Key Points
p18 played an important role in human HSC maintenance and expansion.
Targeting p18 by small molecule compound 005A enhanced self-renewal of human long-term HSCs.

Abstract
The use of umbilical cord blood transplant has been substantially limited by the finite number of hematopoietic stem and progenitor cells in a single umbilical cord blood unit. Small molecules that not only quantitatively but also qualitatively stimulate enhancement of hematopoietic stem cell (HSC) self-renewal ex vivo should facilitate the clinical use of HSC transplantation and gene therapy. Recent evidence has suggested that the cyclin-dependent kinase inhibitor, p18INK4C (p18), is a critical regulator of mice HSC self-renewal. The role of p18 in human HSCs and the effect of p18 inhibitor on human HSC expansion ex vivo need further studies. Here we report that knockdown of p18 allowed for an increase in long-term colony-forming cells in vitro. We then identified an optimized small molecule inhibitor of p18, 005A, to induce ex vivo expansion of HSCs that was capable of reconstituting human hematopoiesis for at least 4 months in immunocompromised mice, and hence, similarly reconstituted secondary recipients for at least 4 more months, indicating that cells exposed to 005A were still competent in secondary recipients. Mechanistic studies showed that 005A might delay cell division and activate both the Notch signaling pathway and expression of transcription factor HoxB4, leading to enhancement of the self-renewal of long-term engrafting HSCs and the pool of progenitor cells. Taken together, these observations support a role for p18 in human HSC maintenance and that the p18 inhibitor 005A can enhance the self-renewal of long-term HSCs.
Introduction
Hematopoietic stem cells (HSCs) give rise to all types of blood cells throughout life and have been widely used in transplantation to treat human diseases such as hematopoietic malignancies and bone marrow failure.1,2 Allogeneic HSC transplantation (allo-HSCT) offers superior overall survival compared with autologous HSCT (auto-HSCT), largely because of potentially curative graft-versus-leukemia/graft-versus-tumor effects. Although allo-HSCT mainly depends on the availability of human leukocyte antigen (HLA)-matched adult donors, its application has been limited by the low probability of finding such HLA-matched donors and the risk of developing severe graft-versus-host disease.3 Alternatively, umbilical cord blood (UCB) represents an essential alternate source of hematopoietic stem and progenitor cells (HSPCs) for HSCT because of its extensive availability and reduced need for HLA matching.4 For most of the preclinical trials for expansion conditions, a double UCB transplant was required to ensure the presence of long-term engrafting HSC in the UCB unit,5 which would substantially increase the cost for HSC therapy and the risk of graft-versus-host disease. Research has made it clear that double cord blood unit hematopoietic cell transplantation is no more efficient than single cord blood unit hematopoietic cell transplantation for children or adults.6 However, the limited number of hematopoietic cells in a single cord blood unit results in delayed hematopoietic recovery and higher mortality and prevents its widespread use in recipients with larger body mass.7
Small molecules have been considered as an attractive therapeutic tool because of their flexible and reversible effects that can be achieved by adjusting the concentrations and combinations of the molecules administered.8 Several compounds have been identified as agonists for UCB HSPC expansion. However, most reported culture conditions optimized for HSC expansion were accompanied with increased differentiation, leading to loss of primitive long-term HSC (LT-HSC) activity and late graft failure.9 For example, the aryl hydrocarbon receptor antagonist StemRegenin 1 (SR1) was found to preferentially expand short-term HSCs (ST-HSC).10 A recent study by Fares et al11 identified the small molecule UM171 as a potential agonist for UCB LT-HSC expansion, although the precise mechanism of action of UM171 on LT-HSC remains unclear. Therefore, innovative approaches that not only quantitatively but also qualitatively expand HSPCs with long-term full lineage reconstitution properties during ex vivo culture are still in critical need.
Much progress has been made in recent years in identifying intrinsic regulators that can support the expansion of ST-HSCs and LT-HSCs,12 and targeting of these critical molecules could represent a strategy for inducing the expansion of HSCs ex vivo.13,14 A study by Broxmeyer et al15 reported a decrease in the number of hematopoietic progenitor cells in p18KO mice. Cell cycle regulators, including cyclin-dependent kinases (CDKs) and CDK inhibitor (CKI) proteins, also have been found to contribute to HSC self-renewal.16,17 It is thought that members of the INK4 family of CKIs compete with cyclin D and selectively target CDK4/6 to inhibit its kinase activity and stop cell cycle progression.18 Among INK4 family members, p18INK4C (p18) has been identified as a unique target for HSC self-renewal.16 In murine models, deletion of p18 prominently improved long-term hematopoietic engraftment primarily by increased primary cell division.19 Other CKIs have been found to regulate HSC behaviors as well, including p21, p27, and p57.19-21 However, only p18 has exhibited the potential to facilitate hematopoietic reconstitution. Thus, the role of p18 in human HSCs and the effect of p18 inhibitor on human HSCs expansion ex vivo need further studies.
In our previous studies, we demonstrated that small molecule inhibitors targeting p18 could expand functional mouse HSCs in a short-term culture, and we performed primary screening of a series of analogs using human HSCs in vitro.22,23 In the present study, p18 expression was first knocked down in human UCB HSCs, and then the HSPC cell number and hematopoietic functions of these cells were studied. A sulfamoyl benzoate derivative p18 inhibitor, 005A, was developed in silico and found to be capable of prompting self-renewal of human HSCs. LT-HSCs function was also assessed by performing primary and secondary transplantation. 005A-cultured HSCs achieved efficient long-term engraftment in immunocompromised mice. The mechanistic details of these effects were further investigated with gene analysis and Numb staining analysis. Overall, these observations support that 005A could enhance human long-term HSC self-renewal ex vivo.
Materials and methods
Cell culturing
The cells were resuspended in Iscove's Modified Dulbecco's Medium (IMDM, Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 ng/mL human stem cell factor (PeproTech), 100 ng/mL human thrombopoietin (TPO, PeproTech), and 100 ng/mL human Flt3L (PeproTech) at a concentration of 1 × 105 cells/mL. 005A was added in a concentration of 20 nM without special description.
Xenotransplant assay
Animal experiments were conducted according to institutional guidelines for the use of laboratory animals and after being granted permission from Chinese Academy of Medical Sciences Animal Ethical Committee for animal experimentation.
Other methods or details can be found in the supplemental Information.
Results
Effect of p18 knockdown on human hematopoietic cells
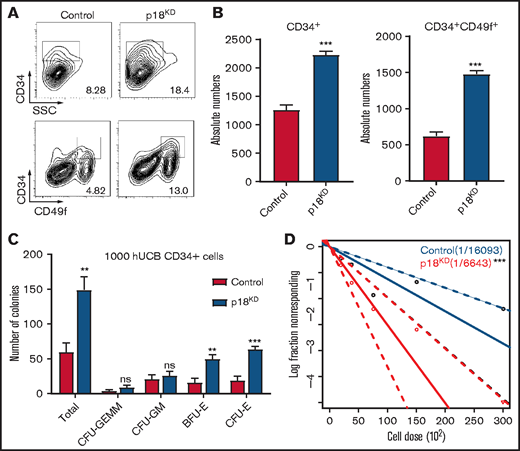
Our previous studies have demonstrated that p18 is a critical regulator of mice HSC maintenance.22 To determine whether p18 has a similar role in human HSCs, lentivirus carrying 3 different short hairpin RNAs targeting p18 were used to infect freshly isolated CD34+ HCB cells, and we chose the most efficient one to perform subsequent experiments (supplemental Figure 1A-B). The sorted GFP+ cells were then cultured ex vivo for 7 days, and flow cytometry analysis demonstrated that the cultured p18 knockdown/GFP+ cells exhibited increased expression of the primitive cell surface HSC markers CD34 and CD49f (Figure 1A). Specifically, the absolute numbers of CD34+ cells and CD34+CD49f+ cells in the p18 knockdown group were higher than those in the control group, and this tendency could be observed in different short hairpin RNA–induced p18 knockdown cells (Figure 1B; supplemental Figure 1C). In contrast, the absolute number of total cells after cultured in the p18KD group was lower than that in the control group (supplemental Figure 1D).
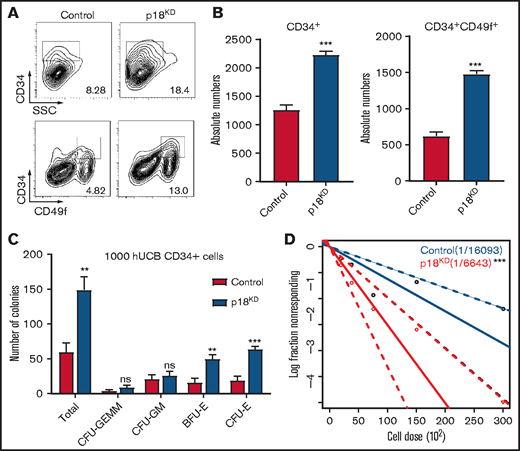
Effect of p18 knockdown on human hematopoietic cells. Representative images (A) and absolute numbers (B) of the flow cytometry data of human CD34+ and CD34+CD49f+ cell populations of hUCB CD34+ cells after infection of lentivirus carrying hairpin RNA targeting p18 were measured at 7 days (n = 3); 1 × 104 CD34+GFP+ human UCB cells were seeded at the beginning. (C) Total CFC contents of control and p18 knockdown human CD34+ cells at 2 weeks (n = 3). GEMM, colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte; CFU-GM, colony-forming unit-granulocyte, macrophage; BFU-E, burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid. (D) Frequencies of CAFCs of control and p18 knockdown human CD34+ cells (n = 10). Doses of 30 000, 15 000, 7500, 3750, and 1875 cells per well were used. The experiment was repeated 3 times. All data represent the means ± standard deviation (SD). Compared with control unless specified: *P < .05, **P < .01, ***P < .001 by 2-tailed unpaired t test.
Effect of p18 knockdown on human hematopoietic cells. Representative images (A) and absolute numbers (B) of the flow cytometry data of human CD34+ and CD34+CD49f+ cell populations of hUCB CD34+ cells after infection of lentivirus carrying hairpin RNA targeting p18 were measured at 7 days (n = 3); 1 × 104 CD34+GFP+ human UCB cells were seeded at the beginning. (C) Total CFC contents of control and p18 knockdown human CD34+ cells at 2 weeks (n = 3). GEMM, colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte; CFU-GM, colony-forming unit-granulocyte, macrophage; BFU-E, burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid. (D) Frequencies of CAFCs of control and p18 knockdown human CD34+ cells (n = 10). Doses of 30 000, 15 000, 7500, 3750, and 1875 cells per well were used. The experiment was repeated 3 times. All data represent the means ± standard deviation (SD). Compared with control unless specified: *P < .05, **P < .01, ***P < .001 by 2-tailed unpaired t test.
To determine the short-term hematopoietic function of the p18 knockdown cells, a colony-forming cell (CFC) assay was performed. All kinds of colonies could be found in the semisolid media based on morphology (supplemental Figure 1E), suggesting knockdown of p18 would not influence the multilineage differentiation potential of HSCs. However, the number of total colonies, colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte; colony-forming unit-granulocyte, macrophage; burst-forming unit-erythroid; and colony-forming unit-erythroid in the p18KD group were increased relative to the control group (Figure 1C; supplemental Figure 1). These results suggest that knockdown of p18 enhances the maintenance and expansion of human progenitor cells ex vivo. Interestingly, in our previous study,22 the p18 knockout mouse was characterized by decreased progenitor cell numbers in bone marrow.
A modified cobblestone area–forming cell (CAFC) assay limiting dilution analysis assay22-24 was used to analyze the long-term colony formation ability. The frequency of HSCs in the p18 knockdown cells (1 in a fraction of cells equivalent to 6614 starting CD34+ cells) was 2.44-fold that of the control cells (1 in a fraction of cells equivalent to 16118 starting CD34+ cells; P < .01; Figure 1D). In addition, a slight increase of S-phase cells percentage (supplemental Figure 1G) and increase of cell apoptosis (supplemental Figure 1H) were observed in the p18 knockdown cells. These results indicate that knockdown of p18 could significantly expand HSCs in vitro culture. Taken together, these results suggest that targeting of p18 has the potential to significantly expand HSC populations in ex vivo cultures.
Specificity of 005A, a small molecule inhibitor of p18
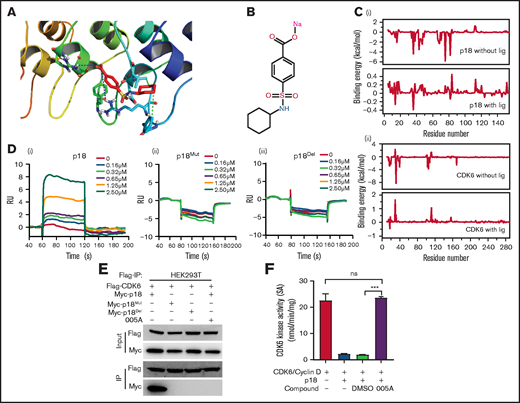
Virtual molecular docking studies and molecular dynamics (MD) simulations were performed for the p18 inhibitor 005A, a sulfamoyl benzoate derivative that was previously developed in silico.23 A model of 005A binding to p18 is shown in Figure 2A. The structure of 005A is shown in Figure 2B. Both Arg39 and Arg79 of p18 form strong electrostatic interactions with 005A, while Val44 and Phe37 in p18 form strong hydrophobic interactions with 005A.
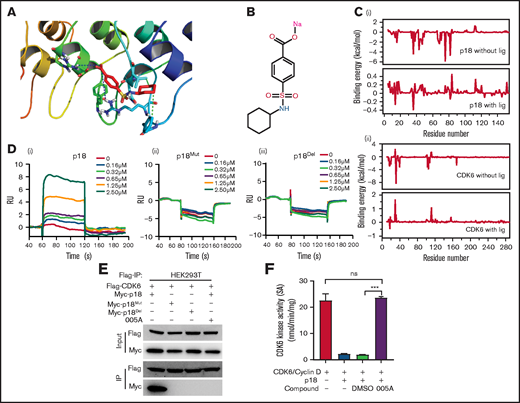
Specificity of 005A, a small molecule inhibitor of p18. (A) Predicted 005A and p18 protein binding interactions at the amino acid level. (B) The structure of 005A. (C) Binding energy changes for p18 (i) and CDK6 (ii) residues in the presence and absence of ligand 005A. (D) Surface plasma resonance (SPR) results of the binding activity of p18 (i), p18Mut (ii), and p18Del (iii) at various concentrations of compound 005A. (E) Co-immunoprecipitation (Co-IP) assay using Flag taged CDK6 and Myc taged p18 and its mutants. HEK293T cells were treated with 005A for 24 hours after 48-hour transient transfection. (F) In vitro CDK6 kinase activity assays were performed in the indicated conditions, Rb phosphating levels indicate the CDK6 kinase activity in these assays (n = 3). All data represent the means ± SD. Compared with control unless specified: ***P < .001 by 2-tailed unpaired t test.
Specificity of 005A, a small molecule inhibitor of p18. (A) Predicted 005A and p18 protein binding interactions at the amino acid level. (B) The structure of 005A. (C) Binding energy changes for p18 (i) and CDK6 (ii) residues in the presence and absence of ligand 005A. (D) Surface plasma resonance (SPR) results of the binding activity of p18 (i), p18Mut (ii), and p18Del (iii) at various concentrations of compound 005A. (E) Co-immunoprecipitation (Co-IP) assay using Flag taged CDK6 and Myc taged p18 and its mutants. HEK293T cells were treated with 005A for 24 hours after 48-hour transient transfection. (F) In vitro CDK6 kinase activity assays were performed in the indicated conditions, Rb phosphating levels indicate the CDK6 kinase activity in these assays (n = 3). All data represent the means ± SD. Compared with control unless specified: ***P < .001 by 2-tailed unpaired t test.
The roles of these key residues were further validated in MD simulations. First, 2 sets of MD simulations of the p18–005A complex with or without CDK6 were performed. All 3 structures remained stable during the simulation, with root mean square deviation values of ∼1.88, 0.97, and 2.03 Å, respectively. The MD results also showed that the energy contributions of Arg39/Arg79 ranked first (−2.53 and −1.54 kcal/mol, respectively), whereas Val44 (−0.83 kcal/mol) and Phe37 (−0.36 kcal/mol) in p18, and Arg31 (−3.76 kcal/mol), Arg39 (−0.66 kcal/mol), and Val16 (−0.25 kcal/mol) in CDK6 were also important residues for the recognition of p18 by 005A (supplemental Figure 2A and B).
The energy contributions of these key residues, as well as the total binding energies between p18 and CDK6 with or without ligand (005A), were also determined. Figure 2C shows the changes in energy contribution for each residue. Several residues in both p18 and CDK6 exhibited lower binding energies (indicating better binding interactions). Moreover, the overall binding energy of the p18/CDK6 complex exhibited a large change from −77.56 (without the ligand) to −1.76 kcal/mol (with the ligand).
We constructed the expression vector of mutation (to Ala, p18Mut) or deletion (p18Del) of Arg39, Arg79, Val44, and Phe37 of p18. Then, the interaction of 005A with wild-type p18 and the mutants plus p16 and p19 proteins were validated through in vitro surface plasma resonance (SPR) assays. Our data showed that 005A binds to p18 in a dose-dependent manner, but with an irregular response with p18 mutants or p16 or p19 (Figure 2D; supplemental Figure 2C). On the other hand, co-immunoprecipitation (co-IP) assays showed 005A blocked the interaction of p18 with CDK4/6 in HEK293T cells, whereas the p18 mutants failed to bind to CDK6, perhaps because the structure of these mutants were changed (Figure 2E; supplemental Figure 2D). Moreover, the 005A blocked interaction of p18 with CDK4/6 could be partly reversed in 7-day culture (supplemental Figure 2E). Thus, 005A can disrupt interactions between p18 and CDK4/6 by inhibiting p18 activity.
To further investigate whether p18 can selectively inhibit CDK4/6’s activity in phosphorylating Rb proteins via direct binding of Rb, an in vitro CDK6 activity assay was performed as previously described.23 A mixture of p18 and 005A (2 µM) was added to a mixture of CDK6 and Rb, whereas a mixture of CDK6 and Rb was prepared as a control. After adding [γ-32P]ATP to each mixture, the reactions were spotted onto polyvinylidene difluoride membrane papers and washed with phosphoric acid. In the presence of p18, CDK6 activity decreased approximately fourfold (Figure 2F), whereas the addition of 005A restored the activity of CDK6. As a control, dimethyl sulfoxide at the same concentration did not have the same effect as 005A. Taken together, these results suggest that 005A can target p18 and rescue CDK6 activity.
005A treatment retained the stemness of human primary HSCs
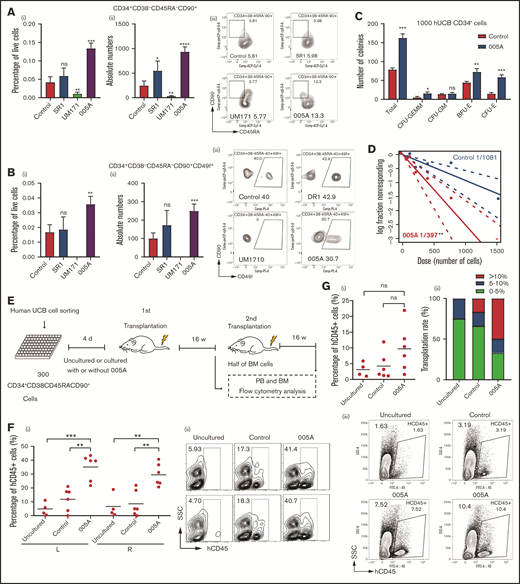
To optimize and maximize the expansion effect of 005A, a dose-response assay was performed. Treatment with 005A significantly increased both the absolute number and relative percentage of both CD34+ (eg, phenotypically defined HSCs and HSPCs) and CD34+CD49f+ cells (eg, phenotypically defined primitive HSCs25). We found that the best concentration of 005A was about 20 nM. An increase of approximately 20% was observed for the CD34+ proportion compared with the control group (supplemental Figure 3A), and the proportion of the CD34+CD49f+ cells was increased >50% (supplemental Figure 3B). Serum has also been identified as a possible factor in the loss of stemness in ex vivo culture.26 When CD34+ cells were cultured in serum-free medium, a greater expansion of CD34+CD49f+ cells were observed in 005A group compared with the control group (supplemental Figure 3C). Furthermore, we used other HSC markers, such as CD45RA, CD90, and CD201, to examine the effects of 005A and 2 other reported compounds (SR1 and UM171) on HSCs. The treatment of SR1 and 005A led to an increase of total cell numbers, whereas treatment of UM171 decreased it (supplemental Figure 3D). We further showed the percentage of live cells and absolute number of CD34+, CD34+CD38−, CD34+CD38−CD201+, CD34+CD38−CD90+CD45RA−, and CD34+CD38−CD90+CD45RA−CD49f+ cell populations. 005A treatment could significantly increase the CD34+CD38−, CD34+CD38−CD90+CD45RA−, and CD34+CD38−CD90+CD45RA−CD49f+ cell populations (Figure 3A-B; supplemental Figure 3F), but failed to increase the percentage of CD34+and CD34+CD38−CD201+ cell populations (supplemental Figure 3E,G). These results indicate that 005A has the potential to expand human phenotypically defined HSC ex vivo.
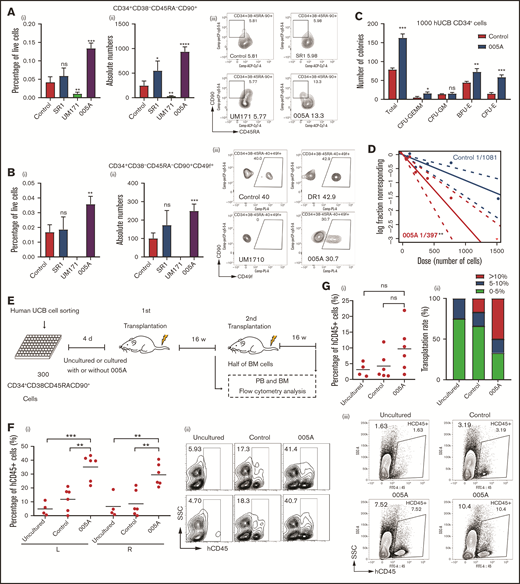
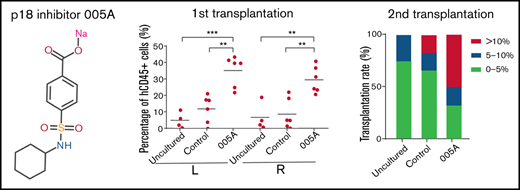
005A treatment retained the stemness of human primary HSCs. (A) The percentage (i) of live cells and absolute numbers (ii) per well of human CD34+CD38−CD45RA−CD90+ cells were measured (n = 4). Representative images (iii) of the flow cytometry data of CD34+CD38−CD45RA−CD90+ cell population and percentage in CD34+CD38− cells. (B) The percentage (i) of live cells and absolute numbers (ii) per well of human CD34+CD38−CD45RA−CD90+CD49f+ cells were measured (n = 4). Representative images (iii) of the flow cytometry data of CD34+CD38−CD45RA−CD90+CD49f+ cell population and percentage in CD34+CD38−CD45RA−CD90+ cells; 1 × 105 CD34+ UBCs were planted in 24-well plate, and cell populations were analyzed by flow cytometry after 7-day treatment with vehicle, SR1(1 µmol/L), UM171 (35 nmol/L), or 005A (20 nmol/mL) in panels A and B. (C) CFU statistical analysis of colony formation data for cells treated with or without 005A for 7 days (n = 3). GEMM, colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte; CFU-GM, colony-forming unit-granulocyte, macrophage; BFU-E, Burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid. (D) Frequencies of CAFCs of vehicle and 005A-treated human CD34+ cells (n = 10). Doses of 1500, 750, 375, 186, and 92 cells per well were used. The experiment was repeated 3 times. (E) An overview of the experimental setup used to perform xenotransplantations in immunodeficient NOG mice. (F) Levels of human engraftment (i) and representative images of the flow cytometry data (ii) in intrafemoral injected (R, right) or none injected (L, left) bone marrow from primary recipients of human CD34+CD38−CD45RA−CD90+ cells that were directly transplanted (uncultured) or cultured with or without 005A (n = 4 for uncultured group, n = 6 for control group and 005A group). Each symbol represents the results from an individual mouse. (G) Levels of human engraftment (i) of the secondary NOG recipients and chimerism were assessed after 4 months. (ii) Transplantation rate of mice at different engraftment levels (<5%; 5%−10%; >10%) in each group are shown. n = 4 for uncultured group, n = 6 for control group and 005A group. (iii) Representative fluorescence-activated cell sorter profiles. All data represent means ± SD. Compared with control unless specified: *P < .05, **P < .01, ***P < .001 by 2-tailed unpaired t test.
005A treatment retained the stemness of human primary HSCs. (A) The percentage (i) of live cells and absolute numbers (ii) per well of human CD34+CD38−CD45RA−CD90+ cells were measured (n = 4). Representative images (iii) of the flow cytometry data of CD34+CD38−CD45RA−CD90+ cell population and percentage in CD34+CD38− cells. (B) The percentage (i) of live cells and absolute numbers (ii) per well of human CD34+CD38−CD45RA−CD90+CD49f+ cells were measured (n = 4). Representative images (iii) of the flow cytometry data of CD34+CD38−CD45RA−CD90+CD49f+ cell population and percentage in CD34+CD38−CD45RA−CD90+ cells; 1 × 105 CD34+ UBCs were planted in 24-well plate, and cell populations were analyzed by flow cytometry after 7-day treatment with vehicle, SR1(1 µmol/L), UM171 (35 nmol/L), or 005A (20 nmol/mL) in panels A and B. (C) CFU statistical analysis of colony formation data for cells treated with or without 005A for 7 days (n = 3). GEMM, colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte; CFU-GM, colony-forming unit-granulocyte, macrophage; BFU-E, Burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid. (D) Frequencies of CAFCs of vehicle and 005A-treated human CD34+ cells (n = 10). Doses of 1500, 750, 375, 186, and 92 cells per well were used. The experiment was repeated 3 times. (E) An overview of the experimental setup used to perform xenotransplantations in immunodeficient NOG mice. (F) Levels of human engraftment (i) and representative images of the flow cytometry data (ii) in intrafemoral injected (R, right) or none injected (L, left) bone marrow from primary recipients of human CD34+CD38−CD45RA−CD90+ cells that were directly transplanted (uncultured) or cultured with or without 005A (n = 4 for uncultured group, n = 6 for control group and 005A group). Each symbol represents the results from an individual mouse. (G) Levels of human engraftment (i) of the secondary NOG recipients and chimerism were assessed after 4 months. (ii) Transplantation rate of mice at different engraftment levels (<5%; 5%−10%; >10%) in each group are shown. n = 4 for uncultured group, n = 6 for control group and 005A group. (iii) Representative fluorescence-activated cell sorter profiles. All data represent means ± SD. Compared with control unless specified: *P < .05, **P < .01, ***P < .001 by 2-tailed unpaired t test.
The CFC assays showed that the total number of colonies significantly increased by 005A treatment (Figure 3C). In addition, the number of colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte; burst-forming unit-erythroid; and colony-forming unit-erythroid colonies increased significantly after treatment with 005A compared with the control group (Figure 3C). These results suggest that 005A can increase the number of progenitor cells and maintain their multilineage differentiation potential. To determine the long-term colony formation capacity of 005A-treated HSCs, a CAFC limiting dilution analysis assay was conducted. Poisson statistics revealed that the frequency of HSCs among the 005A-treated cells (1 in a fraction of cells equivalent to 397 starting cells) was 2.72-fold higher than that of the control cells (1 in a fraction of cells equivalent to 1081 starting cells; P < .01; Figure 3D). These results suggest that 005A can increase the frequency of primitive HSCs.
To determine the functional HSC frequency of 005A-cultured human cells, we performed xenografts into NOG mice (Figure 3E). Flow cytometry analysis of 12-week peripheral blood showed that 005A-treated cells achieved higher engraftment than the cells cultured with vehicle (supplemental Figure 3H). Notably, after unilateral interfemoral injection of cultured HSCs, we observed 005A increased human hematopoietic engraftment in both injected or noninjected femurs relative to the control or untreated group (Figure 3F), indicating that 005A enhanced HSCs without influencing homing. 005A-cultured human HSCs could give rise to all major lineages, including human B (CD19+), T (CD3+), myeloid (CD33+), natural killer (CD56+), and erythroid (CD235a+) cells and progenitors (CD34+CD38−; supplemental Figure 3I-K), suggesting that the 005A-expanded HSCs retain multilinage potential. In addition, our data showed that combi use of 1 μM SR1 + 20 nM 005A did not enhance the engraftment of 005A in NOG mice, perhaps because the mechanism of SR1 and 005A was antagonistic or we had to screen a proper concentration of combi use of SR1 and 005A (supplemental Figure 3L). To identify the function of human LT-HSCs, we also performed secondary transplantation. Although there were no significant differences between the 005A group and control or uncultured groups, one-half of the 005A group mice achieved engraftment rates >10%, and only one-sixth of control group mice and none of uncultured group mice could achieve more than 10% engraftment rates. The secondary transplantation results supported that the numbers of LT-HSC with self-renewal activity was increased by 005A treatment. These results suggest that 005A promotes expansion of human UCB-derived HSCs, contributing to both early and sustained engraftment.
Increase of human long-term HSC self-renewal induced by 005A is characterized by delayed cell division and activation of Notch signaling
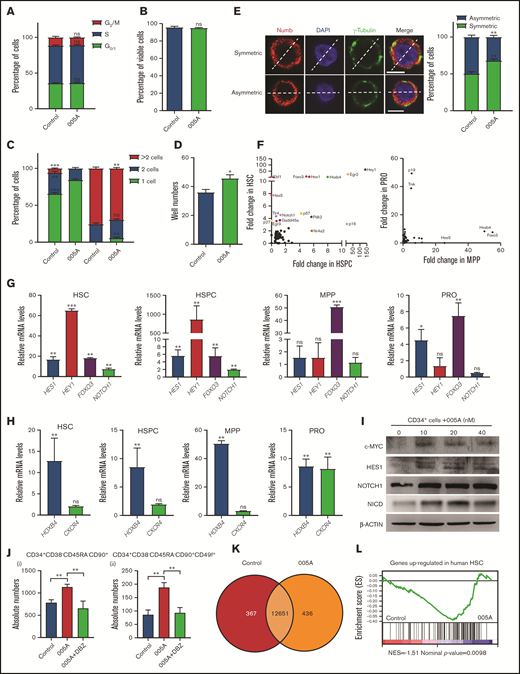
To explore the mechanism by which 005A increased human HSC self-renewal, we examined the cell cycle and survival of 005A cultured cells. No significant difference was found between 005A and vehicle-treated CD34+ cells in cell cycle and apoptosis assays (Figure 4A-B). Here, we sorted single CD34+CD38−CD45RA−CD90+ cells into individual wells with or without 005A and then evaluated their potential to divide and expand for 48 and 72 hours. For the 005A-treated cells, 16.67% of the wells had undergone cell division to form 2 cells, whereas 27.59% of the control cells had undergone a first round of cell division. In addition, none of the 005A-treated cells had undergone 2 rounds of cell division, whereas 6.90% of the wells containing control cells did. At the 72-hour time point, the percentage of wells that contained single cells had decreased to 3.33% for the 005A-treated wells and 0% for the control group wells (Figure 4C). However, the total number of wells containing cell colonies was also higher for the 005A group than the control group 14 days after 005A or vehicle treatment (Figure 4D). These results together indicate that 005A treatment delayed cell division. Because 005A treatment did not affect cell cycle or cell apoptosis, we hypothesized that 005A may prompt the self-renewal of HSCs by improving symmetric self-renewal cell division. Two proteins, γ-tubulin and Numb, were stained as representative determinants of cell division.27,28 We assessed the distribution of these 2 proteins relative to the dividing line that was drawn between each set of centrosomes. In our experiment, approximately 50 cells were found in the mitosis stage for both 005A-treated and control cells. The proportion of 005A-treated cells that showed symmetric Numb and γ-tubulin distribution was higher than that of the control cells (Figure 4E). These observations suggested that 005A might delay cell division and regulate the symmetric cell division to promote human long-term HSC self-renewal.
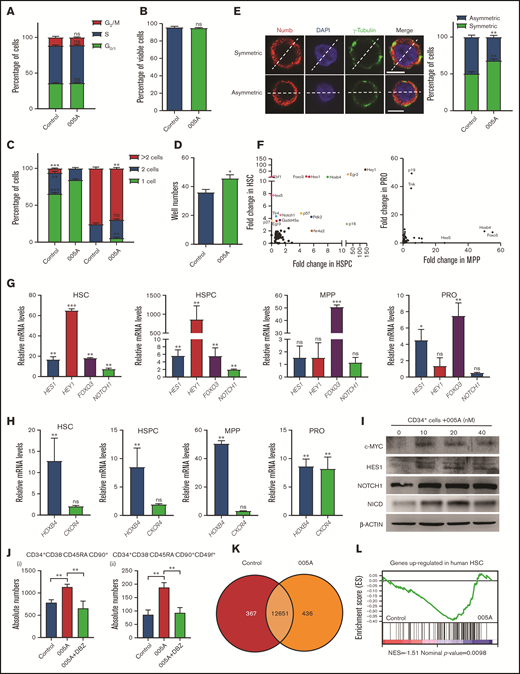
Increase in human long-term HSC self-renewal induced by 005A is characterized by delayed cell division and activation of Notch signaling. (A-B) Cell cycle status and percentage of viable cells were detected for cells cultured in the presence or absence of 005A (n = 3 in panel A, n = 4 in panel B). CD34+ UBCs were planted in 24-well plates, and cell populations were analyzed by flow cytometry after 7-day treatment with vehicle or 005A (20 nmol/mL). (C) Analysis of single cell division status at 48 and 72 hours after 005A or vehicle treatment. (D) The total number of wells containing cell colonies at 14 days after 005A or vehicle treatment. Cells and colonies were counted under a microscope, and the 3 categories indicated were recorded, and 60 wells were measured in each group (n = 3) in panels C and D. (E) The percentage of cells for each cell division type after 72-hour treatment with and without 005A (005A and control, respectively) are presented. One hundred cells were randomly measured. Representative pictures of Numb staining are shown on the left. Scale bar = 6 μm. Original magnification, 60×. (F) Differential expression analysis of the genes closely related to stem cell fate determination in cultured HSCs, HSPCs, MPPs, and progenitors (PROs) with or without 005A treatment by single-cell PCR. The genes exhibiting the greatest changes in gene expression compared with the control group are labeled. (G-H) Expression of Notch signaling genes, HOXB4, and CXCR4 in 005A-cultured HSCs, HSPCs, MPPs, and PROs by real-time PCR (n = 3). Data were normalized to the expression levels in control group. (I) Western blot analysis for c-MYC, HES1, NOTCH1, and Notch intracellular cytoplasmic domain (NICD) in human CD34+ cells treated with vehicle control or various concentrations of 005A for 4 days. Actin was used as a loading control. (J) Absolute numbers per well of human CD34+CD38−CD45RA−CD90+ (i) and CD34+CD38−CD45RA−CD90+CD49f+ (ii) cells were measured (n = 4); 1 × 105 CD34+ UBCs were planted in 24-well plate, and cell populations were analyzed by flow cytometry after 7-day treatment with vehicle, 005A (20 nmol/mL), or 005A (20 nmol/mL) + DBZ (10 µmol/L). (K) Venn diagram shows the numbers of genes expressed differentially between 005A and control group. (L) The gene set enrichment in vehicle or 005A treated HSCs by gene set enrichment analysis. The experiment was repeated 3 times. All data represent means ± SD. Compared with control unless specified: *P < .05, **P < .01, ***P < .001 by 2-tailed unpaired t test.
Increase in human long-term HSC self-renewal induced by 005A is characterized by delayed cell division and activation of Notch signaling. (A-B) Cell cycle status and percentage of viable cells were detected for cells cultured in the presence or absence of 005A (n = 3 in panel A, n = 4 in panel B). CD34+ UBCs were planted in 24-well plates, and cell populations were analyzed by flow cytometry after 7-day treatment with vehicle or 005A (20 nmol/mL). (C) Analysis of single cell division status at 48 and 72 hours after 005A or vehicle treatment. (D) The total number of wells containing cell colonies at 14 days after 005A or vehicle treatment. Cells and colonies were counted under a microscope, and the 3 categories indicated were recorded, and 60 wells were measured in each group (n = 3) in panels C and D. (E) The percentage of cells for each cell division type after 72-hour treatment with and without 005A (005A and control, respectively) are presented. One hundred cells were randomly measured. Representative pictures of Numb staining are shown on the left. Scale bar = 6 μm. Original magnification, 60×. (F) Differential expression analysis of the genes closely related to stem cell fate determination in cultured HSCs, HSPCs, MPPs, and progenitors (PROs) with or without 005A treatment by single-cell PCR. The genes exhibiting the greatest changes in gene expression compared with the control group are labeled. (G-H) Expression of Notch signaling genes, HOXB4, and CXCR4 in 005A-cultured HSCs, HSPCs, MPPs, and PROs by real-time PCR (n = 3). Data were normalized to the expression levels in control group. (I) Western blot analysis for c-MYC, HES1, NOTCH1, and Notch intracellular cytoplasmic domain (NICD) in human CD34+ cells treated with vehicle control or various concentrations of 005A for 4 days. Actin was used as a loading control. (J) Absolute numbers per well of human CD34+CD38−CD45RA−CD90+ (i) and CD34+CD38−CD45RA−CD90+CD49f+ (ii) cells were measured (n = 4); 1 × 105 CD34+ UBCs were planted in 24-well plate, and cell populations were analyzed by flow cytometry after 7-day treatment with vehicle, 005A (20 nmol/mL), or 005A (20 nmol/mL) + DBZ (10 µmol/L). (K) Venn diagram shows the numbers of genes expressed differentially between 005A and control group. (L) The gene set enrichment in vehicle or 005A treated HSCs by gene set enrichment analysis. The experiment was repeated 3 times. All data represent means ± SD. Compared with control unless specified: *P < .05, **P < .01, ***P < .001 by 2-tailed unpaired t test.
Because cell fate decision making in HSCs is driven by gene expression changes in key signaling pathways, genes that contribute to enhanced self-renewal were assayed. Human UCB-derived CD34+ cells were cultured for 7 days and then were sorted into 4 populations: HSCs (CD34+CD38−CD45RA−CD90+ cells), HSPCs (CD34+38−), multipotent progenitors (MPPs) cells (CD34+CD38−CD45RA−CD90−), and progenitor (PROs) cells (CD34+38+).29 Each sample was assayed by single cell polymerase chain reaction (PCR) to detect expression levels of 73 genes involving Notch, p53, and other pathways. Among the most upregulated genes by 005A treatment in HSCs or HSPCs, we found several Notch pathway genes such as HEY1, HES1, HES5, NOTCH1, and FOXO3 (Figure 4F). Real-time PCR assays also showed that 005A-treated HSCs exhibited significantly higher expression levels of Notch targets. In the HSPCs, 005A treatment resulted in a similar upregulation of Notch targets. In contrast, Notch targets in more mature populations of MPP cells and progenitor cells were largely unaffected by 005A (Figure 4G). Another critical gene for HSC self-renewal, HOXB4,30 was found to be upregulated in all 4 005A-treated groups, whereas CXCR4 was not affected by 005A treatment in HSCs, HSPCs, and MPPs (Figure 4H). In addition, similar results showed that p18 knockdown increased the Notch targets’ expression in HSC and HSPC (supplemental Figure 4A-B). Western blot analysis also confirmed that the expression of c-MYC, HES1, Notch1, and Notch intracellular cytoplasmic domain (NICD) were dose dependently increased by 005A treatment (Figure 4I). Using a reported Notch signaling inhibitor, DBZ,31 we found the 005A-induced increase of CD34+CD38−CD90+CD45RA−CD49f+ cell number could be abolished by DBZ (Figure 4J; supplemental Figure 4C) and decreased colony numbers by DBZ in CFC assays (supplemental Figure 4D). Furthermore, sorted CD34+ cells were used for RNA sequencing. By gene set enrichment analysis, we observed significant enrichment of gene sets characteristic of “genes up-regulated in human HSC enriched populations compared to committed progenitors and mature cells” in 005A-treated cells (Figure 4K-L; supplemental Figure 4E). Although there is no change of Notch signaling (data not shown) detected by RNA-seq, perhaps because amounts in formation of differentiated cells had covered those of primitive cells, these results still supported that 005A treatment enhanced human HSCs. Overall, the results obtained demonstrate that 005A can modulate the fate determination of HSCs at least in part by regulating Notch signaling.
Discussion
HSCs are extensively used for clinical treatment of various hematologic malignancies. However, major obstacles to the clinical use of HSCs remain, largely because of a lack of well-matched human leukocyte antigen sources. Human UCB cells have exhibited promising potential as an alternative source for HSCs. However, the number of HSCs that can be recovered from a single UCB unit is not enough for adult transplantation.32,33 Thus, there is great interest in the development of conditions for ex vivo expansion of HSCs.
Cell cycle regulation is a key target as well. In murine studies, several CKIs, including p18, p21, p27, and p57, have been shown to alter the behavior of HSCs or HSPCs.20-22 In this study, the role of p18 in human HSCs was similar to that in mouse models, in which knockdown of p18 led to the production of a greater number of HSCs and promoted hematopoietic function.22 However, in contrast to mouse HSCs lacking p18 exhibiting no phenotypic difference in colony forming assays, human HSCs lacking p18 exhibited a significant increase in the number of colony-forming units. These results suggest that the role of regulatory function of p18 differ between mouse and human HSCs, and these findings were consistent with the noted differential responses of mouse vs human immature hematopoietic cells to small molecules.34-36
Small molecules with explicit targets may be of great utility in stem cell research. Hou et al37 recently reported the use of multiple small molecule compounds, including C6FZ, CHIR99021, 616452, FSK, and DZNep, to achieve complete reprogramming by targeting glycogen synthase kinase 3 β, transforming growth factor-β receptor 1, cAMP, and S-adenosylhomocysteine hydrolase, respectively. Recently, a pyrimidoindole derivative (UM171) was reported to be an agonist of human HSCs and increased the number of HSCs.11 Subramaniam et al38 reported that lysine-specific demethylase 1A was a target of UM171, and EPCR (CD201) marked UM171-expanded cord blood stem cells.39 In our study, both UM171 and 005A triggered expansion of CD34+ cells. However, 005A preferred to expand the CD90+CD45RA− and CD49f+ cell populations but could not expand the CD201+ cells.
In this study, 005A was found to affect several signaling pathways in addition to targeting the p18 protein. The signaling pathway mainly affected was the Notch signaling pathway. This pathway has long been recognized as a critical factor for the self-renewal and fate determination of stem cells.40 Incubation of human UCB-derived progenitors with immobilized Notch ligands increased the number of CD34+ cells and enhanced hematopoietic reconstitution in an immunodeficient mouse model.41,42 Furthermore, preliminary results from a phase 1 clinical trial suggest that targeting of the Notch pathway may represent a safe and efficacious cell-based therapy.43 In contrast, downregulation of Notch targets has been found to be associated with enhanced differentiation commitment of HSCs.44 In particular, the Notch targets HES1 and FOXO3 have been studied for their roles in the hematopoietic system.43,44 These results indicate that 005A has the potential to significantly enhance the expression of several Notch signaling pathway factors, as well as other related factors. Furthermore, upregulation of NOTCH1, HEY1, HES1, HES5, and FOXO3 gene expression may mediate the mechanism(s) by which 005A induces a self-renewal effect in HSCs. Correspondingly, 005A appears to fulfill these criteria and represents a promising compound for HSC expansion.
Laurenti et al45 demonstrated that CDK6 levels are critical for the exit of HSCs from a quiescent state, and they can also trigger cell division. In LT-HSCs, this action is delayed because of the absence of CDK6, and this is possibly related to maintenance of the integrity of internal populations of HSCs that are needed for long-term hematopoiesis. In this study, 005A was shown to selectively block the activity of p18 and act as an agonist of CDK6, thereby facilitating the exit of ST-HSC from a G0 phase of quiescence. Accordingly, 005A delayed cell cycle progression in the initial stage of ex vivo HSC culture and a higher frequency of HSCs were present in the 005A-treated wells. Similarly, 005A treatment enhanced the activation of CDK6 activity. In combination with the cell division assay results, it is hypothesized that the p18 inhibitor may regulate ST-HSCs and LT-HSCs via distinct mechanisms. The p18 inhibitor might increase symmetric cell divisions of LT-HSCs to expand the pool of HSCs and induce ST-HSCs, as well as progenitor cells, to enter the cell cycle and produce a greater number of functional cells. These 2 pools of cells would be helpful to avoid both early mortality and long-term hematopoietic failure of UCB transplantation. More importantly, this potent strategy targeting p18 for human HSC expansion was not found to be involved in tumorigenic processes in our xenograft model and previous p18 knockout mice.19
Our data showed that the optimized small molecule inhibitor of p18, 005A, could prompt self-renewal of human HSCs to induce ex vivo expansion that were capable of reconstituting human hematopoiesis for at least 8 months when we performed primary and secondary transplantation in immunocompromised mice. Mechanistic studies suggested that 005A might increase the frequency of symmetric cell division. Meanwhile, 005A activated both the Notch signaling pathway and HoxB4 expression in human HSCs. These observations supported roles of p18 in human HSC self-renewal and 005A in facilitating the clinical use of HSC therapy. Further studies to optimize strategies for HSC expansion may involve the additive and synergistic activities of 005A with other compounds.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of China (2017YFA0104900 and 2016YFA0100600), the National Natural Science Foundation of China (92068204, 81870083, 81970105, and 81800165), the Tianjin Science and Technology planning project (18ZXXYSY00010), and a State Key Laboratory of Experimental Hematology pilot research grant.
Authorship
Contribution: Y.G., X.-Q.X., T.C., and W.Y. designed the research and analyzed data; Y.L., Y.Z., X.L., and Y.G. wrote the paper; Z.F. and X.-Q.X. contributed vital new reagents; Y.L., W.Z., J.G., Y.Z., Y.D., M.Y., X.L., M.H., Y.L., J.Y., S.X., H.G., H.S., H.X., Q.J., S.M., and W.Y. performed research and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Yingdai Gao, State Key Laboratory of Experimental Hematology; e-mail: ydgao@ihcams.ac.cn; Tao Cheng, State Key Laboratory of Experimental Hematology; e-mail: chengtao@ihcams.ac.cn; and Xiang-Qun Xie, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center; e-mail: xix15@pitt.edu.