

Abstract
The accuracy of pharmacokinetic (PK)-guided dosing depends on the clinical and laboratory data used to construct a population PK model, as well as the patient’s individual PK profile. This review provides a detailed overview of data used for published population PK models for factor VIII (FVIII) and factor IX (FIX) concentrates, to support physicians in their choices of which model best suits each patient. Furthermore, to enhance detailed data collection and documentation, we do suggestions for best practice. A literature search was performed; publications describing prophylactic population PK models for FVIII and FIX concentrates based on original patient data and constructed using nonlinear mixed-effect modeling were included. The following data were collected: detailed demographics, type of product, assessed and included covariates, laboratory specifications, and validation of models. Included models were scored according to our recommendations for best practice, specifically scoring the quality of data documentation as reported. Respectively, 20 models for FVIII and 7 for FIX concentrates were retrieved. Although most models (22/27) included pediatric patients, only 4 reported detailed demographics. The wide range of body weights suggested that overweight and obese adults were represented. Twenty-six models reported the assay applied to measure factor levels, whereas only 16 models named reagents used. Eight models were internally validated using a data subset. This overview presents detailed information on clinical and laboratory data used for published population PK models. We provide recommendations on data collection and documentation to increase the reliability of PK-guided prophylactic dosing of factor concentrates in hemophilia A and B.
Introduction
Patients with moderate and severe hemophilia receive replacement therapy with factor VIII (FVIII) and factor IX (FIX) concentrates to prevent spontaneous bleeding and bleeding after minor trauma.1 Ahlberg et al reported fewer joint bleeds in patients with trough levels >0.01 IU/mL.2 Therefore, clinicians historically use prophylactic replacement therapy to maintain trough factor levels of ≥0.01 IU/mL or higher in case of a severe bleeding phenotype by body weight-based dosing. However, body weight-based dosing does not account for inter-individual variability in pharmacokinetics (PK) of respective factor concentrates, that affect the achieved factor levels.3 As an example, body weight-based dosing may lead to higher dosing than necessary in obese patients because FVIII concentrate is distributed in the blood plasma, which does not increase proportionally with body weight.4 To address these inter-individual differences, PK-guided dosing using a posteriori Bayesian estimation is increasingly applied.5 This methodology relates an individual’s measured factor levels to the PK observed in a population to obtain more reliable individual PK parameters.6 To perform PK-guided dosing, accurate and validated population PK models are obligatory to enable the ability to prospectively predict data reliably.7
The accuracy of PK-guided dosing of factor concentrates is both dependent on the clinical and laboratory data used to construct the individual PK profile of the patient and on the quality of the underlying population PK model if Bayesian forecasting is applied. The clinical characteristics of the individual patient undergoing PK-guided dosing should preferably correspond to the population used to develop the population PK model because allometric exponents may otherwise not be accurate. If the characteristics do not correspond, the model may need to be further validated or enriched with such a specific patient group. However, population PK models are generally constructed with data from drug trials that often do not include specific patient populations such as children or older and obese patients. Consequently, estimation of their PK characteristics may be less accurate. In the same way, if factor levels of an individual patient are measured with different assays than with which the factor levels were measured within a certain population PK model, this may affect estimation of the individual PK parameters. As an example, a specific B-domain depleted recombinant FVIII should be measured by the chromogenic substrate assay (CSA) because factor levels are significantly lower when measured by one-stage assay (OSA), therefore affecting PK parameters and subsequently population PK modeling.8 Discrepancies between results obtained by CSA and OSA have been extensively discussed in literature and are especially relevant when extended half-life (EHL) FVIII and FIX concentrates are considered.9-11 In addition, variation in assay results may also be caused by variation in reagents used within an assay. Moreover, the source of the applied factor-deficient plasma, and instruments such as analyzers and the material used to calibrate, may be of influence on measured factor activity levels, in particular levels when measuring below 0.10 IU/mL.12,13
Therefore, clinicians and clinical pharmacologists should be aware of the specifications with regard to clinical and laboratory data of the individual patient undergoing PK-guided treatment, as well as the data with which the population PK model has been constructed. Recently, Preijers et al discussed available population PK models for FVIII and FIX concentrates, focusing on the methods used to construct these models, key features, and established covariate relationships.14 Our review aims to provide a detailed overview of the clinical and laboratory data used to construct available population PK models for both standard half-life (SHL) and EHL FVIII and FIX concentrates, as reported in literature. In this way, we aim to support physicians in their choices that model best suits each individual patient. Moreover, we propose some suggestions for best practice with regard to data collection and documentation after evaluating these models in detail to increase the reliability of PK-guided dosing and to aid in future population PK model construction.
Methods
Literature search strategy
We performed the following literature search to identify publications found on PubMed: (search date January 26, 2021): (haemophilia* OR hemophilia*) AND (“VIII”[Tiab] OR “IX”[Tiab]) AND (“population pharmacokinetic*”[Tiab] OR “pharmacokinetic model*”[Tiab] OR “population pharmacokinetic analysis”[Tiab] OR “population PK”[Tiab]) NOT (“monkey*”[Tiab] OR “mice*”[Tiab] OR “dog*”[Tiab] OR “rabbit*”[Tiab] OR “rat*”[Tiab]).
First, publications were selected by title and abstract. Reading full text, we included publications that described a new population PK model for prophylactic treatment with both SHL and EHL FVIII or FIX concentrates. In addition, models were required to be based on original patient data and had to be constructed using nonlinear mixed-effect modeling. Backward citations were screened to include additional studies. Population PK models for von Willebrand factor (VWF) containing concentrates were excluded. The screening process was performed by 2 independent authors (M.G., L.B.).
Data collection
We collected data from both the publications describing the development of the population PK model as well as from publications of the underlying clinical trials. The following clinical data of the study populations were collected: total number of patients, number of children (age <18 years), age; morphometric variables: body weight, body mass index (BMI), ideal body weight (IBW), lean body weight (LBW), body surface area (BSA), fat-free mass (FFM); endogenous factor level; factor concentrate; and assessed and included covariates and number of patients used for validation by data splitting. The collected laboratory data included the following items: type of assay used (OSA/CSA) and further specifications, namely reagent, calibrator, deficient plasma, and analyzer. Missing data were kindly requested from respective pharmaceutical companies or by correspondence with authors of the included publications.
Criteria to establish best practice
Based on our expert opinion and literature, we defined 12 clinical criteria of data documentation that should be reported in publications of population PK models. Thereafter, our included models were evaluated accordingly as complete (+), incomplete (±), absent (−), or not applicable. To specifically evaluate the quality of data documentation of included publications, only data were used as reported in the respective publications for this separate best practice evaluation. This is in contrast to the overview tables (Tables 1-4), in which all available and requested data are included. Documentation of 3 main demographic characteristics (number, age, and body weight) were evaluated for both the total population and pediatric populations separately. The first criterion (number) was scored as complete when the exact number of included (pediatric) patients was reported. If a minimum of pediatric patients was reported, we evaluated this item as incomplete. The criterion’s age, body weight, and other morphometric variables such as BMI, LBW, FFM, and IBW were scored “complete” if both measures of location (eg, median, mean) and dispersion (eg, range, standard error) were available as is scientifically common practice, and “incomplete” and “absent,” respectively, if 1 or both of those elements were lacking. In addition, if no exact age of the included children was given but only numbers of patients per age group (eg, between 6 and 12 years) was reported, age was defined as incomplete. As data splitting is a powerful method for model evaluation,15 we scored the criterion validation as complete if a data subset was used (comprising patients not used in the calibration population) to validate the constructed model. If other internal validations were executed, such as simulations and/or goodness-of-fit plots, we labeled the criteria as absent. Next, covariate analysis was evaluated as complete if the performance of a covariate analysis was reported (or the explanation why analysis was not performed), otherwise as absent. “Laboratory assay” was scored as complete, if the applied assay (OSA or CSA) was reported. If the other 4 further laboratory test specifications (applied reagent, calibrator, deficient plasma, and analyzer) were reported, this criterion was assessed as complete. If at least 1 of the 4 or no specification was reported, the publication was scored as incomplete or absent, respectively. Finally, we assessed whether the applied assays and reagents corresponded to the recommendations for that factor concentrate according to UK Haemophilia Centre Doctors' Organization guidelines.16 This guideline describes, for each licensed and available product in the United Kingdom, specific reagents that are regarded as suitable, or should be avoided. If the model was constructed using data obtained, by applying a suitable reagent, or if no specific recommendations were given by the guidelines for the specific factor concentrate, we evaluated this item as complete (+). If specific recommendations were given by Gray et al, but the applied reagent was neither defined as suitable, nor as a reagent that should be avoided, the criterion was evaluated criteria as incomplete (±). If models used factor concentrates that were not included in the UK guideline at time of publication, or if the reagents were not specified, scoring this criterion was not applicable (−).
Results
The initial search yielded 85 publications, of which 26 were included and 59 were excluded based on the set inclusion criteria. In addition, we included 1 supplementary article by backward citation,17 leading to a total of 27 included publications.
FVIII population PK models
A population PK model for FVIII concentrates was reported in 20 publications (Table 1). Of these 20, 6 combined data of multiple concentrates.11,18-22 Eleven publications described a population PK model for a SHL FVIII concentrate, 8 publications for an EHL FVIII concentrate, and 1 publication for both SHL and EHL FVIII concentrates.
Patient characteristics
Pediatric patients (<18 years) were included in most population PK models (16/20), although the exact number of included children could not be found or obtained in 5 population PK models. Details on the age of the children of 12 publications were collected and showed that all pediatric age groups were represented. Of the remaining 4 models that included children, only Nesterov et al restricted inclusions to children ≥12 years of age.23 Strikingly, we were only able to retrieve body weight of pediatric patients in 9/20 models.
Overweight and obese patients seem to be included in most studies based on the total maximum reported body weight. Details on other morphometric variables of the total population were reported in 13/20 publications (Table 1). BMI was mostly presented (11 publications), followed by LBW (3 publications) and BSA (2 publications). Of the 7 studies that did not report other morphometric variables, 3 assessed such a variable as a covariate but did not incorporate it in the model.
Model covariates and validation
Table 3 depicts all evaluated and included covariates. The following other morphometric variables were assessed and included as covariates: BMI by respectively 4 and no publication(s), BSA by 3 and 1 publication(s), LBW by 5 and 3 publications, and FFM by 2 and 2 publications. Remarkably, 5/7 models for EHL factor concentrates evaluated the incorporation of VWF levels, in contrast to 1 model for a SHL factor concentrate. Of these 6 models, 4 models included VWF as a covariate.
Table 3 also shows that 6 models have been validated using a subset of the data, in addition to validation by simulations. The ratio between the number of included patients used to develop the model (calibration dataset) and the number of patients used to validate the model (validation dataset) varied between 0.17 and 1.27. Bukkems et al24 externally validated and enriched the model by Nesterov et al.23
Laboratory data
Laboratory data are presented in Table 4. Both OSA and CSA were used to measure the FVIII levels. Moreover, 4 models applied both methods. The reagents used were described in 12/20 models, resulting in a total of 14 different reagents. Only 4 publications reported all 4 laboratory specifications.
FIX population PK models
Four population PK models for SHL FIX concentrates and 3 population PK models for EHL FIX concentrates were reviewed (Table 2). The models included a single recombinant FIX concentrate, with the exception of 2 models which combine multiple plasma-derived FIX concentrates.
Patient characteristics
Except for the nonacog beta pegol (Refixia) population PK model,25 all other 6 models included pediatric patients (<18 years), of which 3 only included children 12 years and older. However, only 3 of the 6 models reported the exact number children, although 2 models showed the number of included pediatric patients per age group.
Morphometric variables for pediatric patients were only available for the population model reported by Zhang et al. In descriptions of the total population, body weight was presented for all models and BMI for 2 models. Furthermore, only 1 model by Zhang et al assessed the inclusion of other morphometric variables as covariates (Table 3). Despite this lack of data, the parameter “patient’s body weight” showed a wide inter-individual variation in all studies. Strikingly, Zhang et al included patients up to a BMI of 63.1.26
Model covariates and validation
As shown in Table 3, only Collins et al did not report assessed and included covariates. As also depicted in Table 3, only 2 of the 7 models were internally validated by data splitting.27,28 The number of patients included in the calibration and validation dataset in the model by Suzuki et al were, respectively, 201 and 72, and 135 and 100 in the model by Diao et al.
Laboratory data
All FIX levels were measured using the OSA for both SHL and EHL FIX concentrates (Table 4). The used reagent was stated in 4 of the 7 population PK models. Only Collins et al reported all 4 laboratory specifications.
Best practice
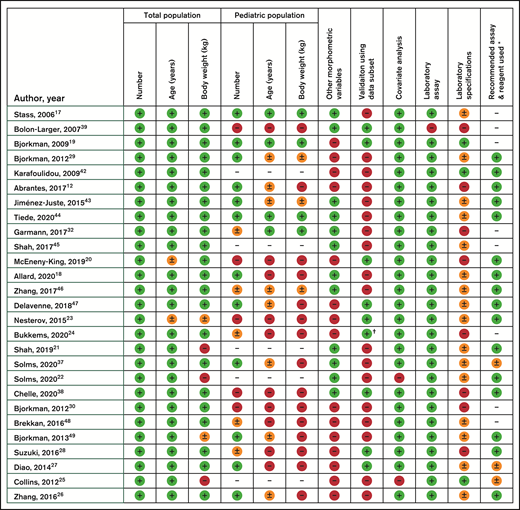
Figure 1 shows the evaluation of best practice of the included publications in this review. The scored items have been discussed previously. As is clearly visible, most publications cannot be scored as complete according to our strict set of criteria because of a lack of detailed information in several domains.
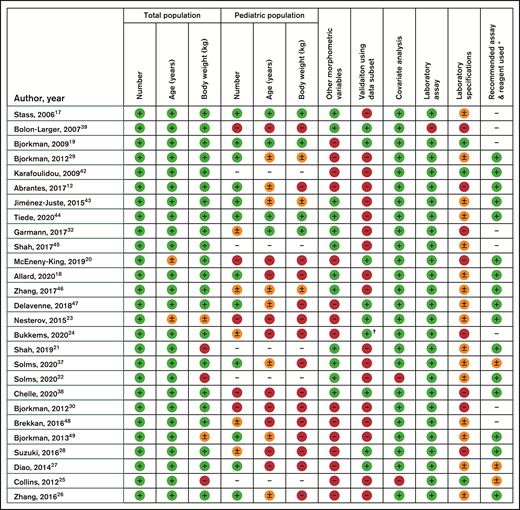
Evaluation of included models according to our personal recommendations of best practice, specifically on the quality of data documentation in the publications. Results presented as complete (green,  ), incomplete (orange,
), incomplete (orange,  ), absent (red,
), absent (red,  ) or not applicable ( – ). * According to UK guidelines by Grey et al. Haemophilia 2020. † Bukkems et al. have externally validated the model by Nesterov et al.
) or not applicable ( – ). * According to UK guidelines by Grey et al. Haemophilia 2020. † Bukkems et al. have externally validated the model by Nesterov et al.
Evaluation of included models according to our personal recommendations of best practice, specifically on the quality of data documentation in the publications. Results presented as complete (green, ), incomplete (orange, ), absent (red, ) or not applicable ( – ). * According to UK guidelines by Grey et al. Haemophilia 2020. † Bukkems et al. have externally validated the model by Nesterov et al.
Discussion
In this overview, we have collected detailed information on the demographic, clinical, and laboratory data used to construct 27 published population PK models for both SHL and EHL FVIII and FIX concentrates. We also introduce recommendations for best practices of data collection and documentation after evaluating the publications of included population PK models accordingly.
Patient characteristics
Importantly, because children have other PK characteristics than do adults, pediatric patients (mostly) of all ages were included in 22/27 PK populations. Body weight adjusted clearance decreases during normal development from child until adulthood, automatically affecting elimination half-life time because it is inversely related to clearance.29 Notably, approximately one-half of these age-related differences disappear when an identical sampling strategy is performed in children as in adults, instead of a reduced sampling strategy. Therefore, we advise, if possible, measuring FVIII and FIX levels 15 to 30 minutes after infusion.3 When both pediatric and adult patients are included in the data collection on the basis of which a population PK model is constructed, an exponential relationship between body weight, both total body weight and/or LBW, and clearance is introduced. This is also called allometric scaling. As a result, a population PK model can be used for patients of all ages.24,26,30 Allometric scaling is applied in all published models in this review, which is a positive finding. Historically, Bjorkman et al illustrates the importance of sufficient variation and volume of the included pediatric participation, as a poor population prediction for children of ages 3 to 10 years was observed with predicted FVIII levels 31% lower than observed FVIII levels because of inclusion of only 4 children from this age group.19 In Bukkems et al, it was shown that population clearance and central volume of distribution were underpredicted in patients aged <12 years when applying a population PK model based solely on patients 12 years and older.24
Overweight and obese adults are represented in most models. This is clinically relevant because of the increasing prevalence of overweight/obesity in 31% of hemophilia patients in Europe and North American, with concomitant risks of under- or overdosing of factor concentrates.31,32 With increasing BMI, studies show an increasing in vivo recovery of FVIII concentrate both in children and adults, with a decreasing steady state distribution volume.33-35 Unfortunately, no guidelines yet exist to optimize factor concentrate dosing in this patient group.
Van Moort et al reported that IBW-based dosing most accurately calculates doses of FVIII concentrate in comparison with other morphometric variables.36 This approach led to both better targeting of FVIII levels, as well as a mean reduction of 48.9% in prophylactic FVIII concentrate consumption during a 3-month period.4 Interestingly however, in the included population PK models in this review, IBW was neither evaluated nor included as a covariate. Contrastingly, LBW was evaluated and included as a covariate in 4 and 5 publications in our review, respectively. LBW showed a linear correlation with central volume of distribution and a positive nonlinear relationship with clearance.11,17,21,31,37 Stass et al reported that scaling to LBW is superior to scaling to actual body weight. A third morphometric variable that was included in several models as a covariate was FFM, which seemed to correlate better to plasma volume than total body weight.20,38
Model covariates and validation
Covariate analysis was described in all models, among which we specifically highlight the inclusion of VWF in the FVIII population PK models. Overall, a negative association between VWF and FVIII clearance is reported.23,24,37 In other words, the higher VWF level, the lower FVIII clearance. This is explained by the fact that VWF protects FVIII from proteolytic degradation and rapid clearance from the blood circulation.23 In our review, VWF was evaluated and included as covariate in 6 and 4 FVIII population PK models, respectively. Stass et al reported VWF as an important covariate, although it was not identified as one in the performed analyses. This contradiction can be attributed to the fact that only sparse data were available to adequately test covariate analysis,17 as also reported by Chelle et al.38 Therefore, when possible, we advise measurement of VWF antigen (VWF:Ag) levels during PK profiling and monitoring of FVIII concentrates to improve both PK guidance and model development.
Interestingly, only 8 models were validated by data splitting, a powerful, advanced method of internal validation. Although, the decision to split available data into a calibration and a validation set of course depends on the number of included study patients.7 Notably, in the FVIII population PK models by Bolon-Larger and Bjorkman et al, the number of included patients in the calibration dataset was 33 and 34, respectively. These were relatively small numbers compared with the patients in the validation sets of these studies (eg, 18 and 16 patients). In contrast, McEneny-King et al included 310 and 394 patients in the calibration and validation dataset, respectively.
Laboratory data
All publications described which assay was used to measure the factor levels, except for the model by Bolon-Larger et al.39 Because of the discrepancies between OSA and CSA and the effects on PK parameter estimation, it is essential that population PK models include results of both assays when both are used in clinical practice. Solms et al constructed separate population PK models based on OSA and CSA results and described that PK parameters were similar.37 However, application of correction factors for OSA and CSA is also generally applied when other assays are used.24,38 For example, Bukkems et al calculated an exponential function of 1.06 to correct for FVIII levels in an enriched FVIII-Fc Fusion protein population PK model. This resulted in an increase of FVIII levels from 100 IU/dL when measured by OSA compared with 131 IU/dL when measured by CSA.24
Because reagents have been identified as the most important cause of assay variability,16 it is striking that we were not able to identify the used reagent of 11/27 models. Interestingly, we identified 16 different documented reagents. Five models used several reagents, therefore increasing accuracy of PK guidance because of the included inter-reagent variability. To illustrate the importance of this covariate, external quality organizations for laboratories have reported variation between FIX measurements when using different OSA reagents between 2.0 and 19.0 IU/mL and when applying a golden standard of spiked plasma with recombinant FIX Fc fusion protein level of 6.0 IU/ML FIX.40 These variations may lead to underestimations that amount to up to 75%.16,40 Understandingly, these differences may lead to errors in PK guidance of dosing, especially when targeting FVIII or FIX trough levels, if reagent variability is not captured in the population PK model.
Therefore, UK Haemophilia Centre Doctors' Organization guidelines provide helpful advice on the appropriate choice of assays and reagents for each factor concentrate, which is licensed for use in the United Kingdom.16 Recommendations are based on safeguarding of assay results within 20% and 30% of FVIII and/or FIX target levels based on potency label in samples spiked as >30 IU/dL and <10 IU/dL. In addition, with each recommendation, it is noted if sufficient data are available to substantiate the guidelines. No population PK models in this review used assays or reagents that were advised to avoid.16
Strengths and limitations
The importance of more insight into the construction of population PK models is highlighted by the comparative study by Preijers and van Moort et al. For identical patients, 3 available PK tools produced significantly different PK parameters and clinically relevant variation in doses of recombinant FVIII concentrate. This was due to the influence of the patient data on which the population PK model was based and the resulting PK parameters that affected individual estimations.41
Our review has some limitations. First, we may have missed population PK models because we only performed the literature search using PubMed and reference searches; however, we do believe that we have included most population PK models used in clinical practice with this approach. Second, we did not succeed in contacting all authors, which may have affected the completeness of presented results. Next, in the formulation of best practice, we did not integrate the number of patients in the validation dataset relative to the number patients in the calibration set because experts have not reached consensus on aspect of validation.7 Finally, we did not apply ranking to the items incorporated in our recommendations for best practice. Despite these limitations, we believe this review provides a valuable and objective overview of the most relevant aspects needed to ensure reliability and feasibility of PK guidance of factor concentrates in hemophilia care.
Recommendations
Clinicians or clinical pharmacologists, who treat patients with PK-guided dosing, should evaluate whether the population PK model is suitable for each individual patient, based on the data used to construct the model. According to our evaluation of best practice in hemophilia, improvements can be made on data collection and documentation. If a factor concentrate is approved and registered for all age groups, we recommend that the corresponding population PK model includes pediatric patients and that their characteristics are described. Also, patients with both underweight and overweight/obesity should be included with documentation of morphometric variables such as BMI, LBW, IBM, or FFM. Next, new population PK models should be validated by a subset of the dataset or even more optimally by external validation to ensure optimal safety in standard clinical practice. Also, when performing PK guidance, it is essential that laboratory specifications, both of the patient populations used to construct the models as well as the patients who receive PK guidance, are reported and taken into account, preferably in accordance with international guidelines.
These recommendations with regard to detailed information on patient characteristics, laboratory assays and reagents, and validation strategies may of course also apply to PK guidance of novel and upcoming therapies in hemophilia with alternative administration routes, as well as expected combined treatment modalities such as emicizumab and on-demand FVIII concentrate, gene therapy, and on-demand FVIII or FIX concentrates.
Conclusion
This overview presents the wide variation in detail of included clinical and laboratory data used to construct 27 population PK models. By providing detailed information on these population PK models, the applicability and reliability of PK-guided prophylactic dosing of factor concentrates in hemophilia A and B can be investigated by (pediatric) hematologists. We also recommend best practice with regard to data collection to enable reliable PK guidance of treatment in patients with bleeding disorders as a whole for current and future therapies.
Acknowledgments
The SYMPHONY consortium, which aims to orchestrate personalized treatment in patients with bleeding disorders, is a unique collaboration between patients, health care professionals, and translational and fundamental researchers specializing in inherited bleeding disorders, as well as experts from multiple disciplines. It aims to identify best treatment choice for each individual based on bleeding phenotype. To achieve this goal, work packages (WP) have been organized according to 3 themes (eg, Diagnostics [WPs 3 and 4], Treatment [WPs 5-9], and Fundamental Research [WPs 10-12]). This research received funding from the Netherlands Organization for Scientific Research (NWO) in the framework of the NWA-ORC Call grant agreement NWA.1160.18.038. Principal investigator: M.H. Cnossen; project manager: S.H. Reitsma. Beneficiaries of the SYMPHONY consortium: Erasmus MC and Erasmus MC Sophia Children’s Hospital, University Medical Center Rotterdam, project leadership and coordination, Sanquin Diagnostics, Sanquin Research, Amsterdam University Medical Centers, University Medical Center Groningen, University Medical Center Utrecht, Leiden University Medical Center, Radboud University Medical Center, Netherlands Society of Hemophilia Patients, Netherlands Society for Thrombosis and Hemostasis, Bayer B.V., CSL Behring B.V., and Swedish Orphan Biovitrum (Belgium) BVBA/SPRL. This study was also performed as part of the OPTI-CLOT international multicenter research consortium, “Patient Tailored Pharmacokinetic (PK) Guided Dosing of Clotting Factor Concentrates and Desmopressin in Bleeding Disorders,” which is currently WP 6 within the SYMPHONY consortium. This paper is written on behalf of the international multicenter OPTI-CLOT and To WiN studies that aim to implement a PK-guided approach for the treatment of bleeding disorders using population PK models for desmopressin, factor concentrates, and other alternative drugs. OPTI-CLOT and To WiN study group members are: Steering committee: M.H. Cnossen (principal investigator and chair), F.W.G. Leebeek, Erasmus MC Sophia Children’s Hospital and Erasmus MC, University Medical Center Rotterdam, Rotterdam; R.A.A. Mathôt (co-leading investigator), K. Fijnvandraat, M. Coppens, Amsterdam University Medical Center, Amsterdam, University Medical Center, Amsterdam; K. Meijer, University Medical Center Groningen, Groningen; S.E.M. Schols, Radboud University Medical Centre, Nijmegen; H.C.J. Eikenboom, Leiden University Medical Centre, Leiden; R.E.G. Schutgens, University Medical Center Utrecht, Utrecht; E.A.M. Beckers, Maastricht University Medical Center, Maastricht; and P. Ypma, Haga Hospital, The Hague. Principal investigators and local collaborators in the Netherlands: M.J.H.A. Kruip, S. Polinder, Erasmus MC, University Medical Center Rotterdam, Rotterdam; R.Y.J. Tamminga, University Medical Centre Groningen, Groningen; P. Brons, Radboud University Medical Centre, Nijmegen; K. Fischer, K.P.M. van Galen, University Medical Centre Utrecht, Utrecht; F.C.J.I. Heubel-Moenen, Maastricht University Medical Centre, Maastricht; L. Nieuwenhuizen, Maxima Medical Centre, Eindhoven; M.H.E. Driessens, The Netherlands Hemophilia Patient Society; I. van Vliet, Erasmus MC, University Medical Centre Rotterdam, Rotterdam. OPTI-CLOT/To WiNs: J. Lock, H.C.A.M. Hazendonk, I. van Moort, J.M. Heijdra, M..H.J. Goedhart, W. Al Arashi, Erasmus MC, University Medical Center Rotterdam, Rotterdam; T. Preijers, N.C.B. de Jager, L.H. Bukkems, M.E. Cloesmeijer, A. Janssen, Amsterdam University Medical Centers, Amsterdam. Principal investigators and local collaborators in the United Kingdom—P.W. Collins, Arthur Bloom Haemophilia Centre, Institute of Infection and Immunity, School of Medicine, Cardiff University, Cardiff; R. Liesner, Great Ormond Street Haemophilia Centre, Great Ormond Street Hospital for Children NHS Trust, London; P. Chowdary, Katharine Dormandy Haemophilia Centre and Thrombosis Unit, Royal Free London NHS Foundation Trust, London; C.M. Millar, Hammersmith Hospital-Imperial College Healthcare NHS Trust, London; D. Hart, Department of Haematology, The Royal London Hospital Barts Health NHS Trust, London; and D. Keeling, Oxford Haemophilia and Thrombosis Centre, Oxford University Hospitals, Churchill Hospital, Oxford.
Authorship
Contribution: T.M.H.J.G. and L.H.B. performed the literature search and collected data; T.M.H.J.G. analyzed the data and wrote the manuscript; R.A.A.M. and M.H.C. supervised the study and provided critical guidance during the analyses; L.H.B., C.M.Z., R.A.A.M., and M.H.C. critically reviewed the paper and provided comments; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: M.H.C. has received grants from governmental and societal research institutes such as NWO, ZonMW, Innovation fund, from private funding institutions, institutional grants and unrestricted investigator research grants/educational and travel funding from the following companies over the years: Pfizer, Baxter/Baxalta/Shire, Bayer Schering Pharma, CSL Behring, Sobi Biogen, Novo Nordisk, Novartis, and Nordic Pharma, and has served as a member on steering boards of Roche and Bayer. All grants, awards, and fees go to the Erasmus MC as an institution. R.A.A.M. has received governmental and societal research institutes such as NWO, ZonMW, and Innovation Fund and unrestricted investigator research grants from Baxter/Baxalta/Shire/Takeda, Bayer, CSL Behring, and Sobi. He has served as an advisor for Bayer, CSL Behring, Merck Sharp & Dohme, and Baxter/Baxalta/Shire/Takeda. All grants and fees were paid to the institution.
Correspondence: Marjon H. Cnossen, Erasmus MC Sophia Children’s Hospital, University Medical Center Rotterdam, PO Box 2060, 3000 CB Rotterdam, The Netherlands; e-mail: m.cnossen@erasmusmc.nl.

 ), incomplete (orange,
), incomplete (orange,  ), absent (red,
), absent (red,  ) or not applicable ( – ). * According to UK guidelines by Grey et al. Haemophilia 2020. † Bukkems et al. have externally validated the model by Nesterov et al.
) or not applicable ( – ). * According to UK guidelines by Grey et al. Haemophilia 2020. † Bukkems et al. have externally validated the model by Nesterov et al.
