indicated the median and interquartile range. (B) Overview of management strategies of hematological toxicity prior to stem cell rescue. (C) Cumulative incidence rate of neutrophil (left) and platelet (right) engraftment in the first 30 days after CAR infusion. *An additional patient (#7) engrafted on day 124. **Two additional patients engrafted after day 30 (#8, 11). (D) Spider plot depicting neutrophil (left) and platelet (right) recovery for the first 45 days following stem cell boost. Each patient is color coded. Patient #7 (light blue) received a second autologous stem cell boost due to persistent thrombocytopenia. (E) Kaplan-Meier estimates of OS (gray) and PFS (dark gray) for the entire study cohort (n = 12). Median survival and 1-year survival rates are depicted. (F) Summary of best overall response at day 90. CR, complete response; PD, progressive disease; PR, partial response.
Stem cell boost for severe CAR T-related hematotoxicity is safe and clinically feasible. (A) Median cumulative duration of severe thrombocytopenia (platelet count < 50 G/L) and severe neutropenia (ANC/µ) in days between CAR infusion and stem cell rescue. Box and whiskers indicated the median and interquartile range. (B) Overview of management strategies of hematological toxicity prior to stem cell rescue. (C) Cumulative incidence rate of neutrophil (left) and platelet (right) engraftment in the first 30 days after CAR infusion. *An additional patient (#7) engrafted on day 124. **Two additional patients engrafted after day 30 (#8, 11). (D) Spider plot depicting neutrophil (left) and platelet (right) recovery for the first 45 days following stem cell boost. Each patient is color coded. Patient #7 (light blue) received a second autologous stem cell boost due to persistent thrombocytopenia. (E) Kaplan-Meier estimates of OS (gray) and PFS (dark gray) for the entire study cohort (n = 12). Median survival and 1-year survival rates are depicted. (F) Summary of best overall response at day 90. CR, complete response; PD, progressive disease; PR, partial response.
Median day of stem cell boost was 69 days (range, 35-617) (Table 1). The indication was severe pancytopenia in 6 cases and persistent neutropenia or thrombocytopenia in 6 cases. Two patients received >1 stem cell boost. The stem cell source was an autologous product in most cases (9/12), although 3 patients received stem cells from a previous allogeneic donor with no subsequent evidence of graft-versus-host disease. None of the treated patients received conditioning chemotherapy. CD34+selection was performed in 5 patients whereas the other patients received an unmanipulated (whole) product. The median day-of-transplant ANC was 0.5 (range, 0-7.9), while the median transplant-day platelet count was 27 (range, 2-133). The median total number of infused CD34+ cells was 3.1 × 106/kg (range, 1.7-7.5). Neutrophil engraftment was eventually noted in all patients, and the 30-day cumulative engraftment rate was 82% (Figure 1C). Median time to neutrophil engraftment in evaluable patients was 15 days (range, 6-124). Platelet engraftment was eventually observed in 7/9 patients, with 1 patient ultimately engrafting after a second autologous stem cell boost (patient #7). Median time to platelet engraftment in evaluable patients was 21 days (range, 12-34) with a 30-day engraftment rate of 60% (Figure 1C). Individual neutrophil and platelet recovery curves are outlined in Figure 1D. In general, day-of-transplant platelet counts were inversely correlated with the day-of-platelet engraftment (r = −0.57; P = .05; supplemental Figure 4). A histopathologic example of engraftment is displayed for a patient with BCP-ALL with underlying BM involvement (patient #12; supplemental Figure 2A). Of note, the patient with mantle cell lymphoma developed post-boost leukemic relapse (patient #8), raising concern for potential contamination. However, flow cytometric analysis of the apheresis product demonstrated no evidence of lymphoma. When studying survival outcomes, we observed 1-year PFS and OS of 46% and 55%, respectively (Figure 1E), comparable to other real-world reports.5,6 Median PFS was 6 months and median OS was 17 months. Best overall and complete response rates were 92% and 50%, respectively (Figure 1F). One-year NRM was 8% (1 infection; supplemental Figures 2 and 3).
In conclusion, our study indicates that stem cell boost represents a clinically feasible strategy for persistent cytopenia after CD19 CAR-T with high engraftment rates, resolution of cytopenia in most cases, and encouraging survival outcomes. Median engraftment times were comparable to published reports for stem cell transplantation in general, even without conditioning chemotherapy.18-21 Engraftment supports the notion that prolonged and/or persistent cytopenia after CD19 CAR-T is primarily driven by dysfunction of the hematopoietic stem and progenitor cell compartment, as opposed to primarily immunogenic phenomena driven by CAR or non–CAR-bearing immune cells. In contrast, autologous grafts are rarely successful in acquired BM deficiency syndromes caused by persistent T-cell–mediated BM suppression such as aplastic anemia.22 Notably, 4 patients displayed underlying BM infiltration prior to CD19 CAR-T, suggesting that changes to the microenvironment may persist even after tumor cell lysis and can be overcome by stem cell engraftment (supplemental Figure 2). Underlying BM disease may predispose for local inflammatory processes that propagate the functional suppression of hematopoietic stem and progenitor cells via cytokines or chemokines.9 In sepsis, inflammatory signatures can facilitate hematopoietic stem cell exhaustion23 and induce remodeling of the BM niche.24 In such a setting, stem cell rescue may provide the necessary reset, enabling the BM niche to recover from the inflammatory stress inherent to cytokine storm.13,25 This study has several relevant limitations. It was retrospective, uncontrolled, and limited to small patient numbers, which restricts drawing firm conclusions. The potential benefit of earlier stem cell rescue remains unclear, although shortening the phase of critical neutropenia likely prevents severe infectious events.8 Future prospective studies must address the question of optimal timing and patient selection in larger cohorts, including the use of preemptive stem cell apheresis in select high-risk patients for potential later use.
Acknowledgments: The authors thank the patients and their families for their participation in this study.
This work was supported by a fellowship from School of Oncology of German Cancer Consortium (DKTK) (K.R.) and was funded by the Else Kröner Forschungskolleg; a Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) research grant provided within the Sonderforschungbereich (SFB-TRR 388/1 2021–452881907) and DFG research grant (451580403) (M.S.); the Bavarian Elite Graduate Training Network (M.S.), the Wilhelm-Sander Stiftung (project no. 2018.087.1) (M.S.), the Else-Kröner-Fresenius Stiftung (M.S., K.R., V. Bücklein, V. Blumenberg), and the Bavarian Center for Cancer Research (BZKF).
Contribution: K.R. and M.S. conceived the study; K.R., A.B., K.S., G.I., P.S., L.F., V. Bücklein, C.T., R.H., R.N., J.S., V. Blumenberg, C.S., K.S., M.v.B.-B., E.B., P.B., and M.S. conducted the investigation; K.R. performed formal analysis and visualization; K.R. and M.S. conceived the methodology; K.R. and M.S. wrote the original draft; K.R., A.B., G.I., P.S., L.F., V. Bücklein, C.T., R.H., R.N., J.S., V. Blumenberg, C.S., K.S., M.v.B.-B., E.B., P.B., A.B., and M.S. reviewed and edited the writing; all authors read and approved the final manuscript.
Conflict-of-interest disclosure: K.R. received research funding and travel support from Kite/Gilead and honoraria from Novartis. K.S. received honoraria and travel support from Kite/Gilead. G.I. received consultancy fees and honoraria from Novartis, Roche, Kite/Gilead, Bristol-Myers Squibb, AbbVie, Janssen, Sandoz, Miltenyi, and AstraZeneca. P.S. received research funding from Chugai, Novartis, and Kite/Gilead. R.H. received honoraria from Kite/Gilead, Novartis, Merck Sharp & Dohme, Janssen, and Celgene and research funding from Kite/Gilead. R.N. received consultancy fees and honoraria from Amgen, AstraZeneca, Bristol-Myers Squibb, Celgene, Cellex Patient Treatment, GlaxoSmithKline, Janssen-Cilag, Kite/Gilead, Oncopeptides, Sanofi, and Takeda. V. Bücklein received honoraria from AMGEN, research funding from Celgene, honoraria from Pfizer, research funding from Kite/Gilead, and honoraria from Novartis. V. Blumenberg received honoraria and research funding from Novartis; consultancy fees, honoraria, and research funding from Gilead; research funding from Celgene; and research funding and honoraria from Janssen. C.S. received consultancy fees and honoraria from Celgene, Kite/Gilead, and Novartis. M.v.B.-B. received consultancy fees, research funding, and honoraria from MSD Sharp & Dohme, Novartis, Roche, Kite/Gilead, Bristol-Myers Squibb, Astellas, Mologen, and Miltenyi. P.B. received honoraria from Amgen, BMS, Gilead, Incyte, Miltenyi Biotec, Novartis, and Pfizer not related to the present article. A.B. received honoraria and served on the advisory board for Kite/Gilead, BMS, Pfizer, and Incyte and received research support from AOP Health. M.S. received research funding from Morphosys; consultancy fees and research funding from Novartis; consultancy fees from Janssen; research funding from Seattle Genetics; consultancy fees, honoraria, and research funding from AMGEN; consultancy fees and honoraria from Celgene; consultancy fees, honoraria, and research funding from Kite/Gilead; and consultancy and research funding from Roche AG. The remaining authors declare no competing financial interests.
Correspondence: Marion Subklewe, Department of Medicine III–Hematology/Oncology, University Hospital, LMU Munich, Marchioninistrasse 15/81377 Munich, Germany; e-mail: marion.subklewe@med.uni-muenchen.de.