Key Points
Stem cell transplantation (SCT) has a positive impact on several quality-of-life domains in patients with SCD and thalassemia.
Quality-of-life outcomes should be reported in all ongoing SCT, gene therapy, and gene-editing trials in SCD and thalassemia.

Abstract
Patients with sickle cell disease (SCD) and thalassemia experience several complications across their lifespan that lead to impairment in different health-related quality of life (HRQOL) domains. There is increasing interest in curative therapies for patients with SCD and thalassemia, including hematopoietic stem cell transplant (HSCT) and gene therapy; however, the effect of these therapies on various HRQOL domains remains unclear. Our objective was to systematically evaluate the most recent evidence for the effect of HSCT and gene therapy on HRQOL in patients with SCD and thalassemia. A systematic search of medical literature databases was conducted. A total of 16 studies (thalassemia, n = 9; SCD, n = 6; both, n = 1) involving 517 participants met inclusion criteria (thalassemia, n = 416; SCD, n = 101). HSCT was associated with a small to large positive effects in most HRQOL domains (Cohen’s d; mean = 0.47; median = 0.37; range, 0.27-2.05). In thalassemia, HSCT was frequently associated with large positive effects in physical and emotional HRQOL domains (median d = 0.79 and d = 0.57, respectively). In SCD, HSCT was associated with large positive effects in all HRQOL domains. Emerging data suggest improvement in HRQOL outcomes across different domains following gene therapy in thalassemia and SCD. The quality of evidence was moderate in 13 studies (81%). HSCT has a positive impact on several HRQOL domains in patients with SCD and thalassemia; however, more longitudinal studies are warranted to assess the sustainability of these effects. Reporting HRQOL outcomes from ongoing gene therapy or gene-editing trials in SCD and thalassemia is key to better understand the benefits of such therapies.
Introduction
The curative therapies of hematopoietic stem cell transplant (HSCT) and gene therapy or editing are increasingly used to treat patients with hemoglobinopathies, including thalassemia and sickle cell disease (SCD).1-4 It is estimated that 5% of the world’s population carries at least 1 variant globin allele for thalassemia.5 Additionally, birth prevalence for homozygous SCD is estimated at 112 per 100 000 live births globally, along with 4230 per 100 000 heterozygous SCD births.6
Thalassemia occurs as a result of an imbalance in the synthesis of either α- or β-globin chains, leading to excess production of 1 chain within the red blood cells, which in turn results in chronic hemolysis and ineffective erythropoiesis.7 Patients with severe form of thalassemia are transfusion dependent and are at increased risk for several complications, including iron overload,8 growth impairment,9 bone abnormalities,9,10 osteoporosis,11-14 endocrinopathies,9,15 pulmonary hypertension,16 splenomegaly,8 and hypercoagulability.17
SCD results from the replacement of glutamic acid with valine in the β-globin subunit of hemoglobin. This genetic alteration leads to the formation of the new hemoglobin S molecule (HbS), which in turn leads to Hb polymerization and sickling of the red blood cells, causing increased hemolysis and vaso-occlusion.18 Vaso-occlusion is a multifactorial process that includes activation of endothelial and blood cells, small and large blood vessel involvement, hypoxia/reperfusion injury, ischemic tissue damage, and chronic inflammation.19 Several genetic variants of SCD exist, including homozygous HbSS and common compound heterozygous forms such as HbSC, HbSbeta+, and HbSBeta0 thalassemia.20,21 Patients with SCD suffer from several complications, including debilitating acute and chronic pain, acute chest syndrome, pulmonary hypertension, chronic anemia, strokes, kidney disease, gall stones, and other chronic end-organ damage.22
Disease-related complications across the lifespan, in both thalassemia and SCD, significantly impair physical, mental, and psychosocial domains of health-related quality of life (HRQOL).23 Chronic blood transfusion with iron chelation is the standard treatment of transfusion-dependent thalassemia (TDT) patients,8 and is usually needed for SCD patients with or at increased risk of stroke or cerebrovascular events.22 Chronic transfusion has been associated with improvement in HRQOL outcomes in both TDT and SCD.23-25 Hydroxyurea is the most commonly used disease-modifying therapy in SCD26-29 and has been also associated with benefits for HRQOL outcomes, in particular physical and psychosocial domains.30-32 More therapies have been recently approved by the US Food and Drug Administration, including luspatercept for TDT,33 as well as l-glutamine, crizanlizumab, and voxelotor for SCD34,35 ; nevertheless, their potential benefits for HRQOL outcomes is not well established. Although HSCT and gene therapy or editing remain the only curative options for these patients,36-38 the cumulative effects of these therapies on different HRQOL domains are unclear.
The objective of this systematic review was to evaluate recent available longitudinal and cross-sectional data for evidence of the effects of both HSCT and gene therapy or gene-editing on HRQOL outcomes in patients with TDT or SCD. We also compared HRQOL outcomes in these populations vs various control groups, including healthy controls and TDT or SCD patients receiving other therapies, as well as reported correlates of different HRQOL domains.
Methods
This systematic review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines.39 The PRISMA checklist is included as supplemental Table 1. Studies were included if they (1) involved patients with thalassemia or SCD (HbSS, HbSC, HbS/β0/+ thalassemia); (2) examined the role of curative therapies (intervention), including HSCT or gene therapy in these populations; (3) included HRQOL outcomes using reliable and validated measures or instruments; and (4) were cross-sectional or longitudinal studies with 5 or more participants as a minimum number of observations that could provide meaningful insight into HRQOL outcomes in SCD or thalassemia following HSCT. We only included studies that used reliable and validated HRQOL instruments to: (1) have more consistency in the measured outcomes; (2) allow comparisons across studies; and (3) provide an opportunity to calculate effect sizes of different HRQOL domains in SCD and thalassemia following HSCT. Case reports of fewer than 5 participants, reviews, viewpoints, editorials, letters to the editor, animal studies, and studies of laboratory investigations were excluded. Primary outcomes were patient- and/or proxy-reported HRQOL scores in physical, emotional, social, and other reported domains using validated measures. Secondary outcomes were (1) correlates of patient- and/or proxy-reported HRQOL domain scores and (2) intervention type (HSCT, gene therapy, or gene-editing).
The literature search was conducted in the following databases: PubMed MEDLINE and Cochrane Central Register of Controlled Trials on the Wiley platform. Database searches included articles indexed as of May 20, 2019, without publication date restriction. The search terms used were a combination of (1) thalassemia OR sickle cell AND (2) stem cell transplant OR bone marrow transplant OR gene therapy AND (3) health related quality of life OR quality of life. The search was limited to human data, without language restrictions. We also scanned reference lists of included studies for any additional articles. Two independent reviewers carried out title and abstract screening, full-text screening, and quality assessment. Disagreements were resolved by discussion and consensus. Data extraction for studies meeting inclusion criteria using a standardized form. We used Grades of Recommendation, Assessment, Development, and Evaluation (GRADE) criteria to evaluate the quality of the evidence for the included studies.40,41 GRADE criteria include 5 key domains: study design, directness, consistency of results, imprecision of results, and risk of bias.
We analyzed the evidence quantitatively and qualitatively. Our primary outcome was the mean change in patient- and caregiver-reported HRQOL scores across different domains using baseline (eg, pre-HSCT) and follow-up (eg, post-HSCT) values. We were able to calculate Cohen’s d effect size (ie, standardized mean differences) with 95% confidence intervals in 8 of the included studies, using means and standard deviations, to evaluate the effect of HSCT or gene therapy on improving various domains of patient- and caregiver-reported HRQOL outcomes.42 These Cohen’s d effect sizes reflected the differences in HRQOL domain scores for HSCT recipients vs healthy controls or patients receiving other therapies (n = 6, 75%), or pre- vs post-HSCT (n = 2, 25%). When standard deviation of HRQOL scores were not reported, Cohen’s d effect size was calculated using the recommended range rule for standard deviation or the formula for standard deviation of means without reported standard deviations.43 We contacted the corresponding authors of the longitudinal studies included in our review to obtain additional information related to their HRQOL scores in all follow-up visits; however, we received no response and were not able to calculate the change in Cohen’s d effect size over time. We analyzed data using Microsoft Excel.
Results
Literature search
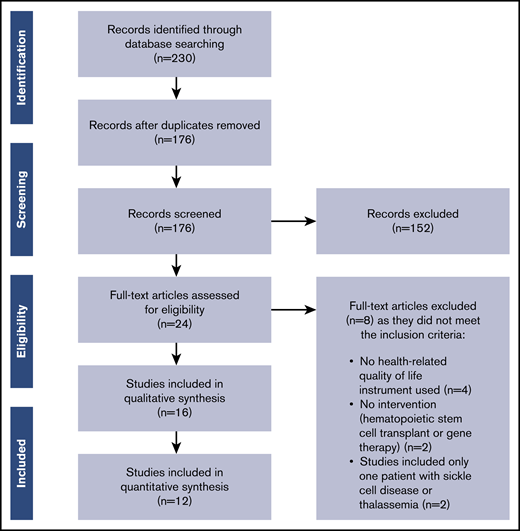
The PRISMA flow diagram detailing selection of studies included in this review is provided in Figure 1. The literature search identified 230 articles. After removing duplicates, 176 articles remained; of these, 152 articles were excluded based on the title and abstract screening, and 24 articles were retrieved for full-text screening. Sixteen articles met all inclusion criteria and were included in this review.
Description of all included studies
The majority of the studies that met the inclusion criteria assessed patients with thalassemia (n = 9, 56.3%),44-52 1 study covered both thalassemia and SCD patients (n = 1, 6.25%),53 and the remaining studies were focused on SCD patients (n = 6, 37.5%).54-59 A total of 517 patients and/or their parents took part in the included studies and reported HRQOL outcomes44-59 ; of these, 101 (19.5%) had SCD53-59 and 416 (80.5%) had thalassemia.44-53 Participants were either patients only (n = 9, 56.3%), parents or proxy only (n = 1, 6.3%), or both (n = 6, 37.5%). Seven studies (43.8%) included both pediatric and adult populations, whereas others included either pediatric (n = 4, 25%) or adult (n = 5, 31.3%) ones. All of the included studies used at least 1 validated HRQOL instrument.
Description of study characteristics in thalassemia
Table 1 summarizes the characteristics for all studies that included thalassemia patients post-HSCT. Half of the studies (n = 5) used the Pediatric Quality of Life (PedsQL) instrument to evaluate HRQOL outcomes following HSCT,44,47,49,51,52 3 (30%) used the Short Form 36 (SF-36) survey,46,48,50 2 (20%) used World Health Organization Quality of Life Brief Version,47,52 1 (10%) used the Child Health Rating Inventories,53 1 (10%) used the European Organization for Research Treatment of Cancer Quality of Life Questionnaire,45 1 (10%) used European Quality of Life 5D (EQ-5D),48 and 1 study (10%) used the Functional Assessment of Cancer Therapy-Bone Marrow Transplant survey.50 Four studies used multiple HRQOL instruments.47,48,50,52
Seven studies (70%) enrolled patients from multiple centers,44-48,50,53 and 3 (30%) were single centers.49,51,52 There were 8 (80%) cross-sectional45-52 and 2 (20%) longitudinal studies.44,53 There was a control arm for comparison of HRQOL outcomes in 6 of the 8 (75%) cross-sectional studies46-48,50-52 and 1 of the 2 (50%) longitudinal studies.53 Details for each of these control arms are described in supplemental Table 2. Four studies were conducted in Italy,44-46,50 2 in Turkey,49,52 and 1 each in Hong Kong,47 India,51 Iran,48 and the United States.53 All studies reported HRQOL outcomes for patients who had undergone allogenic HSCT.44-47,49-53
One-half of the studies (n = 5, 50%) did not report the specific thalassemia diagnosis of the patients included in the study.45,46,48,49,53 The other one-half (50%) reported that patients with β thalassemia major were included,44,47,50-52 and 1 study included a patient with hemoglobin Bart’s disease.47 Patients’ age ranged from 2 to 48 years, with the reported means ranging from 10 to 33 years and medians ranging from 8 to 34 years.44-53 The average number of thalassemia participants per study was 42, with a median of 34 participants (range, 6-109).44-53 Most studies reported the details of their conditioning regimens (n = 6, 60%),44,45,50-53 but 3 studies (30%) did not.46-48
The duration between HSCT and time of evaluating HRQOL outcomes varied in the cross-sectional studies and was less than a year from HSCT in 4 studies45,46,48,49 and at least 1 year after HSCT in the other 4 studies.47,50-52 In the longitudinal studies, surveys were taken at baseline, 3, 6, and 18 months post-HSCT in the study by Caocci and colleagues,44 and at baseline, 45 days, 3, 6, and 12 months post-HSCT in the study by Kelly et al.53 No studies reported HRQOL outcomes for patients with thalassemia following gene therapy.
Description of study characteristics in SCD
Table 2 summarizes the characteristics for all studies that included SCD patients post-HSCT. Three of the studies (42.9%) used the PedsQL instrument to evaluate HRQOL outcomes following HSCT54,55,57 ; 2 (28.6%) used the SF-36 survey56,59 ; 1 (14.3%) used the Child Health Rating Inventories survey53 ; 1 (14.3%) study used EQ-5D54 ; and1 (14.3%) study used the Patient Reported Outcome Measurement Information System-57 (PROMIS-57) survey.58 One study used multiple HRQOL instruments.54 Five studies (71.4%) were single-center,54-57,59 and 2 studies (28.6%) were multicenter.53,58 Two studies (28.6%) were cross-sectional54,56 ; of these, Arnold et al reported an average of 6 years since transplant to the time of survey along with study controls (healthy siblings and SCD patients) receiving other therapies (ie, non-HSCT),54 whereas Gallo et al reported a median of 2.71 years with no control.56 Five studies (71.4%) were longitudinal,53,55,57-59 of which all but 257,58 (40%) collected data from patients from baseline to 1 year post-HSCT, and only 1 (20%) used a control.53 All 7 studies that included SCD patients were conducted in the United States,53-59 and all studies reported outcomes for SCD patients who had undergone allogenic HSCT.53-59
SCD genotype varied across studies. Five studies (71.4%) reported including HbSS patients,54,55,57-59 4 (57.1%) included HbSC patients,54,55,58,59 2 (28.6%) included HbS-β0 or HbS/β+ Thal,55,58 2 (28.6%) included HbS-β0 Thal,54,57 and 1 study (14.3%) included HbS-β+ Thal.54 Two studies did not report the specific SCD genotype for included patients.53,56 Patients’ ages ranged from 2 to 52 years old, with the reported means ranging from 8 to 28 years and medians ranging from 7 to 31 years.53-59 The average number of SCD participants per study was 14, with a median of 16 participants (range, 7-18). Most studies enrolled patients who had undergone myeloablative (n = 3, 42.9%)53,54,57 or reduced-intensity (n = 4, 57.1%)54,55,57,58 conditioning regimens. In addition, 2 studies (28.6%) only enrolled patients who had undergone nonmyeloablative regimen.56,59
The duration between stem cell transplant and time of survey varied in the cross-sectional studies. The minimum time reported by Gallo et al was 1 year post-HSCT,56 whereas Arnold and colleagues reported a mean time of 6 years, but did not report a range.54 All but 157 of the longitudinal studies reported data from baseline to 1 year post-HSCT,53,55,58,59 and Green et al reported HRQOL outcomes that were as far out as 7 years post-HSCT.57 No studies reported HRQOL outcomes for patients with SCD following gene therapy.
HRQOL outcomes post-HSCT in thalassemia
Table 3 summarizes HRQOL outcomes and correlates for all studies that included thalassemia patients post-HSCT. Most studies reported significant improvement in several domains of HRQOL following HSCT, especially using the PedsQL and/or the SF-36 instruments. In 2 studies, parents reported worse HRQOL scores in different domains compared with their children44,53 ; in another study, patients’ total and psychosocial health scores correlated with parental Beck's Depression Index scores.49 Additionally, 2 studies reported a negative association between having acute or chronic graft-versus-host disease (GVHD) and HRQOL scores, in particular physical domains45,50 as well as worse overall HRQOL scores in patients who underwent HSCT after the age of 15 years.50
Correlates of HRQOL outcomes post-HSCT in thalassemia
Different studies reported several factors that correlated with better or worse HRQOL scores following HSCT. Cheuk et al found that longer time after transplant was associated with better emotional and psychosocial domain scores as well as less dependence on medical care and feeling more safe and secure as more time passed after transplant.47 The most commonly reported factor that was significantly associated with worse HRQOL scores was GVHD as reported by one-half of the included studies (5/10, 50%).44,45,49,50,52 Other correlates that were significantly associated with worse HRQOL scores included: other comorbidities such as cardiac dysfunction,47,49 risk class III for transplant,47 number of daily and long-term medications,47 urban residence,48 parental depression,49 age at time of transplant older than 15 years,50 and living alone.50 Factors that were significantly associated with better HRQOL scores included the absence of GVHD,50 risk class II for transplant,47 1 or no daily long-term medications,47 rural residence,48 and higher education status.49 Factors that were not significantly associated with better or worse HRQOL scores were age at time of transplant,51 gender,52 welfare status,52 other demographics,48,49 type of transplant,51 and ferritin levels.52
HRQOL outcomes post-HSCT in SCD
Table 4 summarizes HRQOL outcomes and correlates for all studies that included SCD patients post-HSCT. Most studies reported improvement in HRQOL outcomes in SCD patients following HSCT.53,55,57-59 In particular, 4 studies reported significant improvement in physical, emotional, psychosocial, general health, bodily pain, pain interference, and vitality domains.53,55,58,59 Studies that examined HRQOL outcomes over the course of HSCT reported early drop or worsening in various domain scores followed by improvement in these scores by day 30 in 1 study59 and day 45 in another.53 In contrast, Arnold et al reported no significant differences in the HRQOL outcomes among HSCT patients, SCD patients (no HSCT), and their unaffected siblings.54 The authors cited the high usage of hydroxyurea among SCD patients not undergoing HSCT for this lack of difference in SCD patients who did vs did not undergo HSCT.54
Correlates of HRQOL outcomes post-HSCT in SCD
One study reported an association between insurance type and HRQOL outcomes, demonstrating Medicaid patients had better HRQOL domain scores at 1 year compared with those with private insurance.55 Comorbidities, such as GVHD and avascular necrosis, were associated with worse HRQOL scores,55,56 and HSCT recipients without GVHD reported better emotional and psychosocial domain scores at 1 year post-HSCT.55 Moreover, Green and colleagues reported an association between normal baseline brain imaging (ie, magnetic resonance imaging, fluid-attenuated inversion recovery) and better HRQOL post-HSCT, when compared with SCD patients with abnormal pretransplant brain magnetic resonance imaging.57
Effect size for HRQOL outcomes post-HSCT in thalassemia and SCD
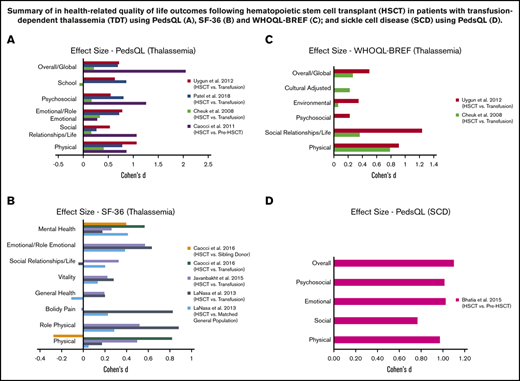
We calculated the effect size (Cohen’s d) for HRQOL outcomes among thalassemia HSCT recipients compared with their respective controls (Table 5) (Figure 2A-C). All studies reported a small (Cohen’s d < 0.2) to large (Cohen’s d > 0.8) significant positive effect (improvement) across almost all domains among HSCT recipients, compared with thalassemia patients receiving chronic transfusions.44,46-48,50-52 The only domains with no significant improvement in HSCT recipients, compared with their controls, included school, bodily pain, and social functioning in 3 different studies, 1 domain in each.47,48,50 When compared with matched healthy general population or sibling controls,46,50 almost all domains had a small to medium positive effect size with HRQOL scores among HSCT recipients, except physical domain in 1 study and general health in another.46,50 We were not able to calculate the effect size for 3 studies where the authors did not include a control group for comparison or the required data were missing.45,49,53
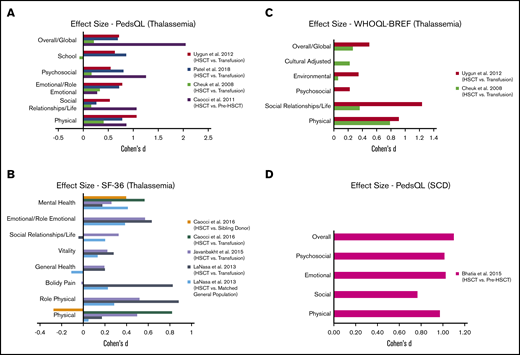
Summary of in health-related quality of life outcomes in patients. Patients with thalassemia using PedsQL (A), SF-36 (B), and World Health Organization Quality of Life Instruments (C). (D) Patients with SCD using PedsQL.
Summary of in health-related quality of life outcomes in patients. Patients with thalassemia using PedsQL (A), SF-36 (B), and World Health Organization Quality of Life Instruments (C). (D) Patients with SCD using PedsQL.
We were able to calculate effect size (Cohen’s d) using data from only 1 study that included SCD following HSCT55 ; other studies either had missing data or did not include a control group. Bhatia et al showed large positive effects and improvement in HRQOL outcomes across all domains at 1 year following HSCT (Table 5) (Figure 2D).
HRQOL outcomes following gene therapy in thalassemia and SCD
Limited, yet promising, data are emerging to support potential benefits and improvement of HRQOL outcomes in TDT and SCD following gene therapy.60-63 Using the EQ-5D-3L or EQ-5D-Y instrument, a report of 8 TDT patients who underwent gene therapy and achieved transfusion independence suggested an improvement in HRQOL outcomes, with health state today score increasing from 65 to 96 (baseline) to 90 to 100 (24 months).60 Similarly, among TDT patients younger than 18 years of age who achieved transfusion independence following gene therapy, using EQ-5D-Y, health state today score improved from 67 (range, 50-96) at baseline to 92.5 (range, 85-95) at 12 months.63 In 2 reports, by Kanter et al and Thompson et al, the authors evaluated HRQOL outcomes in SCD patients following gene therapy using PROMIS-57 and Pain Intensity numeric rating scale of 0 to 10.61,62 The authors reported clinically meaningful improvement in the mean HRQOL scores across different domains from baseline to 12 months, including: numeric rating scale pain intensity (6 to 2.4) as well as PROMIS-57 for pain interference (63 to 48), anxiety (62 to 48), depression (62 to 44), satisfaction with social roles (39 to 60), fatigue (50 to 43), and physical function (40 to 56), respectively. We found no studies reporting on HRQOL outcomes following gene-editing in TDT or SCD.
Discussion
HSCT and gene therapy are the only curative therapies for both thalassemia and SCD. In this systematic review, we identified 16 studies with 517 participants and of low to high quality, that evaluated HRQOL in these populations post-HSCT. Several studies demonstrated significant improvements in various domains of HRQOL after HSCT for patients with SCD and thalassemia, regardless of the conditioning regimen (ie, myeloablative or nonmyeloablative). The PedsQL and the SF-36 instruments were the most commonly used tools. Emerging data suggest improvement in HRQOL outcomes across different domains following gene therapy in thalassemia and SCD. The included studies provided evidence to support the short- and long-term benefits of these curative therapies and to help health care providers as well as patients in their decision-making process.
We found moderate quality evidence to suggest that HRQOL domain scores following HSCT in patients with SCD or thalassemia may be comparable to that measured in the general population.46,50,54 Using the PedsQL instrument, baseline HRQOL domain scores among SCD participants before HSCT are worse than that expected for children with other chronic health conditions.64 This may be due to physical symptoms related to SCD, such as pain and fatigue, as well as anxiety associated with anticipation of HSCT.55 The significant improvement in physical domain scores, in both SCD and thalassemia, after HSCT may be attributed to achieving better disease control and resolution of their chronic anemia and its associated symptoms. There was no significant improvement in social domain scores in a number of studies. This could be due to the prolonged hospitalization during HSCT and/or the isolation period that patients have to go through after transplant because of the risk of infection and missing school attendance.55,65 Other possible explanations include stigma related to the underlying illness (ie, SCD or TDT), the perceptions of being different, their dependence on parents in different daily activities, and losing social interactions with friends at and outside school for an extended period.61 Nevertheless, it is also possible that HSCT may not have an expected impact on relational or social HRQOL outcomes.61 The age at the time of HSCT is an important consideration when evaluating HRQOL outcomes. One study showed that patients with SCD or thalassemia who receive HSCT at an early age reported better HRQOL outcomes compared with those who received HSCT later in life, in particular after the age of 15 years.50 This is related to the fact that younger patients are more likely have less frequent disease-related complications, comorbidities, or end-organ damage before HSCT. In contrast, another study showed no association between age at the time of transplant and HRQOL outcomes.51 Further, HRQOL outcomes varied in pediatric and adult studies with no clear demonstration of additional benefits in one over the other. Time from HSCT seems to be an important variable to consider when evaluating HRQOL outcomes in these populations. In our review, the time of HRQOL assessment in relation to HSCT varied widely, regardless of the age (pediatric vs adult), population (TDT, SCD, or both) and study design (cross-sectional vs longitudinal). HRQOL outcomes were more likely to improve as patients are further away from the time of HSCT in both longitudinal and cross-sectional studies; nevertheless, this observation was not consistent across all domains in HRQOL outcomes.
There is a well-established literature that describes the difficulty parents have in rating HRQOL outcomes of their children, particularly internalizing symptoms (eg, anxiety, depressive symptoms, anger, stress).66-70 In 2 studies, patients were more likely to report better HRQOL outcomes compared with their parents,44,53 whereas in another study, both reported comparable or similar HRQOL scores.57 Differences in HRQOL outcomes reported by patients and parents could be related to the psychosocial distress parents experience post-HSCT while providing care for their other children as well, and that the improvement in each domain may not be immediately apparent to the parents. Additionally, parental depression may also affect their children’s HRQOL scores.49
In our review, we found limited, yet promising, data that suggest potential improvement of HRQOL outcomes in TDT and SCD following gene therapy60-63 ; however, we found no studies reporting on HRQOL outcomes following gene-editing in these populations. In particular, TDT patients who achieved transfusion independence demonstrated more benefits in their HRQOL outcomes. Long-term follow-up studies following gene therapy in TDT are ongoing and more data are expected to be available in the next few years as more patients complete study assessments.60,63 In SCD, patients reported remarkable improvement in their HRQOL outcomes following gene therapy when their baseline scores were worse than the general population.61,62
Our systematic review has several strengths.71 First, we followed the recommendations for rigorous systematic reviews methodology.39-41,72-74 Second, we used a sensitive search strategy guided by a librarian (ie, information specialist) with no language or publication date restriction to identify the largest possible number of relevant studies and minimize publication bias.75 Third, we searched bibliographies of all included articles and relevant systematic reviews for any potentially eligible studies. Finally, the entire systematic review process was completed by 2 independent authors/reviewers, including title and abstract screening, full text screening, data extraction, and grading the quality of the evidence.39,72
Some potential methodological limitations of our systematic review are worth mentioning.71 First, similar to other published systematic literature reviews, although we used a sensitive and comprehensive search strategy, it is possible that we may have still missed some relevant articles or unpublished studies in abstract form in the gray literature.71 Second, we only included published studies in peer-reviewed journals to focus on the strongest available evidence in the literature; therefore, publication bias cannot be excluded. For example, positive study results are more likely to be published.75 Third, some studies had a small sample size and others lacked a control group to compare HRQOL outcomes, which limited our interpretation of their findings. Many studies were cross-sectional and only a few longitudinal studies had limited pre- and post-HSCT data available, some of which also had a short follow-up period. Additionally, the included studies measured different HRQOL domains using a number of validated instruments. These issues limited our ability to perform a meta-analysis of HRQOL outcomes71 ; nevertheless, we were able to calculate a summary Cohen’s d or effect sizes for various HRQOL domains across studies that used the same validated instruments.42,43 Finally, the number of studies that met our predefined criteria was relatively small and we found no published gene therapy studies with reported HRQOL outcomes; however, this is probably because of gene-based therapies representing a new field of investigation with fewer published data available.
Conclusion
In our review, we found that HRQOL improves in patients with SCD or TDT after HSCT. We identified several patient- and HSCT-related factors across studies that were associated with either improved or impaired HRQOL after HSCT; nevertheless, sample size, age groups, and study design in the included studies varied, which are important considerations in our interpretation of their findings. Emerging data suggest improvement in HRQOL outcomes across different domains following gene therapy in thalassemia and SCD. By understanding HRQOL outcomes after HSCT and gene therapy, patients can be offered more insight to help them in the decision-making process regarding interventions. Therefore, assessing HRQOL outcomes using reliable and valid generic and/or disease-specific measures in all future prospective HSCT, gene therapy or gene-editing trials is essentially needed.
For original data used in this systematic review article, please contact sbadawy@luriechildrens.org.
Acknowledgments
The authors thank Midwestern University Library and Galter Health Sciences Library for their support with literature search as well as Mary Therese Forsyth (UCD School of Medicine, University College Dublin, Belfield, Dublin) for her help with article screening.
This project was supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (K23HL150232) (principal investigator: S.M.B.). The content is solely the responsibility of the authors and does not necessarily represent the National Institutes of Health.
Authorship
Contribution: S.M.B. and U.B. designed the review protocol study; S.M.B. and U.B. carried out the screening processes for inclusion, exclusion, and grading; U.B. extracted and analyzed the data; S.M.B., U.B., R.I.L., S.C., and A.A.T. interpreted the data; S.M.B. and U.B. drafted the paper; R.I.L., S.C., and A.A.T. critically revised the paper; and all authors approved the submitted final version of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sherif M. Badawy, Ann & Robert H. Lurie Children’s Hospital of Chicago, Northwestern University Feinberg School of Medicine, 225 E Chicago Ave, Box #30, Chicago, IL 60611; e-mail: sbadawy@luriechildrens.org.