

Key Points
Mismatched SIRPα is frequently present in HSCT from HLA-matched related donors.
Mismatched SIRPα is associated with a higher risk for chronic GVHD and improved relapse-free survival.

Abstract
Recent compelling evidence showed that innate immune effector cells could recognize allogeneic grafts and prime an adaptive immune response. Signal regulatory protein α (SIRPα) is an immunoglobulin superfamily receptor that is expressed on myeloid cells; the interaction between SIRPα and its ubiquitously expressed ligand CD47 elicits an inhibitory signal that suppresses macrophage phagocytic function. Additional studies showed that donor-recipient mismatch in SIRPα variants might activate monocytic allorecognition, possibly as the result of non-self SIRPα-CD47 interaction. However, the frequency of SIRPα variation and its role in hematopoietic stem cell transplantation (HSCT) remains unexplored. We studied 350 patients with acute myeloid leukemia/myelodysplastic syndrome who underwent HLA-matched related HSCT and found that SIRPα allelic mismatches were present in 39% of transplantation pairs. SIRPα variant mismatch was associated with a significantly higher rate of chronic graft-versus-host disease (GVHD; hazard ratio [HR], 1.5; P = .03), especially de novo chronic GVHD (HR, 2.0; P = .01), after adjusting for other predictors. Those with mismatched SIRPα had a lower relapse rate (HR, 0.6; P = .05) and significantly longer relapse-free survival (RFS; HR, 0.6; P = .04). Notably, the effect of SIRPα variant mismatch on relapse protection was most pronounced early after HSCT and in patients who were not in remission at HSCT (cumulative incidence, 73% vs 54%; HR, 0.5; P = .01). These findings show that SIRPα variant mismatch is associated with HSCT outcomes, possibly owing to innate allorecognition. SIRPα variant matching could provide valuable information for donor selection and risk stratification in HSCT.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a potentially curative therapy for many hematologic malignancies and nonmalignant hematologic disorders. The allorecognition in transplantation is predominantly attributed to the adaptive immune response in which lymphoid cells express rearranging receptors for “non-self” antigens.1 Potent alloreactivity is initiated by T cells recognizing the non-self HLA molecules on the graft, as well as the peptides derived from the mismatched HLA molecules.2 Therefore, matching the HLA genotype and restraining lymphocyte alloreactivity are key steps in current allo-HSCT interventions.
Conventionally, it has been believed that, unlike in the adaptive immune response, innate immune cells that express nonrearranging receptors recognize necrotic cells or “danger” molecules released from dying graft cells and then prime adaptive immunity.3 However, recent studies have provided compelling evidence that the innate immune system could specifically distinguish the allogeneic graft and sequentially enhance the adaptive immune response. Oberbarnscheidt et al showed that allogeneic grafts induced persistent maturation of dendritic cells (DCs) derived from host monocytes in a murine model that lacked adaptive immune effector cells, including T and B lymphocytes and natural killer cells. Mature DCs generated interleukin-12 and stimulated T-cell proliferation and interferon-γ production ex vivo. In contrast, monocytes provoked by syngeneic grafts were not capable of stimulating interleukin-12 or interferon-γ production.4 Distinct from allorecognition by the adaptive immune cells, monocytic allorecognition is independent of major histocompatibility complex mismatch and perhaps is determined by the mismatches at non–major histocompatibility complex genomic loci.5
Signal regulatory protein α (SIRPα) is an immunoglobulin superfamily receptor expressed on myeloid cells, including macrophages, DCs, and neutrophils. The SIRPα molecule interacts with its ubiquitously expressed ligand CD47 and elicits an inhibitory signal upon engagement, leading to the suppression of macrophage phagocytic function.6 In solid organ transplants, SIRPα/CD47 interaction facilitates the transplant tolerance, and blocking the SIRPα/CD47 axis elicited an innate response and increased the risk of graft rejection.4,7 In patients who have undergone allo-HSCT, involvement of SIRPα/CD47 in alloimmunity could be complex, because inhibitory SIRPα and stimulatory CD47 are expressed concurrently on cells from the donor or recipient. Notably, inhibition of the CD47/SIRPα axis with anti-CD47 antibody did not impair bone marrow engraftment by normal human stem cells.8 However, CD47−/− recipients demonstrated phagocytic tolerance to xenotransplantation with no signs of graft-versus-host disease (GVHD).9 Moreover, a recent study suggested that donor monocyte-derived macrophages stimulate donor T cells and enhance acute GVHD (aGVHD), and SIRPα was found to be remarkably upregulated in GVHD macrophages.10 Whether SIRPα signaling is involved in the pathogenesis of GVHD in human allo-HSCT remains unknown.
A recent murine study using marrow plug transplantation showed that recipient monocytes detect variations in the SIRPα molecule, and mismatches of SIRPα variants between the donor and recipient regulate the allorecognition response.11 It was believed that the mismatched SIRPα molecule introduced by the allograft may be recognized as “non-self,” owing to unbalanced signals resulting from the interaction between CD47 and variant SIRPα, and this will result in enhanced monocyte activation and DC differentiation.12 Although specific variations in human SIRPα have been identified,13 the prevalence and impact of SIRPα variant mismatch on allo-HSCT clinical outcomes, especially on the risk of GVHD, have not been studied. We hypothesize that the mismatch between donor and recipient SIRPα regulates the innate alloimmune response and contributes to GVHD pathogenesis in allo-HSCT. Herein, we evaluated the clinical impact of SIRPα variant mismatch in a retrospective cohort of 350 patients with acute myeloid leukemia (AML)/myelodysplastic syndrome (MDS) who had undergone hematopoietic stem cell transplantation (HSCT) from HLA-matched related donors with the intention of minimizing the confounding alloreactivity caused by HLA mismatch. Results of our study may provide insight into the underlying role of innate systems in allo-HSCT, in the context of adaptive immunity.
Methods
Patient population
This retrospective analysis included adult patients (with the exception of a 16-year-old patient) with MDS or primary AML who underwent allo-HSCT at The University of Texas MD Anderson Cancer Center between January of 2008 and November of 2019. All patients in our analysis received peripheral blood stem cells from an HLA-matched adult sibling donor, and donor and patient DNA samples were available for SIRPα testing. Patients who failed to engraft were excluded from outcome analyses. A minority of patients had received post-HSCT cyclophosphamide as GVHD prophylaxis; they were excluded from the study because of the profound effect that this treatment has on GVHD outcomes compared with conventional GVHD-prevention regimens. All patients provided written informed consent for transplant following the Declaration of Helsinki. The study protocol was approved by the Institution Review Board of The University of Texas MD Anderson Cancer Center.
SIRPα typing
SIRPα typing was performed with 3 sets of SIRPα-specific targeting primers. Primer recognition sites are shown in Figure 1. The primer set (21376 + 21380) was needed only to confirm ambiguous results with the other 2 sets of primers. Each 20-µL polymerase chain reaction included 2 µL of tested DNA (20 ng/mL), 4 µL of primer mix, 13.9 µL of LABType Primer Set Dmix (LTPDMX-B; One Lambda), and 0.1 µL of Tag polymerase. Polymerase chain reaction was conducted at 96°C for 2 minutes, at 10× (96°C for 10 seconds, 63°C for 1 minute) and at 20× (96°C for 10 seconds, 59°C for 50 seconds, 72°C for 30 seconds). A total of 20 µL of the product was run on a 2% agarose gel by electrophoresis, along with controls. Typing was determined by the presence or absence of specific amplicons (Figure 1B). SIRPα variants were identified and separated into 2 categories with different CD47 binding interfaces. The SIRPα VI category included SIRPα v1, v4, v5, v6, and v9, whereas SIRPα VII consisted of 5 other specific alleles (SIRPα v2, v3, v7, v8, and v10).

Variants of SIRPα and mismatch detection in the current study. (A) Sequence alignment of the SIRPα immunoglobulin V domain variants. The sequences are from Takenaka et al.13 SIRPα v1 and v2 are the most common human variants. The residuals involved in CD47 interaction are under the red dots. The blue dots represent the specific hydrogen bond formed between CD47 and SIRPα v1.37 The differences in interaction interfaces of SIRPα and CD47 on the DE and FG loops are highlighted in yellow.37 Primer recognition sites are shown as blue arrows. Primer set 21376 + 21377 detects the presence of VI-type alleles, including v1, v4, v5, v6, and v9, and primer set 21376 + 21379 detects the presence of VII-type alleles, including v2, v3, v7, v8, and v10. (B) Sequence-Specific Primer (SSP) analysis of patient samples and controls. (C) SIRPα variants and mismatches detected in the current study. The mismatches were subclassified as G when more SIRPα VII allele is detected in the graft, or as H if more SIRPα VII variant is detected in the host.
Variants of SIRPα and mismatch detection in the current study. (A) Sequence alignment of the SIRPα immunoglobulin V domain variants. The sequences are from Takenaka et al.13 SIRPα v1 and v2 are the most common human variants. The residuals involved in CD47 interaction are under the red dots. The blue dots represent the specific hydrogen bond formed between CD47 and SIRPα v1.37 The differences in interaction interfaces of SIRPα and CD47 on the DE and FG loops are highlighted in yellow.37 Primer recognition sites are shown as blue arrows. Primer set 21376 + 21377 detects the presence of VI-type alleles, including v1, v4, v5, v6, and v9, and primer set 21376 + 21379 detects the presence of VII-type alleles, including v2, v3, v7, v8, and v10. (B) Sequence-Specific Primer (SSP) analysis of patient samples and controls. (C) SIRPα variants and mismatches detected in the current study. The mismatches were subclassified as G when more SIRPα VII allele is detected in the graft, or as H if more SIRPα VII variant is detected in the host.
Clinical endpoints
The primary outcomes were the incidence of grade 2-4 aGVHD and chronic GVHD (cGVHD). Secondary outcomes were cGVHD requiring systemic therapy treatment, overall survival (OS), incidence of relapse, nonrelapse mortality (NRM), relapse-free survival (RFS), and time to neutrophil engraftment. aGVHD was defined and graded according to conventional criteria, as published by Przepiorka et al,14 and cGVHD was graded based on conventional criteria, as published by Sullivan et al.15 Cases with strictly aGVHD features occurring after day +100 were differentiated from cGVHD and coded as late aGVHD. Ablative and nonmyeloablative HSCTs were defined according to the Center for International Blood and Marrow Transplant Research operational guidelines.16 AML risk groups were defined according to the European LeukemiaNet (ELN) guidelines published in 2010,17 and MDS risk groups were defined according to the International Prognostic Scoring System risk groups.18
Statistical methods
Patient-, disease-, and HSCT-related baseline factors were compared using the χ2 or Fisher’s exact test, as appropriate, for categorical variables; the Wilcoxon rank-sum test was used for continuous variables. The clinical endpoints included GVHD, disease relapse, NRM, RFS, OS, and neutrophil engraftment. The analysis was conducted to evaluate the association between SIRPα variant match or mismatch status with these outcomes. The cumulative incidences of GVHD, relapse, and NRM were estimated accounting for competing risks. Competing risks included death or disease relapse for estimation of GVHD, death of any cause before relapse for estimation of disease relapse, and disease relapse or disease-related death for estimation of NRM. In addition, a diagnosis of grade 1-4 aGVHD was considered a competing risk for de novo cGVHD. RFS and OS were estimated using the Kaplan-Meier method. RFS was defined as the time from HSCT to relapse or death from any cause. OS was defined as the time from HSCT to death from any cause. Surviving patients were censored at the time of last follow-up. Relapse was defined as evidence of recurrence or progression of malignancy. NRM was defined as death without evidence of persistence or relapse of malignancy. Time to neutrophil engraftment was defined as the first of 3 consecutive days with an absolute neutrophil count > 500 per microliter.
Predictors of outcomes were evaluated in univariable and multivariable analyses using competing risk regression for GVHD, disease relapse, and NRM, and Cox proportional hazards regression was used to evaluate predictors of RFS and OS. The proportional hazards assumption and interaction between the effect of SIRPα variant matching status and time were evaluated and adjusted for, when indicated. Bootstrap analysis (based on 3000 resamplings with replacement) was performed to estimate the bias-corrected confidence intervals around the relative risk, evaluating the association between SIRPα allele variant and outcomes. Results were consistent with estimates derived from the original data set. SIRPα variant matching status was forced in all multivariable models, irrespective of statistical significance on univariable analysis. All other predictors that were significant in univariable analysis were included in the multivariable analysis. Backward elimination was used to develop multivariable prognostic models. First-degree interaction effects between SIRPα variant match or mismatch status and predictors that were found to be significant in the univariable analysis were evaluated and accounted for when indicated. In addition to SIRPα variant matching status, the following factors were evaluated for their association with outcomes: donor-recipient sex, recipient age, HSCT-specific comorbidity index (HSCT-CI), diagnosis (MDS or AML), disease status at HSCT (remission, defined as first or second complete remission or not in remission), ELN risk for AML conditioning regimen (myeloablative or nonmyeloablative), donor-recipient ABO matching, and cytomegalovirus (CMV) status. Statistical significance was defined at the 0.05 level, and statistical analyses were performed using primarily STATA 14.0 (StataCorp, College Station, TX).
Results
Patient and HSCT characteristics
A total of 350 patients met the inclusion criteria for the study. The median age was 54 years (range 16-74 years); 170 (49%) patients were female (Table 1). The pre-HSCT diagnosis was MDS in 101 patients (29%) and primary AML in the other 249 patients (71%). Five patients (1%) had undergone a prior allogeneic HSCT. Most patients (91%) received an ablative conditioning regimen. GVHD prophylaxis consisted of tacrolimus and methotrexate in 99% of patients. SIRPα variant mismatch was found in 136 patients (39%) and was not correlated with patient or disease characteristics prior to HSCT (Table 1). The number of SIRPα mismatches detected in HSCT is summarized in Figure 1C. The direction of mismatches was classified based on the presence of “non-self” SIRP VII in the host (H) or donor (G) genotype.
Demographic and clinical characteristics of patients who underwent allogeneic HSCT for AML or MDS (N = 350)
| Characteristic | Overall (N = 350) | SIRPα mismatched (n = 136) | SIRPα matched (n = 214) | P |
|---|---|---|---|---|
| Age at HSCT, y | .6 | |||
| ≤40 | 68 (19) | 25 (18) | 43 (20) | |
| 41-50 | 61 (17) | 20 (15) | 41 (19) | |
| 51-60 | 124 (35) | 53 (39) | 71 (33) | |
| >60 | 97 (28) | 38 (28) | 59 (28) | |
| Diagnosis | .8 | |||
| Primary AML | 249 (71) | 96 (71) | 153 (71) | |
| MDS | 101 (29) | 40 (29) | 61 (29) | |
| ELN risk status for AML* | .6 | |||
| Favorable | 43 (17) | 16 (17) | 27 (18) | |
| Intermediate | 133 (53) | 56 (57) | 78 (51) | |
| Adverse | 72 (29) | 25 (26) | 47 (31) | |
| Missing | 1 (0.4) | 0 (0) | 1 (1) | |
| IPSS for MDS | .4 | |||
| Good | 30 (30) | 14 (35) | 16 (26) | |
| Intermediate† | 17 (17) | 8 (20) | 9 (15) | |
| Bad | 54 (54) | 18 (45) | 36 (59) | |
| Disease status at HSCT | .2 | |||
| Remission | 162 (46) | 69 (51) | 93 (43) | |
| Not in remission | 188 (54) | 67 (49) | 121 (57) | |
| HSCT-CI | .4 | |||
| <3 | 177 (51) | 65 (48) | 112 (52) | |
| ≥3 | 173 (49) | 71 (52) | 102 (48) | |
| Conditioning intensity | .4 | |||
| Myeloablative | 317 (91) | 121 (89) | 196 (92) | |
| Nonmyeloablative | 33 (9) | 15 (11) | 18 (8) | |
| GVHD prophylaxis | .7 | |||
| Tacrolimus/methotrexate | 347 (99) | 135 (99) | 212 (99) | |
| Tacrolimus/MMF | 3 (1) | 1 (1) | 2 (1) | |
| Donor/recipient sex | .7 | |||
| Male/male | 107 (31) | 38 (28) | 69 (32) | |
| Female/male | 100 (29) | 38 (28) | 62 (29) | |
| Female/female | 70 (20) | 28 (21) | 42 (20) | |
| Male/female | 73 (21) | 32 (23) | 41 (19) | |
| Donor/recipient CMV status | .8 | |||
| NR/NR | 26 (7) | 10 (7) | 16 (7) | |
| R/NR | 12 (3) | 6 (4) | 6 (3) | |
| R/R | 194 (55) | 72 (53) | 122 (57) | |
| NR/R | 117 (33) | 48 (35) | 69 (32) | |
| Missing | 1 (0.3) | 0 (0) | 1 (0.5) | |
| Donor/recipient ABO | .3 | |||
| Matched | 209 (60) | 82 (60) | 127 (59) | |
| Minor mismatch | 64 (18) | 25 (18) | 39 (18) | |
| Major mismatch | 67 (19) | 28 (21) | 39 (18) | |
| Bidirectional mismatch | 10 (3) | 1 (1) | 9 (4) |
| Characteristic | Overall (N = 350) | SIRPα mismatched (n = 136) | SIRPα matched (n = 214) | P |
|---|---|---|---|---|
| Age at HSCT, y | .6 | |||
| ≤40 | 68 (19) | 25 (18) | 43 (20) | |
| 41-50 | 61 (17) | 20 (15) | 41 (19) | |
| 51-60 | 124 (35) | 53 (39) | 71 (33) | |
| >60 | 97 (28) | 38 (28) | 59 (28) | |
| Diagnosis | .8 | |||
| Primary AML | 249 (71) | 96 (71) | 153 (71) | |
| MDS | 101 (29) | 40 (29) | 61 (29) | |
| ELN risk status for AML* | .6 | |||
| Favorable | 43 (17) | 16 (17) | 27 (18) | |
| Intermediate | 133 (53) | 56 (57) | 78 (51) | |
| Adverse | 72 (29) | 25 (26) | 47 (31) | |
| Missing | 1 (0.4) | 0 (0) | 1 (1) | |
| IPSS for MDS | .4 | |||
| Good | 30 (30) | 14 (35) | 16 (26) | |
| Intermediate† | 17 (17) | 8 (20) | 9 (15) | |
| Bad | 54 (54) | 18 (45) | 36 (59) | |
| Disease status at HSCT | .2 | |||
| Remission | 162 (46) | 69 (51) | 93 (43) | |
| Not in remission | 188 (54) | 67 (49) | 121 (57) | |
| HSCT-CI | .4 | |||
| <3 | 177 (51) | 65 (48) | 112 (52) | |
| ≥3 | 173 (49) | 71 (52) | 102 (48) | |
| Conditioning intensity | .4 | |||
| Myeloablative | 317 (91) | 121 (89) | 196 (92) | |
| Nonmyeloablative | 33 (9) | 15 (11) | 18 (8) | |
| GVHD prophylaxis | .7 | |||
| Tacrolimus/methotrexate | 347 (99) | 135 (99) | 212 (99) | |
| Tacrolimus/MMF | 3 (1) | 1 (1) | 2 (1) | |
| Donor/recipient sex | .7 | |||
| Male/male | 107 (31) | 38 (28) | 69 (32) | |
| Female/male | 100 (29) | 38 (28) | 62 (29) | |
| Female/female | 70 (20) | 28 (21) | 42 (20) | |
| Male/female | 73 (21) | 32 (23) | 41 (19) | |
| Donor/recipient CMV status | .8 | |||
| NR/NR | 26 (7) | 10 (7) | 16 (7) | |
| R/NR | 12 (3) | 6 (4) | 6 (3) | |
| R/R | 194 (55) | 72 (53) | 122 (57) | |
| NR/R | 117 (33) | 48 (35) | 69 (32) | |
| Missing | 1 (0.3) | 0 (0) | 1 (0.5) | |
| Donor/recipient ABO | .3 | |||
| Matched | 209 (60) | 82 (60) | 127 (59) | |
| Minor mismatch | 64 (18) | 25 (18) | 39 (18) | |
| Major mismatch | 67 (19) | 28 (21) | 39 (18) | |
| Bidirectional mismatch | 10 (3) | 1 (1) | 9 (4) |
Data are n (%), percentages may not add to 100% because of rounding. Mismatched SIRPα included mismatches in either direction (G vs H).
IPSS, International Prognostic Scoring System; MMF, mycophenolate mofetil; NR, nonreactive; R, reactive.
Two hundred and forty-nine patients were in the overall group: 96 for SIRPα matched, 153 for SIRPα mismatched.
The Intermediate group includes categories intermediate 1 and intermediate 2 of the IPSS.
The median follow-up in surviving patients was 59 months (range, 3-124 months). Most treatment failures (90%) occurred within 3 years after HSCT. Summary outcomes, overall and according to SIRPα variant matching status, are presented in supplemental Table 1. No significant difference was detected for the impact of SIRPα mismatch direction (G vs H) on any of the outcomes; therefore, mismatching in either direction was considered a mismatch in this study. Grade 2-4 aGVHD was diagnosed in 113 patients at a median of 50 days (range, 14-471; interquartile range, 34-92); 79% of cases occurred by day +100. cGVHD was diagnosed in 131 patients at a median of 9 months (range, 2.6-73; interquartile range, 6-15). cGVHD was de novo in 53 patients. Among patients with cGVHD, 76 required treatment with systemic therapy.
Association between SIRPα variant mismatch and aGVHD and cGVHD
Results of the univariate risk factor analyses for 100-day grade 2-4 aGVHD and cGVHD are presented in supplemental Table 2. The impact of SIRPα variant mismatch on the rate of grade 2-4 aGVHD varied over time (supplemental Figure 1). Our data indicated that patients with mismatched SIRPα had higher rates of aGVHD (cumulative incidence, 10% vs 4%; hazard ratio [HR], 2.3; 95% confidence interval [CI], 0.9-5.4; P = .05) within the first 30 days after HSCT. After 30 days, the rates of aGVHD in the matched and mismatched SIRPα groups converged, resulting in similar 100-day incidence between the groups (cumulative incidence, 26% vs 24%; P = .8). Multivariable analysis was not performed because none of the remaining risk factors in the univariable analysis was associated with the rate of grade 2-4 aGVHD. Similarly, the day +180 incidence of grade 2-4 aGVHD did not differ (cumulative incidence, 31% vs 26%; P = .4) by SIRPα variant mismatch.
SIRPα variant mismatch was associated with a significantly higher rate of cGVHD (HR, 1.5; P = .03; Figure 2A). This effect (HR, 1.4; P = .04) remained significant in the multivariable analysis after adjusting for mismatched donor/recipient CMV status (HR, 1.7; P = .004), the only other significant predictor of cGVHD rate in the multivariable analysis (Table 2). Notably, SIRPα variant mismatch association reached significance for de novo cGVHD (cumulative incidence, 23% vs 12%; HR, 2.0; P = .01; Figure 2B); this effect was independent of donor/recipient CMV status. SIRPα variant mismatch was also associated with a higher rate of cGVHD requiring systemic therapy in univariate analysis (HR, 1.8; 95% CI, 1.1-2.8; P = .01; Figure 2C) and in multivariate analysis (HR, 1.8; 95% CI, 1.2-2.9; P = .01) adjusting for donor/recipient CMV status. Consistently, SIRPα variant mismatch showed a stronger association (Figure 2D) with de novo cGVHD requiring systemic therapy (HR, 2.1; 95% CI, 1.1-3.9; P = .03).

Impact of SIRPα mismatch on cGVHD. Cumulative incidence of overall cGVHD (A), de novo cGVHD (B), overall cGVHD requiring systemic therapy (C), and de novo cGVHD requiring systemic therapy (D) according to SIRPα variant matching status.
Impact of SIRPα mismatch on cGVHD. Cumulative incidence of overall cGVHD (A), de novo cGVHD (B), overall cGVHD requiring systemic therapy (C), and de novo cGVHD requiring systemic therapy (D) according to SIRPα variant matching status.
Multivariable analysis of predictors of cGVHD, disease relapse, and survival outcomes in patients who underwent allogeneic HSCT for AML and MDS (N = 350)
| HR | 95% CI | P | |
|---|---|---|---|
| Predictors of cGVHD | |||
| Mismatched SIRPα | 1.4 | 1.02-2.1 | .04 |
| Mismatched donor/recipient CMV status* | 1.7 | 1.2-2.4 | .004 |
| Predictors of disease relapse | |||
| Mismatched SIRPα | 0.6† | 0.3-1.1 | .08 |
| Not in remission at HSCT | 1.9 | 1.4-2.9 | <.01 |
| ELN adverse risk for AML | 1.6 | 1.04-2.3 | .03 |
| Predictors of NRM | |||
| Mismatched SIRPα | 0.97‡ | 0.5-1.8 | .9 |
| HSCT-CI ≥ 3 | 2.1 | 1.2-3.9 | .01 |
| Not in remission at HSCT | 1.9 | 1.03-3.6 | .04 |
| Predictors of RFS | |||
| Mismatched SIRPα | 0.6† | 0.4-1.0 | .05 |
| Not in remission at HSCT | 2.5 | 1.8-3.4 | <.001 |
| ELN adverse risk for AML | 1.6 | 1.1-2.3 | .005 |
| Age > 60 y | 1.5 | 1.1-2 | .02 |
| Predictors of OS | |||
| Mismatched SIRPα | 0.9‡ | 0.6-1.3 | .6 |
| Not in remission at HSCT | 2.4 | 1.7-3.4 | <.001 |
| Age > 60 y | 1.6 | 1.1-2.2 | .01 |
| ELN adverse risk for AML | 1.7 | 1.2-2.4 | .003 |
| Donor and recipient are CMV reactive | 1.4 | 1.02-1.9 | .03 |
| HR | 95% CI | P | |
|---|---|---|---|
| Predictors of cGVHD | |||
| Mismatched SIRPα | 1.4 | 1.02-2.1 | .04 |
| Mismatched donor/recipient CMV status* | 1.7 | 1.2-2.4 | .004 |
| Predictors of disease relapse | |||
| Mismatched SIRPα | 0.6† | 0.3-1.1 | .08 |
| Not in remission at HSCT | 1.9 | 1.4-2.9 | <.01 |
| ELN adverse risk for AML | 1.6 | 1.04-2.3 | .03 |
| Predictors of NRM | |||
| Mismatched SIRPα | 0.97‡ | 0.5-1.8 | .9 |
| HSCT-CI ≥ 3 | 2.1 | 1.2-3.9 | .01 |
| Not in remission at HSCT | 1.9 | 1.03-3.6 | .04 |
| Predictors of RFS | |||
| Mismatched SIRPα | 0.6† | 0.4-1.0 | .05 |
| Not in remission at HSCT | 2.5 | 1.8-3.4 | <.001 |
| ELN adverse risk for AML | 1.6 | 1.1-2.3 | .005 |
| Age > 60 y | 1.5 | 1.1-2 | .02 |
| Predictors of OS | |||
| Mismatched SIRPα | 0.9‡ | 0.6-1.3 | .6 |
| Not in remission at HSCT | 2.4 | 1.7-3.4 | <.001 |
| Age > 60 y | 1.6 | 1.1-2.2 | .01 |
| ELN adverse risk for AML | 1.7 | 1.2-2.4 | .003 |
| Donor and recipient are CMV reactive | 1.4 | 1.02-1.9 | .03 |
Mismatched SIRPα included mismatches in either direction (G vs H).
Mismatched: nonreactive/reactive or reactive/nonreactive.
HR adjusted for interaction with time.
SIRPα variation was forced into the multivariable model.
Association between SIRPα variant mismatch and relapse and NRM
Results of the univariate analysis of risk factors for relapse and NRM are presented in supplemental Table 3. The impact of SIRPα mismatch on relapse varied over time. Adjustment for this time-varying effect in the univariate analysis showed that patients with mismatched SIRPα had a lower relapse rate (HR, 0.6; P = .05; Figure 3A). This trend persisted (HR, 0.6; P = .08) in the multivariable analysis after adjusting for the time-varying effect and other significant predictors of relapse rate, including nonremission status at HSCT (HR, 1.9; P < .01) and ELN adverse-risk status (HR, 1.6; P = .03) (Table 2). Subset multivariable analysis (supplemental Table 4) revealed that the protective impact of SIRPα variant mismatch on disease relapse was most pronounced (HR, 0.5; P = .02) early (within 6 months of HSCT) in patients who were not in remission at HSCT.

Impact of SIRPα mismatch on disease relapse and NRM. Cumulative incidence of disease relapse (A) and NRM (B) according to SIRPα variant matching status.
Impact of SIRPα mismatch on disease relapse and NRM. Cumulative incidence of disease relapse (A) and NRM (B) according to SIRPα variant matching status.
In the univariate analysis, SIRPα variant mismatch was not associated with NRM rate (HR, 0.96; P = .9; Figure 3B). The lack of an association persisted (HR; 0.97; P = .9) in the multivariable analysis after adjusting for HSCT-CI ≥ 3 (HR, 2.1; P = .01) and nonremission status at HSCT (HR, 1.9; P = .04), the only significant predictors of NRM rate.
Association between SIRPα variant mismatch and RFS and OS
Results of the univariate analysis of risk factors for RFS and OS are presented in supplemental Table 5. SIRPα variant mismatch was associated with superior RFS (HR, 0.6; P = .04; Figure 4A). This effect persisted (HR, 0.6; P = .05) in the multivariable analysis (Table 2) after adjusting for the time-varying effect and for significant adverse predictors of RFS, including nonremission status at HSCT (HR, 2.5; P < .001), ELN adverse status in AML (HR, 1.6; P = .005), and recipient age > 60 years (HR, 1.5; P = .02). Mirroring the results described above for disease relapse, subset multivariable analysis (supplemental Table 6) revealed that the protective effect of SIRPα variant mismatch was most pronounced early (within the first 6 months) after HSCT and in patients who were not in remission at HSCT (cumulative incidence, 73% vs 54%; HR, 0.5; P = .01).

Impact of SIRPα mismatch on RFS and OS. Actuarial RFS (A) and OS (B) according to SIRPα variant matching status.
Impact of SIRPα mismatch on RFS and OS. Actuarial RFS (A) and OS (B) according to SIRPα variant matching status.
On univariate analysis, SIRPα variant mismatch was not significantly associated with OS (HR, 0.9; P = .5; Figure 4B). This lack of a significant association persisted (HR, 0.9; P = .6) in the multivariable analysis (Table 2) after adjusting for significant predictors of OS, including nonremission status at HSCT (HR, 2.4; P < .001), age > 60 years (HR, 1.6; P = .01), ELN adverse status in AML (HR, 1.7; P = .003), and CMV reactive donor and recipient status (HR, 1.4; P = .03).
Impact of SIRPα variant mismatch according to donor chimerism
We performed an exploratory subset analysis (supplemental Table 7) to evaluate the impact of SIRPα variant match or mismatch status on cGVHD, disease relapse, and RFS according to the percentage of donor chimerism at 90 days after HSCT. Chimerism data at 90 days were available for 235 of the 350 patients who had not experienced disease relapse, death, or cGVHD before or within 7 days after the 90-day assessment date. Of the 235 eligible patients, 202 (86%) had >95% donor chimerism. Acknowledging the small number of patients with “mixed” chimerism, our data showed that the effect of SIRPα variant mismatch on cGVHD and disease progression was limited to patients with >95% donor chimerism. This effect was also seen for RFS (supplemental Figure 2).
SIRPα variant mismatch was not associated with neutrophil engraftment
The impact of SIRPα variant mismatch on engraftment was also evaluated. In contrast to a previous murine study,13 we did not find any association between SIRPα variant matching status and engraftment incidence or time to engraftment. Four deaths occurred before the engraftment, and no difference was noticed in the early death rate between the matched and mismatched SIRPα groups (1.4% vs 0.7%; P = .5).
Discussion
An increasing body of evidence from experimental models suggests that the innate immune system senses the allogeneic non-self signal and initiates immunity in response to allograft. In a cohort of patients who underwent allogeneic HSCT from HLA-matched related donors, we demonstrated for the first time that mismatched SIRPα, a regulatory protein in the innate immune system, is associated with a higher risk for cGVHD and a lower risk for relapse compared with matched SIRPα. These findings suggest that optimal donor(s) could be selected on the basis of the specific SIRPα variant to mitigate the risk of GVHD or relapse.
Many SIRPα polymorphisms are located at the CD47-binding V-like immunoglobulin domain, and the variants have putatively different binding affinities to CD47.11,19 Therefore, allelic variation in SIRPα between the donor and recipient may change the interaction balance and elicit a “non-self” signal, which leads to increased monocyte activity and alloimmune response.11 Consistently, our data demonstrated that SIRPα donor-recipient mismatches increase GVHD risk, which likely resulted from an innate alloimmune response in which antigen-presenting cells (APCs), including monocytes, macrophages, and DCs, are activated. With the “non-self” signal, APCs are continually stimulated posttransplant, which subsequently provokes and sustains the adaptive alloresponse. Although GVHD is mediated by donor T lymphocytes and host APCs, donor DCs and macrophages have been shown to be likely to contribute to cGVHD development.20,21 Recent data further demonstrated that GVHD can also result from host T cells being stimulated by donor APCs in the form of monocyte-derived macrophages. Moreover, these donor monocyte-derived macrophages can mediate cytopathic effects against resident host T cells.10,22,23 We found that, unlike the mismatches in HLA, the SIRPα mismatch in donor and recipient directions is associated with a trend toward increased cGVHD, indicating that donor and recipient APCs are likely involved in cGVHD pathogenesis, perhaps through perpetuating T-cell–mediated adaptive immune responses.
Our findings are consistent with a graft-versus-leukemia effect,24,25 because mismatched SIRPα correlated with a higher rate of cGVHD and superior RFS that were primarily attributable to reduced relapse.26 It is reasonable to postulate that the innate immune response is elicited by the non-self signal from mismatched SIRPα, which further enhances the adaptive immunity manifested with cGVHD and relapse protection (Figure 5). The impact of SIRPα variant mismatch observed in the current study appeared to vary over time. Transiently enhanced aGVHD was seen within the first 30 days, and relapse protection was more profound within 6 months after HSCT. An oversimplified hypothesis is that the impact of SIRPα variant mismatch and the consequent innate response are partially determined by the time-dependent effector T cells and innate immune cells. The enhanced innate response may initially promote T-cell activation through proinflammatory cytokines or through regulation of recipient APC presentation mediated by host innate immune cells.27,28 The effect might transform later with engraftment when donor-derived myeloid cells and APCs become dominant while the mismatching signal is still present. The donor innate cells with greater activity could contribute to the pathogenesis of cGVHD through macrophage migration/differentiation29 and/or a direct cytopathic effect against host T cells.10
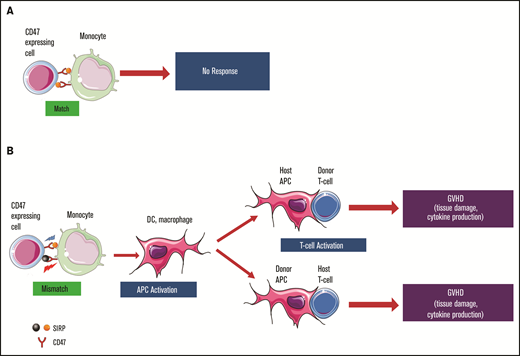
Hypothetical role of SIRPα variant mismatch in HSCT with an HLA-matched donor. (A) Matched SIRPα variants between donor and recipient. The interaction between SIRPα and CD47 remains in the balance of activation and inhibition signals without any “non-self” response from monocytes. (B) Mismatched SIRPα variants between donor and recipient elicit a “non-self” signal for monocyte activation. Donor and recipient APCs, including macrophages and DCs, are activated. The enhanced innate immunity may further promote adaptive immunity through specific effector cells, which will lead to a higher risk for cGVHD and other clinical manifestations.
Hypothetical role of SIRPα variant mismatch in HSCT with an HLA-matched donor. (A) Matched SIRPα variants between donor and recipient. The interaction between SIRPα and CD47 remains in the balance of activation and inhibition signals without any “non-self” response from monocytes. (B) Mismatched SIRPα variants between donor and recipient elicit a “non-self” signal for monocyte activation. Donor and recipient APCs, including macrophages and DCs, are activated. The enhanced innate immunity may further promote adaptive immunity through specific effector cells, which will lead to a higher risk for cGVHD and other clinical manifestations.
Notably and consistent with the lack of a significant effect on day +180 grade 2-4 aGVHD, the rate of de novo cGVHD was significantly higher in the mismatched group, suggesting that the pathogenesis of cGVHD is not simply an overlap or progression from aGVHD and might involve specific donor T cells.30 Hakim et al showed that innate immune activation is essential for the initiation and persistence of cGVHD; induction of interferon by the innate response was upregulated in circulating monocytes from patients with cGVHD.31 The recruitment and infiltration of specific macrophage subsets have been shown to contribute to cGVHD.32 Moreover, a higher plasma concentration of CD163, a scavenger receptor expressed upon monocyte and macrophage activation, is related to the incidence of de novo–onset cGVHD.33 Extended investigation of these aspects is necessary to determine the role of innate cells in the risk for de novo cGVHD related to SIRPα variant mismatch.
The validation of a genetic biomarker of cGVHD is rather complex and requires multiple steps34; an independent series of well-controlled HSCT studies is warranted to verify our findings. Our study has several limitations, including its retrospective nature and a relatively small number of patients. Although we looked at the rate of cGVHD requiring systematic therapy to gauge the severity of cGVHD, we were not able to classify cGVHD severity according to National Institutes of Health criteria.35 This may limit the generalization of our findings and their comparison with future studies. Additionally, because of our relatively small sample size, the primers that we used were designed to allow us to distinguish only two major categories (Figure 1), not the individual variant alleles. In the current study, the direction of mismatches was classified based on the presence of “non-self” SIRPα VII allele in the H or G genotype, considering that the mismatching signal is elicited when self-CD47 recognizes a “non-self” SIRPα molecule. Although no significant outcome difference was identified between the two groups, the positive findings could be overlooked as a result of limited statistical power. Moreover, other confounding factors, such as underlying disease, stem cell source, conditioning intensity, and GVHD prophylactic regimens, could be instrumental in modulating innate and adaptive immune response and remain to be investigated.
Although the specific SIRPα allele frequency varies across different populations,36 SIRPα variant mismatch between the donor and recipient in HSCT does not appear to be uncommon. In our cohort of patients who underwent HSCT from related donors, SIRPα variant mismatch was detected in almost 40% of donor-recipient pairs. The presence of a mismatch is associated with a higher risk for cGVHD and improved RFS. Thus, this finding could be clinically important and potentially useful with regard to donor selection, risk stratification, and even post-HSCT immunotherapy.
Acknowledgments
The authors thank Jar-How Lee (Thermo Fisher Scientific) for helpful discussions and development of the SIRPα typing assay. They also thank Erica Goodoff, Senior Scientific Editor in the Research Medical Library, The University of Texas MD Anderson Cancer Center, for editing this article.
Authorship
Contribution: K.C., R.E.C., and J.Z. designed the study, collected and interpreted data, and wrote the manuscript; R.M.S., U.G., and J.Z. wrote the initial draft of the manuscript; R.M.S. designed and performed statistical analyses, interpreted results, and reviewed and approved the manuscript; L.L. performed and interpreted statistical analyses and reviewed and approved the manuscript; U.G., Q.M., S.A.S., Y.C., M.M., J.W., P.K., and D.L. collected and analyzed data and reviewed and approved the manuscript; G.R. collected data and reviewed and approved the manuscript; B.O., S.O.C., P.K., and D.P. interpreted data and reviewed and approved the manuscript; and U.G., S.A.S., K.R., E.J.S., and R.E.C. treated patients and reviewed, edited, and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jun Zou, Department of Laboratory Medicine, Unit 1060, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: jzou@mdanderson.org; and Richard E. Champlin, Department of Stem Cell Transplantation and Cellular Therapy, Unit 423, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: rchampli@mdanderson.org.

