Key Points
TFPIα is elevated in patients with mild bleeding disorders, especially in those with BUC and PFDs.
No genetic alterations in the F5 gene that are linked to increased TFPIα levels could be identified.

Abstract
High levels of tissue factor pathway inhibitor (TFPI), caused by a longer TFPIα half-life after binding to a factor V splice variant and variants in the F5 gene, were recently identified in 2 families with an as-yet-unexplained bleeding tendency. This study aimed to investigate free TFPIα in a well-characterized cohort of 620 patients with mild to moderate bleeding tendencies and its association to genetic alterations in the F5 gene. TFPIα levels were higher in patients with bleeding compared with healthy controls (median [interquartile range], 8.2 [5.5-11.7] vs 7.8 [4.3-11.1]; P = .026). A higher proportion of patients had free TFPIα levels more than or equal to the 95th percentile compared with healthy controls (odds ratio [OR] [95% confidence interval (CI)], 2.82 [0.98-8.13]). This was pronounced in the subgroup of patients in whom no bleeding disorder could be identified (bleeding of unknown cause [BUC; n = 420]; OR [95% CI], 3.03 [1.02-8.98]) and in platelet function defects (PFDs) (n = 121; OR [95% CI], 3.47 [1.09-11.08]). An increase in free TFPIα was associated with a mild delay in thrombin generation (prolonged lag time and time to peak), but not with alterations in routinely used global clotting tests. We could neither identify new or known genetic variations in the F5 gene that are associated with free TFPIα levels, nor an influence of the single-nucleotide variant rs10800453 on free TFPIα levels in our patient cohort. An imbalance of natural coagulation inhibitors such as TFPIα could be an underlying cause or contributor for unexplained bleeding, which is most probably multifactorial in a majority of patients.
Introduction
Mild to moderate bleeding disorders (MBDs) are characterized by symptoms such as epistaxis, easy bruising, or menorrhagia, but bleeding can also be severe under certain circumstances such as hemorrhage after surgery or birth.1 The most common diagnoses underlying MBDs are von Willebrand disease (VWD), platelet function defects (PFDs) and certain coagulation factor deficiencies (CFDs). Still, despite thorough hemostatic investigations, a majority of 50% to 70% of patients with MBDs lacks a diagnosis, categorized as patients with bleeding of unknown cause (BUC).2-5 Clinical characterizations of BUC patients do not differ from those with an established diagnosis of a bleeding disorder, nevertheless they have reduced thrombin generation and altered plasma clot properties.3,6,7 These observations underline the urgent demand to identify novel underlying mechanisms of bleeding disorders.8
Tissue factor (TF) pathway inhibitor (TFPI) is a pivotal anticoagulant player in hemostasis, regulating TF-induced coagulation. Among the 2 major isoforms, free TFPIα, primarily produced in endothelial cells, is the only anticoagulant isoform found in blood circulation. Its specific molecular structure as a multivalent Kunitz-type protease inhibitor allows it to bind to both, factor Xa (FXa) and TF-FVIIa, resulting in impaired thrombin generation.9,10 Furthermore, recent evidence shows a TFPIα related inhibition of the procoagulant function of truncated FV(a) isoforms and thus the prothrombinase assembly.11 The high-affinity binding of the negatively charged basic C terminus of TFPIα to the highly acidic region of FV-short leads to an increase of the TFPIα concentration. This better binding also assists in the localization of TFPIα to the surface of negatively charged phospholipids where FXa inhibition takes place.12
Only recently, the biological relevance of TFPIα/FVa interaction has been further elucidated, as increased levels of free TFPIα due to enhanced binding to truncated FV isoforms have been revealed as causal for a bleeding tendency in 2 families.13-16 In these subjects, binding of TFPIα to a truncated splice variant of FV led to a prolonged half-life of free TFPIα by protecting it from degradation and cleavage.12,15 Two B-domain variants in exon 13 of the FV-encoding gene resulting in FV splice variants have been identified: in the East Texas bleeding disorder (NM_000130.4:c.2350A>G, NC_000001.10:g.169511978T>C [h19, GRCh37]) the rarely used splice donor site is activated leading to the truncated form of FV (FV-short),15 and in the FV Amsterdam bleeding disorder (NM_000130.4:c.2588C>G, NC_000001.10:g.169511740G>C (h19, GRCh37)) the variant leads to a similar truncated form of FV.16 In both families, an increased free TFPIα-mediated inhibition of coagulation led to a clinical bleeding tendency, prolonged prothrombin (PT), and activated partial thromboplastin times (aPTT) as well as reduced thrombin generation.15,16
Besides these 2 variants, an intronic single-nucleotide variant (SNV) rs10800453 (NC_000001.10:g.169507076T>A [h19, GRCh37]) in the F5 gene was identified as being associated with elevated TFPI levels in a genome-wide association study.17
Based on these recent observations, increased free TFPIα levels might be associated with MBDs. Still, this has not been systematically investigated thus far. The goal of this study was to analyze levels of the biologically active free TFPIα as a possible underlying cause for increased bleeding and reduced thrombin generation in a cohort of thoroughly characterized patients with a mild to moderate bleeding tendency of known or unknown cause in comparison with a group of healthy controls. Genetic high-throughput sequencing data of the F5 gene was analyzed to identify genetic variations with a possible impact on patients’ free TFPIα levels.
Patients and methods
Study design and patients
The Vienna Bleeding Biobank (VIBB) is a single-center cohort of patients aged ≥16 years with a mild to moderate bleeding tendency, without an established diagnosis of a coagulation disorder, who were admitted to our outpatient clinic since 2009.3 Inclusion and exclusion criteria were published recently3 and are shown in supplemental Methods (paragraph 1). The assessment of the bleeding severity is described in supplemental Methods (paragraph 2).
One hundred age- and sex-matched healthy controls without a clinical bleeding tendency were recruited by trained health care personnel for comparison.
The study had approval by the Ethics Committee of the Medical University Vienna (EC no. 603/2009) and all patients and healthy controls had to sign a written informed consent.
Blood sampling and measurement of free TFPIα
Samples were timely processed to routine laboratory assessments and to storage at the biobank facility of the Medical University of Vienna (www.biobank.at; supplemental Methods [paragraph 3]).18 Laboratory tests as well as diagnostic criteria are described in supplemental Tables 1 and 2, respectively.
Quantification of free TFPIα levels was performed in plasma samples using a standardized enzyme-linked immunosorbent assay (Asserachrom Free TFPI-ELISA, Stago, Asniéres, France).
Thrombin-generation assay
Thrombin generation was assessed with a commercially available kit (Technothrombin; Technoclone, Vienna, Austria). Thrombin induced cleavage of a fluorogenic substrate and fluorescence was measured. According to the manufacturer's information the reagent used to initiate thrombin generation (RC low; Technoclone) contains a final concentration of tissue factor of <3 pM and of phospholipids of 3 to 4 µM. Parameters that were measured using a specific software (Technothrombin TGA, Vienna, Austria) were: lag time (time that is required for thrombin burst, minutes), maximum thrombin generation (peak, nmol/L), time to peak (TTP; velocity of thrombin generation, minutes), velocity index (compound index including lag time and TTP; peak/(TTP − lag time), nmol/L/min), and area under the curve (AUC; nmol/L × min).
Identification of FV (short) by western blot analysis
Plasma was loaded on a 4% to 20% SDS-polyacrylamide gel (Bio-Rad). After the gel was blotted on a nitrocellulose membrane, detection of FV was performed with a mouse anti FV-heavy chain, AHV-5146 (Haematologic Technologies, Essex Junction, VT) and a secondary goat anti-mouse antibody (DAKO).
ThromboGenomics and genetic testing
DNA samples were processed as previously described19 (supplemental Methods [paragraph 4]). DNA libraries were captured using ROCHE NimbleGen SeqCap ThromboGenomics capture baits (Roche NimbleGen, Inc, Madison, WI). Final libraries were quantified, 4 samples pooled and sequenced using an Illumina Hiseq 4000 sequencer, 150-bp paired-end (PE) run using the ThromboGenomics assay.19 Chromosomes were phased using the EAGLE2 haplotype phaser software.20 The reads in the demultiplexed paired-end FASTQ files were aligned using BWA21 0.7.10 and possible PCR duplicates marked using Picard (Broad Institute, Cambridge, MA) 1.123. Single-nucleotide variants (SNVs) and insertions/deletions (indels) were called using GATK22 3.3 HaplotypeCaller and filtered using the following VariantFiltration expressions “QD <2.0 || FS >100.0 || MQ <40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0” for SNVs and “QD <2.0 || FS >200.0 || ReadPosRankSum < −20.0” for indels (Broad Institute). The remaining variants were annotated with their predicted impact against Ensembl23 release 100 using Ensembl Variant Effect Predictor.24 The genotypes for rs10800453 were imputed using the pbwt package (Positional Burrows-Wheeler Transform) based on the Haplotype Reference Consortium genotypes.25 Genotyping was performed using the Affymetrix Axiom UK Biobank genotyping array.26
Statistical analysis
Statistical analysis was performed with the Statistical Package for Social Sciences (SPSS IBM Version 24.0) and the free open source software GNU R version 3.5.3.27 Group comparison was performed using the Student t test or the Wilcoxon rank-sum test in case of nonnormal distribution for unadjusted groups and the χ2 test for comparing categorical variables. To evaluate the adjusted differences for free TFPIα levels between patients and healthy controls we applied multiple linear regression analysis (considering age, sex, and body mass index [BMI] as confounding variables). For correlation of free TFPIα with metric variables the bivariate Spearman-ρ test was performed. Comparisons between genotypes and free TFPIα levels were performed using a Kruskal-Wallis test. To account for the number of multiple comparisons performed within the individual secondary research questions, the Bonferroni-Holm correction (BHC) was accordingly applied. All P values are results of 2-sided tests, and P values <.05 were considered as statistically significant.
Results
Patients’ clinical and laboratory characteristics
Six hundred twenty patients were included in the analysis. Clinical characteristics of patients and healthy controls are shown in Table 1. Blood group O was overrepresented in the patient cohort and patients had a higher BMI. Patients had a prolonged aPTT and PT and lower levels of von Willebrand factor antigen (VWF:Ag) and activity (VWF:RCo) than healthy controls, albeit the values were within the normal range.
Tissue factor pathway inhibitor
Free TFPIα levels (ng/mL, median [interquartile range (IQR)]) were higher in patients than in healthy controls (8.2 [5.5-11.7] and 7.8 [4.3-11.1]; P = .017). In patients, free TFPIα levels were higher in male than in female patients (10.0 [7.8-13.2] and 7.6 [5.3-11.3]; P = .002) and associated with higher age (rs = 0.414; P < .001) and higher BMI (rs = 0.224; P < .001). In line, also in healthy controls, free TFPIα levels differed between male and female patients (median [IQR], 9.5 [7.8-12.2] vs 6.6 [3.4-10.9] ng/mL; P = .018) and correlated with age (rs = 0.317; P < .001) and BMI (rs = 0.344; P < .001).
The statistically significant difference of free TFPIα levels between patients and healthy controls prevailed after adjustment for sex, age, and BMI in multivariable linear regression analysis (P = .026, Table 2). There was no difference in free TFPIα levels between patients with blood group O and those with non-O (supplemental Table 4).
In the separate analysis of each established diagnosis and BUC, we found significantly increased free TFPIα levels in patients with BUC and patients with PFDs, whereas there was no difference in patients with VWF antigen and/or activity ≤50 U/mL or coagulation factor deficiencies in multivariable linear regression analysis, adjusted for sex, age, and BMI (Table 2).
To identify outliers of free TFPIα levels in our patients, we next defined a cutoff according to the 95th percentile of free TFPIα in healthy controls (≥15.4 ng/mL). We found an increased number of patients above the predefined cutoff, which only barely missed statistical significance in the overall cohort (odds ratio [OR] [95% CI], 2.82 [0.98-8.13]). Significantly higher odds for being more than or equal to the 95th percentile of healthy controls were found in the group of patients with BUC (OR [95% CI], 3.03 [1.02-8.98]) and PFDs (OR [95% CI], 3.47 [1.09-11.08]) (Table 2; Figure 1).
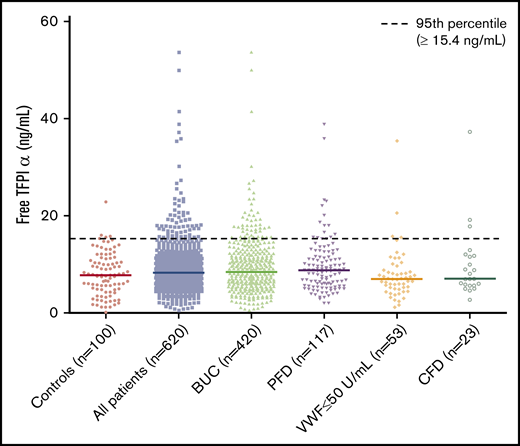
Scatter plot of free TFPIα levels in healthy controls and all patients and according to the established diagnoses.
Scatter plot of free TFPIα levels in healthy controls and all patients and according to the established diagnoses.
Free TFPIα and clinical bleeding phenotype
We next investigated whether higher levels of free TFPIα were associated with a more severe bleeding phenotype in our patients with MBDs. In our patients, the median [IQR] Vicenza Bleeding Score was 5 [4-8] and the median International Society on Thrombosis and Haemostasis (ISTH) Bleeding Score, available of 359 patients (57.9%), was 6 [4-9], respectively (Table 1). Multivariate analysis, by adjustment for sex, age, and BMI did not reveal a correlation of free TFPIα with both the Vicenza Bleeding Score (P = .079) and the ISTH Bleeding Assessment Tool (BAT) score (P = .506). Also in the separate analysis of patients with BUC or PFDs no association between free TFPIα levels and the bleeding scores were identified (Table 3).
Patients with high free TFPIα levels above the 95th percentile of healthy controls had similar bleeding scores compared with those below (supplemental Table 3).
Correlation of free TFPIα levels with global coagulation tests and thrombin generation
To analyze a possible influence of high free TFPIα levels on the aPTT, the PT, and thrombin generation, we calculated correlations in all patients and patients with BUC and PFDs separately (Table 4). There was no clear correlation of free TFPIα with aPTT or PT. In thrombin generation, we found a weak positive correlation between free TFPIα levels and lag time (time that is required for thrombin burst, minutes) in all patients (r = 0.247; P < .001; BHC < .05) and patients with BUC (r = 0.233; P < .001; BHC < 0.05), which was more pronounced in patients with PFDs (r = 0.350, P < .001, BHC = 0.05). TTP showed a weak positive correlation (r = 0.231, P < .05) and AUC a weak negative correlation (r = −0.220; P < .05) in patients with PFDs only (supplemental Table 5).
There was no difference in the aPTT between patients with high free TFPIα levels above the 95th percentile of healthy controls and patients below this cutoff (median [IQR], 35.5 [32.9-39.1] and 35.9 [33.7-38.9]; P = .901), whereas the PT was even prolonged in patients with free TFPIα levels below the cutoff (98.0 [93.0-106.0] and 95.0 [88.0-104.0]; P = .031)
Genetic analysis
Sequencing and genotyping data were available for 465 of the 620 patients (75%). In the sequencing data we did not identify the previously reported gain-of-function mutations East Texas Bleeding Disorder (NM_000130.4:c.2350A>G, NC_000001.10:g.169511978T>C [h19, GRCh37]) or FV Amsterdam (NM_000130.4:c.2588C>G, NC_000001.10:g.169511740G>C [h19, GRCh37]) in the F5 gene in any of our patients, despite both nucleotide positions having good read coverage. Furthermore, we did not find known genetic variations in the F5 gene that could explain increased free TFPIα levels. In total, 15 homozygous known variations in the F5 gene were identified. Of these 15 variants, 1 variant code for FV Leiden is considered as pathogenic, whereas the remaining 14 variants, including 12 likely benign variants and 2 variants of uncertain significance, are not known to be pathogenic (supplemental Table 6).
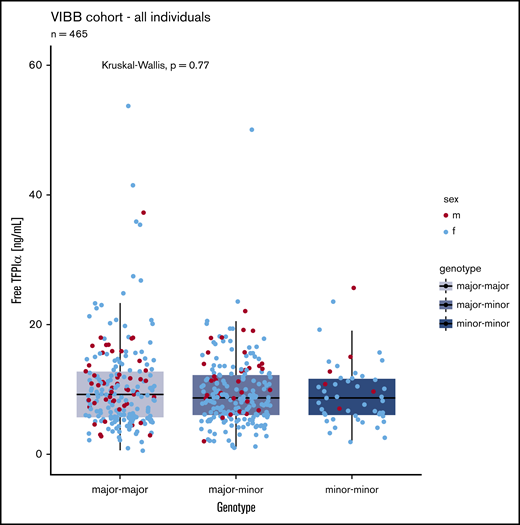
The common SNV rs10800453 was genotyped in 465 patients (49 had 2 alternative alleles, 212 were heterozygous for the alternative allele). We did observe a trend of slightly lower levels in carriers of the minor allele. However, with the sample size of this study, we were not powered to confirm or refute the observation by Sun et al.17 Nevertheless, in this cohort of patients, free TFPIα levels were not significantly associated with the alleles of this SNV in our cohort (Figure 2).
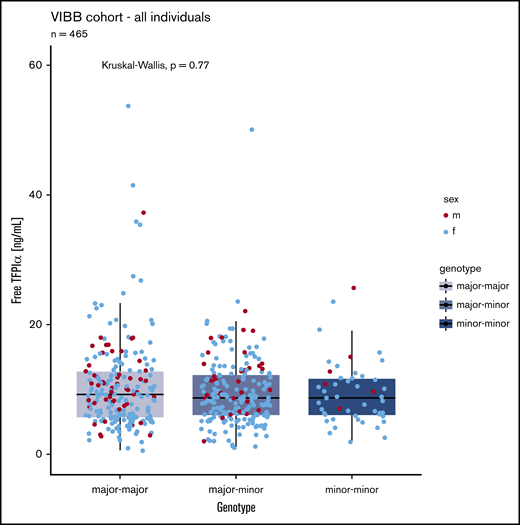
Comparison of free TFPI α levels in patients with 0 (n = 204), 1 (n = 212), or 2 (n = 49) minor (variant) alleles of SNV rs10800453. Blue dots, female (f); red dots, male (m).
Comparison of free TFPI α levels in patients with 0 (n = 204), 1 (n = 212), or 2 (n = 49) minor (variant) alleles of SNV rs10800453. Blue dots, female (f); red dots, male (m).
Western blot analysis
Western blot analysis of plasma of the 3 patients with free TFPIα levels >40 ng/mL and 3 healthy controls with the highest TFPIα levels (22.9, 15.8, 15.1 ng/mL) did not show a FV-short isoform (supplemental Figure 1).
Clinical and laboratory data of the 3 patients are summarized in supplemental Table 7. All were female and categorized as BUC patients. Two patients had postsurgical bleeding, whereas 1 had easy bruising, gastrointestinal bleedings, and menorrhagia. These phenotypes were also observed in patients with the East Texas and FV Amsterdam bleeding disorder, respectively (bleeding after trauma or surgery, menorrhagia, bruising, and epistaxis).
Discussion
In this study, we found that free TFPIα levels are significantly increased in patients with mild to moderate bleeding tendencies compared with healthy controls, especially in the groups of patients with BUC and PFDs. This increase was associated with a mild delay in thrombin generation, but not with prolongations in routinely used global coagulation tests or a clinically more severe bleeding phenotype. We could not identify new genetic variations in the exons of the FV-encoding gene or known variants associated with enhanced free TFPIα levels.15,16 Also, the previously reported SNV rs10800453 did not have a statistically significant association with free TFPIα levels in our patients.17
Our results revealed increased levels of free TFPIα in our patients with MBDs, as well as a higher proportion of patients with significantly higher free TFPIα levels, defined as more than or equal to the 95th percentile of healthy controls. Nevertheless, even those patients with the highest outliers (median [IQR], 18.0 [16.4-22.1] ng/mL) had free TFPIα levels that were much lower than reported for TFPIα-associated bleeding disorders. In the East Texas or FV Amsterdam bleeding disorders, free TFPIα levels were at least 10-fold higher than the normal range (>100 ng/mL).15,16 Systematic data on TFPI levels in patients with MBDs are hardly available. MacDonald et al recently found increased TFPI activity in a cohort of 13 patients with BUC and either a prolonged lag time or a decreased endogenous thrombin potential, and showed a partial correction of thrombin generation with anti-TFPI antibodies.28 In contrast to this study and above reported bleeding disorders, we did not find prolongations of global clotting tests and only a mild delay in thrombin generation in our patients with high free TFPIα levels, even after correction for multiple testing.
Interestingly, in our study increased free TFPIα levels were not only observed in BUC patients, but also in patients with PFDs. It is already well described that megakaryocytes produce TFPIα, still, the exact storage within platelets and the release mechanisms are unknown.10 Upon activation, human platelets secret TFPIα, which can then dampen and control local thrombus growth.10 In our patients, increased platelet activation might have led to TFPIα secretion and reduced platelet activatability upon addition of agonists in performed platelet function tests, resulting in the diagnosis of a PFDs. However, mice experiments have shown that free TFPIα in plasma was not influenced by the platelet secretion.10 In humans, it is still unclear whether and to what extent platelet TFPIα contributes to plasmatic free TFPIα levels and its anticoagulant effect. Whether there is a causal relationship between high free TFPIα levels and the PFDs in our patients or whether these are 2 independent mechanisms resulting in a clinically relevant bleeding disorder still needs to be elucidated.
No association between free TFPIα levels and the bleeding phenotype, which was defined by standardized bleeding scores,29,30 was found. According to recent studies, existing bleeding scores might not be precise enough when analyzing patients with MBDs, especially due its heterogenic subgroups.3,6 We recently found that bleeding scores have a low ability in distinguishing patients with established bleeding disorders from those with BUC.6 Additionally, it was also shown by our group that thrombin generation does not correlate well with the bleeding score in patients with BUC either,7 which also holds true for enhanced levels of free TFPIα and underlines the probably too low sensitivity of the bleeding scores.
The analysis of genetic data did not reveal relevant genetic variations, which are known to result in a FV-short splice variant, that stabilizes free TFPIα. Also, the SNV rs10800453, which has recently been found to be associated with increased total TFPI levels in a genome-wide association study by Sun et al,17 did not significantly correlate with free TFPIα levels in our patients. This might base on the different methods to assess TFPI, as in this study total TFPI levels were measured using a proteomic tool (SomaLogic Inc, Boulder, CO). Furthermore, our study did not provide adequate power to replicate the observation made in this genome-wide association study on 3301 subjects.
In general, data on DNA variants in mild to moderate bleeding tendencies are scarce. We recently showed that a molecular diagnosis was identified in only 3.2% patients with unexplained bleeding disorders using the high-throughput ThromboGenomics gene panel test, which was designed for the diagnosis of inherited bleeding, thrombotic and platelet disorders.19 Therefore, in future, a whole-genome sequencing approach in highly characterized patients could identify further genes or potential aggregations of the effects of variants at hundreds of loci (the so-called polygenic risk scores) that could elucidate new pathophysiological mechanisms for unexplained bleeding tendencies.31,32
Our study was performed in a large cohort of well-characterized patients with MBDs, yet, has some limitations. First, we were only able to analyze rare variants in the F5 gene, but not in the TFPI gene, as it was not sequenced by the ThromboGenomics test. Nevertheless, reported TFPIα-associated bleeding disorders are caused by variants in the B-domain of the F5 gene, which has also been investigated in our study. Further, we did not analyze total TFPI, which includes also TFPIα bound to lipoproteins since it is known that free TFPIα is the main contributor to the anticoagulant effect in plasma.9,28,33 Lastly, as we found a significant, but only discrete alterations in thrombin generation of patients with high free TFPIα levels, we did not investigate whether anti-TFPI antibodies could correct thrombin generation in our patients.
In general, there is an urgent need for a better understanding of underlying causes for bleeding in patients with MBDs. Increased free TFPIα levels in our patients could not be explained on a genetic level. A deeper insight into the pathophysiological mechanism behind increased TFPIα levels and their impact on a patients hemostatic potential is crucial, as targeting TFPI has big therapeutic potential. As it was shown in hemophilia patients, anti-TFPI treatment could be a therapeutic approach to restore thrombin generation and manage bleeding symptoms in patients with MBDs and increased TFPI levels.34
To summarize, we found that free TFPIα is increased in patients with mild to moderate bleeding tendency and was associated with delayed thrombin generation. This could be an underlying cause or a contributor for bleeding, especially in patients with BUC and PFDs. We did not identify genetic variations that could be linked to higher free TFPIα levels in our patients. Based on our findings and existing data, we conclude that TFPI has an important role for hemostatic balance, and alterations could provoke bleeding in patients with MBDs.
For original data, please contact: johanna.gebhart@meduniwien.ac.at.
Acknowledgments
Illustrations used in the visual abstract were created with BioRender.com.
The Vienna Bleeding Biobank was supported by an unrestricted grant of CSL Behring.
Authorship
Contribution: J.G., I.P., C.A., and D.M. designed the study; J.G., I.P., C.A., D.M., S.H., and J.R. recruited patients; H.H. processed and stored the samples; D.M. performed statistical analyses; J.G., I.P., and D.M. analyzed the data; K.D. and W.H.O. provided the genetic data; A.T. and M.H. analyzed the genetic data; I.P., J.G., and D.M. interpreted the data; and D.M., J.G., and A.T. wrote the manuscript, which was reviewed, edited, and finally approved by all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Johanna Gebhart, Clinical Division of Hematology and Hemostaseology, Department of Medicine I, Medical University of Vienna, Waehringer Guertel 18-20, A-1090 Vienna, Austria; e-mail: johanna.gebhart@meduniwien.ac.at.