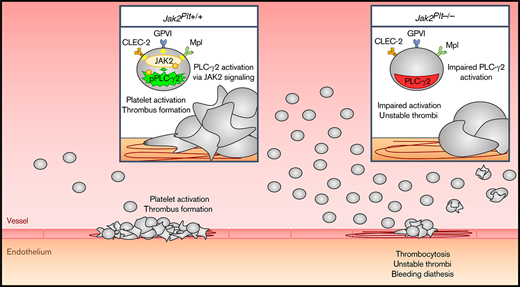
Key Points
Mice lacking the tyrosine kinase JAK2 in platelets develop a bleeding diathesis, despite severe thrombocytosis.
JAK2 is required for immunoreceptor tyrosine-based activation motif signaling and platelet hemostatic function in mice.

Abstract
The tyrosine kinase JAK2 is a critical component of intracellular JAK/STAT cytokine signaling cascades that is prevalent in hematopoietic cells, such as hematopoietic stem cells and megakaryocytes (MKs). Individuals expressing the somatic JAK2 V617F mutation commonly develop myeloproliferative neoplasms (MPNs) associated with venous and arterial thrombosis, a leading cause of mortality. The role of JAK2 in hemostasis remains unclear. We investigated the role of JAK2 in platelet hemostatic function using Jak2fl/fl Pf4-Cre (Jak2Plt−/−) mice lacking JAK2 in platelets and MKs. Jak2Plt−/− mice developed MK hyperplasia and splenomegaly associated with severe thrombocytosis and bleeding. This notion was supported by failure to occlude in a ferric chloride carotid artery injury model and by a cremaster muscle laser-induced injury assay, in which Jak2Plt−/− platelets failed to form stable thrombi. Jak2Plt−/− platelets formed thrombi poorly after adhesion to type 1 collagen under arterial shear rates. Jak2Plt−/− platelets spread poorly on collagen under static conditions or on fibrinogen in response to the collagen receptor GPVI-specific agonist, collagen-related peptide (CRP). After activation with collagen, CRP, or the CLEC-2 agonist rhodocytin, Jak2Plt−/− platelets displayed decreased α-granule secretion and integrin αIIbβ3 activation or aggregation, but showed normal responses to thrombin. Jak2Plt−/− platelets had impaired intracellular signaling when activated via GPVI, as assessed by tyrosine phosphorylation. Together, the results show that JAK2 deletion impairs platelet immunoreceptor tyrosine-based activation motif signaling and hemostatic function in mice and suggest that aberrant JAK2 signaling in patients with MPNs affects GPVI signaling, leading to hemostatic platelet function.
Introduction
The protein tyrosine kinase JAK2 is a member of the Janus kinase family, which includes JAK1, JAK2, JAK3, and Tyk2. JAK2 is most commonly associated with its role in cytokine signaling, which is prevalent in a variety of cell types and is required in numerous physiological processes, including hematopoiesis, immunity, inflammation, and tumorigenesis.1 The interaction between the cytokine receptor Mpl and its cognate ligand, thrombopoietin (TPO), induces its oligomerization followed by JAK2 phosphorylation and activation, instigating the recruitment of signal transducer and activator of transcription (STAT) proteins and their subsequent translocation to the nucleus to induce transcription.2
Essential thrombocythemia (ET), myelofibrosis (MF), and polycythemia vera (PV) are Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) that arise from mutually exclusive somatic mutations in the JAK2, MPL, or CALR genes within the bone marrow hematopoietic stem cell (HSC) compartment and result in constitutive cytokine signaling via the JAK/STAT pathway and clonal expansion of HSCs.3,4 Structurally, JAK2 comprises an N-terminal FERM domain, an atypical SH2 domain, an autoinhibitory pseudokinase domain, and a C-terminal kinase domain, where the pseudokinase domain is critical in autoinhibiting JAK2 kinase activity.5 Activating somatic mutations within the JAK2 gene, including the mutation V617F and mutations in exon 12, which encodes the pseudokinase domain, can be found in more than half of patients with ET and MF and in >90% of patients with PV, illustrating a critical role for JAK2 in the development of MPN.6
Patients with MPNs are at increased risk of thrombosis/thromboembolism and major disease-related bleeding complications.7-11 Because of the morbidity and mortality related to these events, antiplatelet and/or anticoagulant agents are commonly used as primary and/or secondary prophylaxis. Individuals harboring the JAK2 V617F mutation show increased risk of thrombotic events, compared with individuals with CALR mutations,6,11,12 lending credence to the notion of a highly complex role of JAK2 in hemostasis.
Recent studies in knockin mice expressing JAK2 V617F show that hyperactive JAK2 can lead to diminished, normal, or enhanced platelet function and hemostasis, regardless of the number of circulating platelets.13-16 Combined with the clinical observations derived from patients carrying JAK2 V617F, the human and mouse data suggest that JAK2 expression and activity affect the degree of hemostatic potential. However, the cellular mechanisms by which JAK2 expression and activity regulate platelet hemostatic function remain unclear.
In this study, the role of platelet JAK2 was examined in Jak2fl/fl Pf4-Cre (Jak2Plt−/−) mice lacking JAK2 in platelets and megakaryocytes (MKs) and presenting an expansion of MK-affiliated hematopoietic progenitors and subsequent severe thrombocytosis.17 Our data show that Jak2Plt−/− mice developed a bleeding diathesis despite the thrombocytosis. This defect was associated with an impaired ability of Jak2Plt−/− platelets to form thrombi under flow and spread onto type 1 collagen. Jak2Plt−/− platelets displayed decreased responses to the collagen receptor GPVI-specific agonist, collagen-related peptide (CRP), including spreading onto fibrinogen, α-granule secretion, integrin αIIbβ3 activation, aggregation, and protein tyrosine phosphorylation. Jak2Plt−/− platelets also displayed mildly attenuated responses to the CLEC-2 agonist rhodocytin. Taken together, the data illustrate a role for the tyrosine kinase JAK2 in platelet immunoreceptor tyrosine-based activation motif (ITAM)/hemITAM signaling that is required for successful in vivo hemostasis.
Materials and methods
Mice
Jak2fl/fl (Jak2Plt+/+) mice kindly provided by Kay-Uwe Wagner (Wayne State University) were bred with Pf4-Cre mice to generate Jak2fl/fl Pf4-Cre (Jak2Plt−/−) mice lacking JAK2 in platelets and MKs.18–21 Mice were treated according to the National Institutes of Health and Medical College of Wisconsin Institutional Animal Care and Use Committee guidelines (Animal Use Application 5600).
Complete blood counts
Mouse blood was collected from the retroorbital plexus and diluted in Cellpack (Sysmex) supplemented with EDTA and PGE1.17 Complete blood counts were determined with a Sysmex XT-2000i automatic hematology analyzer, including the immature platelet fraction, which was measured by fluorescent flow cytometry, with proprietary platelet-specific oxazine dyes.
Blood smears
Blood smears were performed with Wright-Giemsa stain. Anticoagulated whole blood was thinly smeared across a glass slide and fixed for 3 minutes in methanol, stained for 1 minute with Wright-Giemsa stain, and washed for 5 minutes in phosphate-buffered saline. Imaging was performed on a Nikon Eclipse E600 microscope equipped with a SPOT insight firewire color mosaic camera (SPOT imaging solutions) and Plan Apo 40×/0.75 objective, with SPOT imaging v5.1.3 software.
Immunohistochemistry
For immunofluorescence microscopy, femurs were stained for GPIbα and laminin and imaged on a Nikon Eclipse Ti2-E platform equipped with a DS-Qi2 camera and Plan Apo 10×/0.45 (NIS-Elements AR 5.02.00 software). Data were image processed with Imaris (Bitplane) and Matlab (MathWorks) software. Additional information is available in the supplemental Methods.
Tail-bleeding time
Bleeding time was determined by snipping 2 mm of distal mouse tail and immediately immersing the tail in 37°C isotonic saline.22 A complete cessation of bleeding was defined as the bleeding time.
Ferric chloride–induced thrombus formation
Mice were anesthetized and the carotid artery was exposed.23 A microvascular flow probe (0.5 mm) attached to a transit time perivascular flowmeter (Transonic TS420; AD Instruments) was positioned on the carotid artery to monitor blood flow. Vascular injury was induced by applying a filter paper saturated with 10% ferric chloride (FeCl3) to the top of the vessel for 3 minutes. Blood flow was recorded from immediately after removal of the filter paper until 3 minutes after complete occlusion or a maximal time point of 30 minutes. The time to first occlusion was defined by blood flow of <0.05 mL/min.
Laser-induced thrombus formation
Intravital imaging of the formation of platelet thrombi in mouse cremaster arterioles was performed in the Versiti Blood Research Institute Thrombosis Core Laboratory.24 Laser intensity and duration were adjusted to produce an injury visible in the brightfield image. Fluorescent images were captured with a high-speed camera (Orca Flash4.0; Hamamatsu). Data were collected for at least 3 minutes after vessel injury. Platelet and fibrin accumulation and/or extravasation confirmed consistent injury. Details are provided in the supplemental Methods.
Ex vivo perfusion assay
Platelet adhesion to immobilized collagen under flow was performed with a microfluidic VenaFlux platform and Vena8Fluor+ biochips (Cellix).22 An additional description of the procedure is available in the supplemental Methods.
Plasma VWF measurements
Mouse blood was collected by cardiac puncture and anticoagulated in citrate.25 The plasma Von Willebrand factor (VWF) level was determined by antigen capture enzyme-linked immunosorbent assay with monoclonal anti-mouse VWF antibody 344.3 (Versiti) for capture and biotinylated polyclonal anti-VWF (DAKO) for detection. The VWF propeptide (VWFpp) level was determined by antigen capture enzyme-linked immunosorbent assay, with monoclonal anti-VWFpp antibody 349.3 (Versiti) used for capture and biotinylated monoclonal anti-VWFpp antibody 349.2 (Versiti) for detection. A pool of collected C57BL/6J plasma was used as the standard and assigned a value of 1 U/mL.
Platelet preparation
Mouse blood was collected from the retroorbital plexus and anticoagulated in acid citrate dextrose.22 Platelets were isolated by centrifugation and resuspended at 5 × 108 platelets per milliliter in resuspension buffer (140 mM NaCl, 3 mM KCl, 0.5 mM MgCl2, 5 mM NaHCO3, 10 mM glucose, and 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [pH 7.4]).
Platelet spreading
Samples were imaged on Nikon Structured Illumination Microscopy (N-SIM, NIS-Elements AR v4.40.00 software) and Olympus Confocal FV1000-MPE (FluoView software) platforms under 100× oil objectives. Additional details are available in the supplemental Methods.
Flow cytometry
Platelet surface glycoproteins were investigated by flow cytometry using fluorescently conjugated antibodies: GPIbα, GPIbβ, GPIX, GPV, GPVI (Emfret Analytics), GPIIa, and GPIIIa (BD Biosciences).26 For VWF binding, washed platelets were incubated in 20% mouse platelet-poor plasma, with or without 4 µg/mL botrocetin (Sigma-Aldrich) for 5 minutes at 37°C and stained with fluorescein isothiocyanate (FITC)-labeled rabbit anti-mouse VWF antibody (Emfret Analytics).27 For α-granule secretion and αIIbβ3 activation, washed platelets were activated or not with CRP (Versiti Blood Research Institute Protein Chemistry Core Laboratory), rhodocytin (kindly provided by Johannes Eble, University of Münster), human thrombin (Roche), adenosine diphosphate (ADP; Chrono-log), and/or U46619 (Enzo Life Sciences) for 2 minutes at 37°C and stained with FITC-labeled rat anti-mouse P-selectin antibody (BD Biosciences) or Oregon Green 488-labeled fibrinogen (Thermo Fisher Scientific).22 For αIIbβ3 activation assessment in response to the weak agonists ADP and/or U46619, washed platelets were stimulated for 20 minutes at room temperature in the presence of 1 mM CaCl2 and directly stained with phycoerythrin-labeled anti-active mouse αIIbβ3 antibody JON/A (Emfret Analytics). Fluorescence was quantified with an Accuri C6 flow cytometer (BD Biosciences) and FlowJo software. A total of 20 000 events were analyzed for each sample.
Platelet aggregation
Platelet aggregation was monitored for 5 minutes at 37°C by light transmission under stirring conditions using a Chrono-log Model 490-X aggregometer.
Immunoblot analysis
Platelet proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto an Immobilon-P membrane (EMD Millipore), and probed with antibodies directed against the proteins of interest.22
Statistical analysis
Results were compared by using the unpaired Student t test (mean comparison between 2 groups), 2-way analysis of variance (ANOVA; mean comparison between multiple groups), or the log-rank test (survival distribution comparison between 2 groups), with Prism software (GraphPad). Differences were considered statistically significant when P < .05.
Results
Thrombocytosis in Jak2Plt−/− mice
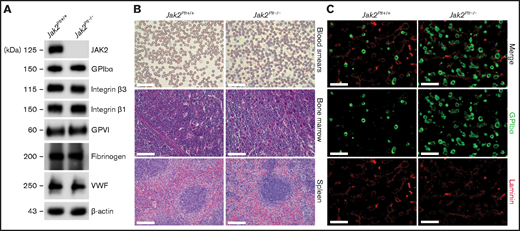
To investigate the role of JAK2 in platelet hemostatic function, we generated Jak2fl/fl Pf4-Cre (Jak2Plt−/−) mice lacking JAK2 in platelets and MKs, as described previously.17 Loss of JAK2 in Jak2Plt−/− platelet lysates was confirmed by immunoblot analysis (Figure 1A). Jak2Plt−/− mice developed a severe thrombocytosis with 7544 × 103 ± 1013 × 103 platelets per microliter (mean ± standard deviation [SD]; n = 9), compared with 1520 × 103± 167 × 103 platelets per microliter in Jak2Plt+/+ littermate controls (n = 10; P < .001), a fivefold increase (Table 1). Although the platelets were of normal size, the thrombocytosis was associated with a slight increase in the immature platelet fraction from 4.55% ± 0.66% to 6.00% ± 1.85% (P = .033). The erythrocyte count was within normal range, and the leukocyte count, although slightly increased, was not statistically significant. Although they confirmed the thrombocytosis phenotype of the Jak2Plt−/− mice, thin blood smears of whole blood did not reveal any notable differences from peripheral blood cell morphology (Figure 1B).
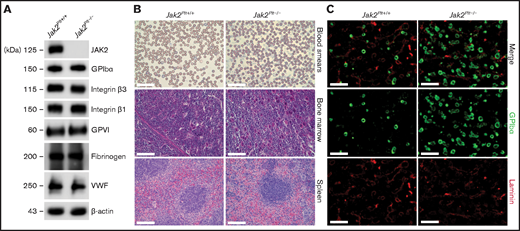
Thrombocytosis and MK hyperplasia in Jak2Plt−/− mice. (A) Jak2Plt+/+ and Jak2Plt−/− platelet lysates corresponding to 2 µg of protein were subjected to SDS-PAGE and probed for the proteins indicated, with β-actin as a loading control. Results are representative of 3 independent experiments. (B) Thin blood smears of Jak2Plt+/+ and Jak2Plt−/− mice. Bars represent 40 µm. H&E staining. Bars represent 80 µm. Sections are representative of 3 mice per genotype. (C) Seven-μm-thick femur bone marrow tissue sections from Jak2Plt+/+ and Jak2Plt−/− mice were immunostained for resident MKs and vasculature using anti-GPIbα (488 nm, green) and anti-laminin (568 nm, red) antibodies, respectively, before fluorescent microscopic analysis. Scale bars, 100 µm. Sections shown are representative of 6 mice per genotype.
Thrombocytosis and MK hyperplasia in Jak2Plt−/− mice. (A) Jak2Plt+/+ and Jak2Plt−/− platelet lysates corresponding to 2 µg of protein were subjected to SDS-PAGE and probed for the proteins indicated, with β-actin as a loading control. Results are representative of 3 independent experiments. (B) Thin blood smears of Jak2Plt+/+ and Jak2Plt−/− mice. Bars represent 40 µm. H&E staining. Bars represent 80 µm. Sections are representative of 3 mice per genotype. (C) Seven-μm-thick femur bone marrow tissue sections from Jak2Plt+/+ and Jak2Plt−/− mice were immunostained for resident MKs and vasculature using anti-GPIbα (488 nm, green) and anti-laminin (568 nm, red) antibodies, respectively, before fluorescent microscopic analysis. Scale bars, 100 µm. Sections shown are representative of 6 mice per genotype.
We assessed the surface and total expression of major glycoproteins on Jak2Plt−/− platelets by flow cytometry and immunoblot analysis (Figure 1A; Table 2). Jak2Plt−/− mice had normal surface expression of all major platelet glycoproteins, and immunoblots of total Jak2Plt−/− platelet lysates showed normal fibrinogen and VWF expression, indicative of a normal α-granule cargo.
MK hyperplasia and splenomegaly in Jak2Plt−/− mice
Megakaryopoiesis was investigated in Jak2Plt−/− and Jak2Plt+/+ littermate controls by hematoxylin and eosin (H&E) staining and immunofluorescence microscopy. H&E staining revealed an MK expansion in bone marrow sections and throughout the length of the spleen, localized in the peripheral-to-marginal zones of Jak2Plt−/− mice (Figure 1B). Jak2Plt−/− bone marrow sections had 219 ± 34 MKs per square millimeter (mean ± SD; n = 6), compared with 77 ± 18 MKs per square millimeter in Jak2Plt+/+ mice (n = 6; P < .001), a 2.8-fold increase, as quantified by immunofluorescence microscopy with an antibody directed against MK GPIbα (Figure 1C; Table 1).
Jak2Plt−/− mice displayed severe splenomegaly. At 8 weeks of age, the spleen/body ratio of Jak2Plt−/− mice was 5.90 ± 1.13 mg/g (mean ± SD; n = 17), compared with 2.72 ± 0.53 (n = 10; P < .001) mg/g in Jak2Plt+/+ littermate controls, a 2.2-fold increase (Table 1), suggesting extramedullary hematopoiesis.
Bleeding and failure to form stable thrombi in vivo in Jak2Plt−/− mice
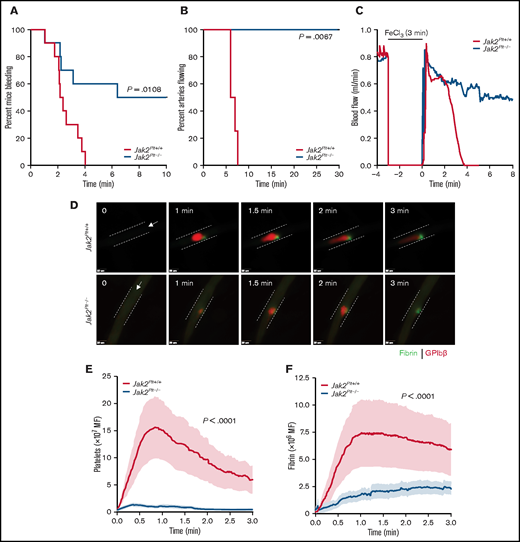
We evaluated the contribution of platelet JAK2 to hemostasis by using the tail-bleeding–time assay (Figure 2A). Despite the thrombocytosis, Jak2Plt−/− mice had a severe bleeding diathesis with a median bleeding time of 8.20 minutes, compared with 2.29 minutes in the Jak2Plt+/+ littermate controls (n = 10 in each group; log-rank P = .0108). In half of the mice, the bleeding continued for 10 minutes, our experimental end point.
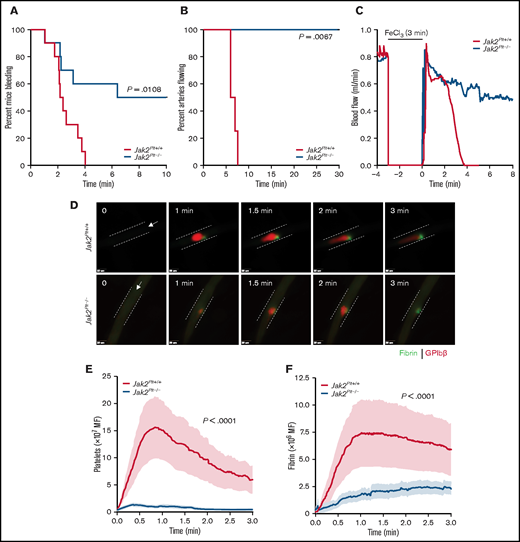
Bleeding and thrombosis defects in Jak2Plt−/− mice. (A) Tail-bleeding time of Jak2Plt+/+ and Jak2Plt−/− mice. Results were estimated by the Kaplan-Meier method and were compared by using the log-rank test (n = 10 in each group; log-rank P = .0108). (B-C) Jak2Plt+/+ and Jak2Plt−/− mice were exposed to 10% FeCl3 for 3 minutes, and arterial flow rates of the carotid artery were measured with a flow probe up to a maximum of 30 minutes to occlusion. (B) Time to occlusion after FeCl3-induced injury. Results were estimated by the Kaplan-Meier method and were compared by using the log-rank test (n = 4 in each group; log-rank P = .0067). (C) Representative blood flow rate graphs. (D-F) Laser-induced injury of the cremaster muscle. Jak2Plt+/+ and Jak2Plt−/− mice were injected with anti-GPIbβ (red) and anti-fibrin (green) antibodies, and the cremaster arteries were interrogated with a 3i Ablate! laser during fluorescence, real-time, intravital video microscopy. (D) Representative still images at 0 to 3 minutes. Arrows represent blood flow direction. Bars represent 25 µm. Platelet (E) and fibrin (F) accumulation at the site of injury was measured by fluorescence intensity. Data are the mean ± standard error of the mean at each time point, compared by using the unpaired Student t test (n = 20 vessels in 4 Jak2Plt+/+ mice and 28 vessels in 5 Jak2Plt−/− mice; platelet and fibrin; P < .0001).
Bleeding and thrombosis defects in Jak2Plt−/− mice. (A) Tail-bleeding time of Jak2Plt+/+ and Jak2Plt−/− mice. Results were estimated by the Kaplan-Meier method and were compared by using the log-rank test (n = 10 in each group; log-rank P = .0108). (B-C) Jak2Plt+/+ and Jak2Plt−/− mice were exposed to 10% FeCl3 for 3 minutes, and arterial flow rates of the carotid artery were measured with a flow probe up to a maximum of 30 minutes to occlusion. (B) Time to occlusion after FeCl3-induced injury. Results were estimated by the Kaplan-Meier method and were compared by using the log-rank test (n = 4 in each group; log-rank P = .0067). (C) Representative blood flow rate graphs. (D-F) Laser-induced injury of the cremaster muscle. Jak2Plt+/+ and Jak2Plt−/− mice were injected with anti-GPIbβ (red) and anti-fibrin (green) antibodies, and the cremaster arteries were interrogated with a 3i Ablate! laser during fluorescence, real-time, intravital video microscopy. (D) Representative still images at 0 to 3 minutes. Arrows represent blood flow direction. Bars represent 25 µm. Platelet (E) and fibrin (F) accumulation at the site of injury was measured by fluorescence intensity. Data are the mean ± standard error of the mean at each time point, compared by using the unpaired Student t test (n = 20 vessels in 4 Jak2Plt+/+ mice and 28 vessels in 5 Jak2Plt−/− mice; platelet and fibrin; P < .0001).
To determine whether the prolonged tail bleeding time of Jak2Plt−/− mice was related to defective thrombus formation, we next performed a series of in vivo injury models examining thrombosis. We assessed occlusion in Jak2Plt−/− mice under high shear flow, by using the FeCl3-induced carotid artery injury model in which the blood flow rate is measured until time to occlusion (Figure 2B-C). In agreement with the observed bleeding diathesis, Jak2Plt−/− mice failed to show occlusion up to the experimental end point of 30 minutes, whereas occlusion occurred in Jak2Plt+/+ littermate controls at a median time of 6.53 minutes (n = 4 in each group; log-rank P = .0067; Figure 2B). The pattern of flow during the FeCl3 injury in Figure 2C suggested that the initial formation of thrombi within the artery appeared to dissipate repeatedly, illustrating instability in the developing thrombi, thereby resulting in occlusion failure.
To investigate the bleeding phenotype in closer detail, we performed a cremaster vessel laser-induced injury model and documented acute thrombus development over 3 minutes (Figure 2D-F; supplemental Videos 1 and 2). The laser-induced injury model enabled us to follow the simultaneous evaluation of platelet accumulation and fibrin deposition in real time. Similar to the FeCl3-induced arterial thrombosis model, platelet accumulation and fibrin deposition were severely decreased at the site of injury in Jak2Plt−/− mice, compared with Jak2Plt+/+ littermate controls (n = 20 vessels in 4 Jak2Plt+/+ mice and 28 vessels in 5 Jak2Plt−/− mice; P < .0001 each; Figure 2E-F). Furthermore, Jak2Plt−/− mice showed repeated loss of clot aggregation compared with Jak2Plt+/+ mice (supplemental Videos 1 and 2). Our data demonstrated that JAK2 expression in platelets plays an essential role in platelet thrombus formation in vivo.
Reduced thrombus formation of Jak2Plt−/− platelets at arterial shear rates
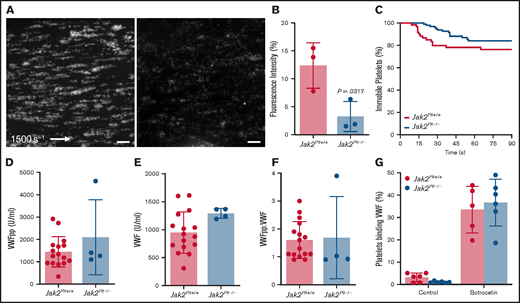
After blood vessel injury and disruption of the vascular endothelium, platelet exposure to basement membrane proteins and soluble agonists initiates platelet adhesion and activation, leading to formation of thrombi. At arterial shear rates, initial platelet adhesion and subsequent stable activation are mediated by collagen-bound VWF binding to the platelet GPIb-IX complex, followed by platelet activation via the collagen receptor GPVI.28,29 Using a VenaFlux microfluidics platform,22 we examined the functionality of fluorescence-labeled Jak2Plt−/− platelets in whole blood in adhering to type 1 collagen and forming thrombi under arterial shear rates (1500 s−1; Figure 3A-B). After 3 minutes, the mean fluorescence intensity measured for Jak2Plt−/− platelets was 3.25% ± 2.69% (mean ± SD; n = 3), compared with 12.38 ± 4.07% in Jak2Plt+/+ controls (n = 3; P = .0317). We analyzed the dwell time of individual platelets under the same experimental conditions (Figure 3C). Jak2Plt+/+ (n = 60) and Jak2Plt−/− (n = 97) platelets dwelled for similar times (log-rank P = .141), suggesting that thrombus formation, rather than initial platelet adhesion, was defective in Jak2Plt−/− mice.
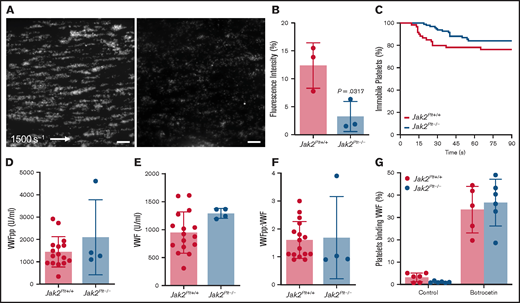
Reduced thrombus formation of Jak2Plt−/− platelets at arterial shear rates. PPACK-anticoagulated whole blood from Jak2Plt+/+ and Jak2Plt−/− mice was labeled and perfused on a type 1 collagen–immobilized surface at an arterial shear rate of 1500 s−1. (A) Representative still images at 3 minutes. Bars represent 100 µm. (B) Fluorescence intensity at 3 minutes. Results represent mean ± SD and were compared by using the unpaired Student t test (n = 3 in each group; *P = .0317). (C) Dwell time of individual Jak2Plt+/+ and Jak2Plt−/− platelets. Results were estimated by the Kaplan-Meier method and were compared by using the log-rank test (n = 60 Jak2Plt+/+ and 97 Jak2Plt−/− platelets; log-rank P = .141). Plasma VWFpp (D) and VWF (E) levels and VWFpp/VWF ratio (F) in Jak2Plt+/+ and Jak2Plt−/− mice. Results are the mean ± SD and were compared by using the unpaired Student t test (n = 16 Jak2Plt+/+ and 4 Jak2Plt−/− mice; VWFpp, P = .223; VWF, P = .0868; VWFpp/VWF, P = .865). (G) Jak2Plt+/+ and Jak2Plt−/− platelets were activated for 5 minutes at 37°C with 4 µg/mL botrocetin, incubated with FITC-labeled anti-mouse VWF antibody, and analyzed by flow cytometry. Results are the mean ± SD and are compared by 2-way ANOVA (n = 6 in each group; control, P = .96; botrocetin, P = .89).
Reduced thrombus formation of Jak2Plt−/− platelets at arterial shear rates. PPACK-anticoagulated whole blood from Jak2Plt+/+ and Jak2Plt−/− mice was labeled and perfused on a type 1 collagen–immobilized surface at an arterial shear rate of 1500 s−1. (A) Representative still images at 3 minutes. Bars represent 100 µm. (B) Fluorescence intensity at 3 minutes. Results represent mean ± SD and were compared by using the unpaired Student t test (n = 3 in each group; *P = .0317). (C) Dwell time of individual Jak2Plt+/+ and Jak2Plt−/− platelets. Results were estimated by the Kaplan-Meier method and were compared by using the log-rank test (n = 60 Jak2Plt+/+ and 97 Jak2Plt−/− platelets; log-rank P = .141). Plasma VWFpp (D) and VWF (E) levels and VWFpp/VWF ratio (F) in Jak2Plt+/+ and Jak2Plt−/− mice. Results are the mean ± SD and were compared by using the unpaired Student t test (n = 16 Jak2Plt+/+ and 4 Jak2Plt−/− mice; VWFpp, P = .223; VWF, P = .0868; VWFpp/VWF, P = .865). (G) Jak2Plt+/+ and Jak2Plt−/− platelets were activated for 5 minutes at 37°C with 4 µg/mL botrocetin, incubated with FITC-labeled anti-mouse VWF antibody, and analyzed by flow cytometry. Results are the mean ± SD and are compared by 2-way ANOVA (n = 6 in each group; control, P = .96; botrocetin, P = .89).
Normal VWF levels in Jak2Plt−/− plasma
Platelet adhesion at arterial shear rates is dependent on GPIbα-VWF interaction, and hemorrhage in patients with MPN-related thrombocytosis has been associated with increased interaction between platelets and circulating and hemostatically active .VWF.8,30 Therefore, plasma VWF levels and GPIbα-VWF interactions were assessed to determine their role in the in vivo and in vitro defects in thrombus formation of Jak2Plt−/− mice. Compared with Jak2Plt+/+ controls, Jak2Plt−/− mice had normal levels of synthesized VWF propeptide (VWFpp), circulating plasma VWF, and VWF turnover rate (VWFpp/VWF; Figure 3D-F).
To measure platelet GPIbα-VWF interactions, we further performed a flow cytometry–based VWF binding assay after treatment with botrocetin (Figure 3G). The data did not reveal any difference in VWF binding between Jak2Plt−/− and Jak2Plt+/+ control platelets. Together, the data indicate that the bleeding diathesis of Jak2Plt−/− mice and failure to form occlusive arterial thrombi was not related to decreased plasma VWF levels or to altered VWF binding to the platelet GPIb-IX complex.
Decreased spreading of Jak2Plt−/− platelets
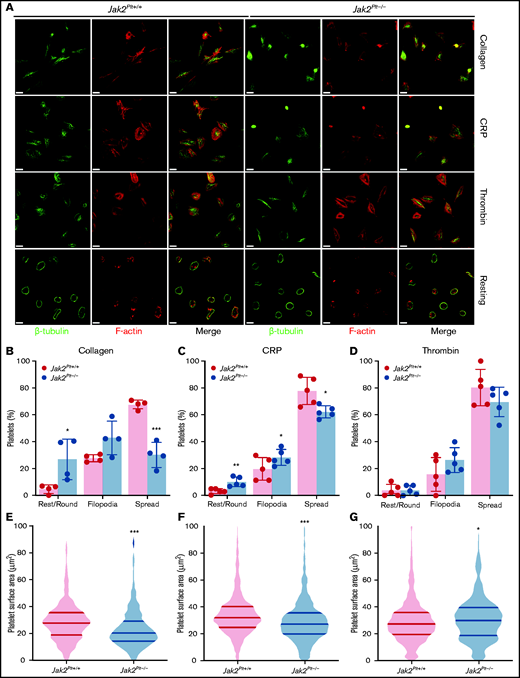
After activation, platelets rapidly change shape from disc-like entities to morphologically distinct forms by rounding, then extending finger-like filopodia and spreading thin sheetlike lamellipodia.31 To identify whether the reduced thrombus formation of Jak2Plt−/− mice was due to defects in platelet shape change, washed platelets were seeded onto glass coverslips coated with 50 µg/mL immobilized type 1 collagen, or were activated with 1 µg/mL of the GPVI agonist CRP or 0.01 U/mL of thrombin and seeded onto 100 µg/mL immobilized fibrinogen (Figure 4A). Unstimulated Jak2Plt−/− platelets maintained a normal discoid shape on fibrinogen, as indicated by the presence of their microtubule coil (Figure 4A). We quantified platelet morphology by immunofluorescence staining and superresolution microscopy analysis. On collagen, Jak2Plt−/− platelets spread fewer lamellipodia compared with the controls (n = 4 in each group; P = .0005), whereas filopodia formation was maintained (Figure 4B). Their surface area moderately decreased to 23.38 ± 14.66 µm2 (mean ± SD; n = 374), compared with 28.75 ± 15.12 µm2 in Jak2Plt+/+ platelets (n = 684; P < .001; Figure 4E). Jak2Plt−/− platelets activated with CRP on fibrinogen also spread less (n = 5 in each group; P = .0464; Figure 4C). Their surface area decreased to 29.68 ± 17.31 µm2 (mean ± SD; n = 1174), compared with 34.62 ± 17.13 µm2 in Jak2Plt+/+ platelets (n = 918; P < .001; Figure 4F). By contrast, Jak2Plt−/− platelets stimulated with thrombin spread normally on fibrinogen (Figure 4D). Their surface area increased slightly to 31.11 ± 17.29 µm2 (mean ± SD; n = 842), compared with 29.34 ± 16.15 µm2 in Jak2Plt+/+ platelets (n = 958; P = .03; Figure 4G). Together, the data suggested a defect downstream of the collagen receptor GPVI.
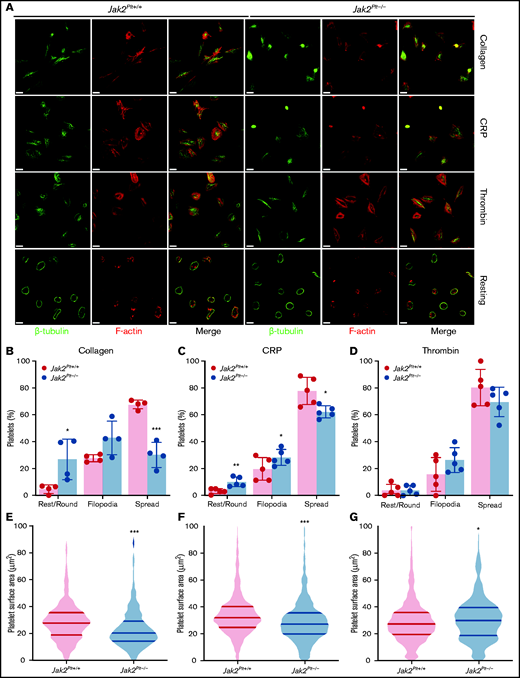
Spreading defects in Jak2Plt−/− platelets.Jak2Plt+/+ and Jak2Plt−/− platelets were left to adhere to 50 µg/mL type 1 collagen or to 100 µg/mL fibrinogen and activated with 1 µg/mL CRP or 0.01 U/mL thrombin glass coverslips for 30 minutes at 37°C as indicated. Fixed platelets were stained for β-tubulin and F-actin (phalloidin) and analyzed by structured illumination microscopy. (A) Representative immunofluorescent micrographs of fixed platelets. Bars represent 3 µm (activated) and 7 µm (resting). (B-D) Morphologic quantification of platelet spreading as determined by resting, extension of defined filopodia, or full spreading in response to collagen (total platelets scored, 549 Jak2Plt+/+ and 311 Jak2Plt−/− in 4 independent experiments) (B); CRP (total platelets scored, 888 Jak2Plt+/+ and 1017 Jak2Plt−/− in 5 independent experiments) (C); or thrombin (total platelets scored, 932 Jak2Plt+/+ and 796 Jak2Plt−/− in 5 independent experiments) (D). Results represent the mean ± SD and were compared by 2-way ANOVA. *P < .05; **P < .01; ***P < .001. (E-G) Violin plots of platelet surface area in response to collagen (total platelets scored, 684 Jak2Plt+/+ and 374 Jak2Plt−/− in 4 independent experiments) (E); CRP (total platelets scored, 918 Jak2Plt+/+ and 1174 Jak2Plt−/− in 5 independent experiments) (F); or thrombin (total platelets scored, 958 Jak2Plt+/+ and 842 Jak2Plt−/− in 5 independent experiments) (G). Results were compared by 2-way ANOVA. *P < .05; ***P < .001.
Spreading defects in Jak2Plt−/− platelets.Jak2Plt+/+ and Jak2Plt−/− platelets were left to adhere to 50 µg/mL type 1 collagen or to 100 µg/mL fibrinogen and activated with 1 µg/mL CRP or 0.01 U/mL thrombin glass coverslips for 30 minutes at 37°C as indicated. Fixed platelets were stained for β-tubulin and F-actin (phalloidin) and analyzed by structured illumination microscopy. (A) Representative immunofluorescent micrographs of fixed platelets. Bars represent 3 µm (activated) and 7 µm (resting). (B-D) Morphologic quantification of platelet spreading as determined by resting, extension of defined filopodia, or full spreading in response to collagen (total platelets scored, 549 Jak2Plt+/+ and 311 Jak2Plt−/− in 4 independent experiments) (B); CRP (total platelets scored, 888 Jak2Plt+/+ and 1017 Jak2Plt−/− in 5 independent experiments) (C); or thrombin (total platelets scored, 932 Jak2Plt+/+ and 796 Jak2Plt−/− in 5 independent experiments) (D). Results represent the mean ± SD and were compared by 2-way ANOVA. *P < .05; **P < .01; ***P < .001. (E-G) Violin plots of platelet surface area in response to collagen (total platelets scored, 684 Jak2Plt+/+ and 374 Jak2Plt−/− in 4 independent experiments) (E); CRP (total platelets scored, 918 Jak2Plt+/+ and 1174 Jak2Plt−/− in 5 independent experiments) (F); or thrombin (total platelets scored, 958 Jak2Plt+/+ and 842 Jak2Plt−/− in 5 independent experiments) (G). Results were compared by 2-way ANOVA. *P < .05; ***P < .001.
GPVI-specific functional defects in Jak2Plt−/− platelets
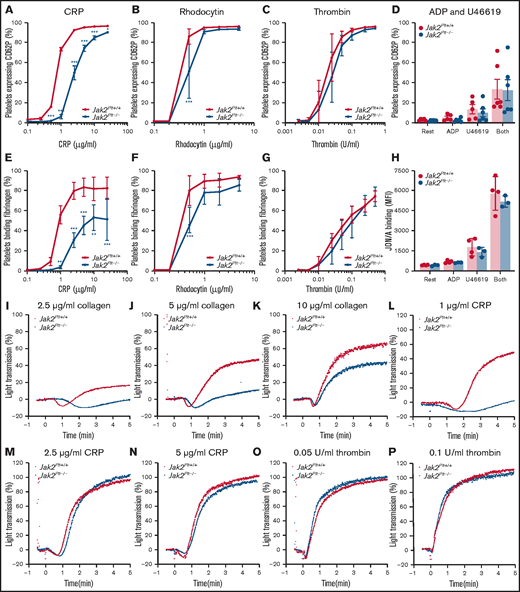
We evaluated the function of Jak2Plt−/− platelets in response to soluble agonists (Figure 5). We determined P-selectin (CD62P) expression, a marker of α-granule secretion, and fibrinogen binding, a marker of integrin αIIbβ3 activation, after platelet stimulation with CRP (Figure 5A,E), the CLEC-2 agonist rhodocytin (Figure 5B,F) or thrombin (Figure 5C,G) in a dose-dependent manner. As expected, >90% of control platelets expressed P-selectin (Figure 5A-C), and >75% bound fibrinogen (Figure 5E-G) after stimulation at the highest doses of CRP (25 μg/mL), rhodocytin (5 µg/mL), or thrombin (0.5 U/mL). P-selectin exposure and fibrinogen binding were delayed in Jak2Plt−/− platelets stimulated with CRP or rhodocytin. Although Jak2Plt−/− platelets reached normal P-selectin levels, there was a decrease in fibrinogen binding at maximum CRP concentrations. By contrast, we did not measure differences in tempo and maximum P-selectin expression and fibrinogen binding after thrombin stimulation (Figure 5C,G).

Functional defects of Jak2Plt−/− platelets downstream of GPVI and CLEC-2.Jak2Plt+/+ and Jak2Plt−/− platelets were activated for 2 minutes at 37°C with CRP (A,E), rhodocytin (B,F), thrombin (C,G), or ADP and U46619 (D,H); incubated with FITC-labeled anti-mouse CD62P antibody (A-D), Oregon Green 488-labeled fibrinogen (E-G), or anti-active αIIbβ3 antibody JON/A (H); and analyzed by flow cytometry. Results are expressed as a percentage of positive platelets (A-G) or mean fluorescence intensity (MFI) (H). Results represent mean ± SD of 5 (CRP), 4 (rhodocytin), 6 (thrombin), and 4 (ADP and U46619) independent experiments and were compared by 2-way ANOVA. *P < .05; **P < .01; ***P < .001. Aggregation of Jak2Plt+/+ and Jak2Plt−/− platelets was determined by light transmission under stirring conditions at 37°C in response to 2.5 (I), 5 (J), or 10 (K) µg/mL collagen; 1 (L), 2.5 (M), or 5 (N) µg/mL CRP; or 0.05 (O) or 0.1 (P) U/mL thrombin. Graphs are representative of 4 independent experiments.
Functional defects of Jak2Plt−/− platelets downstream of GPVI and CLEC-2.Jak2Plt+/+ and Jak2Plt−/− platelets were activated for 2 minutes at 37°C with CRP (A,E), rhodocytin (B,F), thrombin (C,G), or ADP and U46619 (D,H); incubated with FITC-labeled anti-mouse CD62P antibody (A-D), Oregon Green 488-labeled fibrinogen (E-G), or anti-active αIIbβ3 antibody JON/A (H); and analyzed by flow cytometry. Results are expressed as a percentage of positive platelets (A-G) or mean fluorescence intensity (MFI) (H). Results represent mean ± SD of 5 (CRP), 4 (rhodocytin), 6 (thrombin), and 4 (ADP and U46619) independent experiments and were compared by 2-way ANOVA. *P < .05; **P < .01; ***P < .001. Aggregation of Jak2Plt+/+ and Jak2Plt−/− platelets was determined by light transmission under stirring conditions at 37°C in response to 2.5 (I), 5 (J), or 10 (K) µg/mL collagen; 1 (L), 2.5 (M), or 5 (N) µg/mL CRP; or 0.05 (O) or 0.1 (P) U/mL thrombin. Graphs are representative of 4 independent experiments.
Jak2Plt+/+ and Jak2Plt−/− platelets were also treated with the G-protein–coupled receptor (GPCR) agonists (ie, ADP and U46619, a thromboxane A2 analogue), and analyzed by flow cytometry for P-selectin expression (Figure 5D) and JON/A binding (Figure 5H), a marker of integrin αIIbβ3 activation. ADP (10 µM) and U46619 (10 µM) induced comparable α-granule secretion and integrin αIIbβ3 activation in Jak2Plt+/+ and Jak2Plt−/− platelets, alone or combined.
Next, we assessed platelet aggregation by light transmission under stirring conditions. Jak2Plt−/− platelets aggregated poorly in response to collagen (Figure 5I-K) or a low dose (1 µg/mL) of CRP (Figure 5L) but responded normally to higher doses of CRP (Figure 5M-N) or intermediate doses of thrombin (Figure 5O-P). Together, the data indicate that Jak2Plt−/− platelets have impaired α-granule secretion, integrin αIIbβ3 activation, and aggregation when stimulated via the ITAM/hemITAM-containing receptors GPVI and CLEC-2, but not when activated with the GPCR agonists thrombin, ADP, and U46619.
Impaired GPVI signaling in Jak2Plt−/− platelets
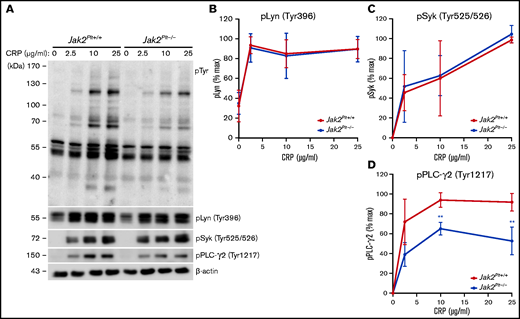
Collagen binding to platelet GPVI initiates an ITAM signaling cascade that involves the stepwise activation and phosphorylation of several of the signaling intermediates, beginning with the proximal Src family tyrosine kinases (SFKs) Fyn and Lyn.32 Activation of SFKs subsequently induces phosphorylation of other signaling intermediates, including the GPVI-associated FcRγ-chain, the tyrosine kinase Syk, and phospholipase C-γ2 (PLC-γ2). We wanted to determine whether the collagen- and CRP-related defects of Jak2Plt−/− platelets were due to impaired ITAM signaling downstream of GPVI. We activated platelets with increasing CRP concentrations and lysed and probed against phosphotyrosine (pTyr; Figure 6A). In control Jak2Plt+/+ platelets, CRP stimulation induced the tyrosine phosphorylation of several proteins, whereas Jak2Plt−/− platelets showed an attenuated response in protein tyrosine phosphorylation.
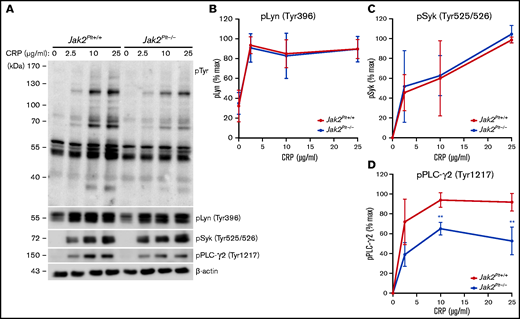
GPVI signaling defects in Jak2Plt−/− platelets. (A) Jak2Plt+/+ and Jak2Plt−/− platelets were activated or not with CRP for 2 minutes at 37°C, as indicated. Platelet lysates corresponding to 2 µg protein were subjected to SDS-PAGE and probed for phosphotyrosine (pTyr), phosphorylated Lyn Tyr 396 (pLyn Tyr396), phosphorylated Syk Tyr525/526 (pSyk Tyr525/526), phosphorylated PLC-γ2 Tyr1217 (pPLC-γ2 Tyr1217), and β-actin as a loading control. Quantification of pLyn Tyr396 (B), pSyk Tyr525/526 (C), and pPLCγ2 Tyr1217 (D) in CRP-stimulated Jak2Plt+/+ and Jak2Plt−/− platelets. Results are the mean ± SD of 4 independent experiments and were compared with the control by 2-way ANOVA. **P < .01.
GPVI signaling defects in Jak2Plt−/− platelets. (A) Jak2Plt+/+ and Jak2Plt−/− platelets were activated or not with CRP for 2 minutes at 37°C, as indicated. Platelet lysates corresponding to 2 µg protein were subjected to SDS-PAGE and probed for phosphotyrosine (pTyr), phosphorylated Lyn Tyr 396 (pLyn Tyr396), phosphorylated Syk Tyr525/526 (pSyk Tyr525/526), phosphorylated PLC-γ2 Tyr1217 (pPLC-γ2 Tyr1217), and β-actin as a loading control. Quantification of pLyn Tyr396 (B), pSyk Tyr525/526 (C), and pPLCγ2 Tyr1217 (D) in CRP-stimulated Jak2Plt+/+ and Jak2Plt−/− platelets. Results are the mean ± SD of 4 independent experiments and were compared with the control by 2-way ANOVA. **P < .01.
To further refine the signaling impairment, we probed CRP-stimulated platelet lysates for activating phosphorylated tyrosine residues on Lyn (Tyr396), Syk (Tyr525/526), and PLC-γ2 (Tyr1217) using phospho-specific antibodies (Figure 6A). Jak2Plt−/− platelets showed comparable phosphorylation of Lyn (Tyr396) (Figure 6B) and Syk (Tyr525/526) (Figure 6C) to control Jak2Plt+/+ platelets. Notably, levels of phosphorylated PLC-γ2 at its activating residue Tyr1217 were reduced by 30% at 10 μg/mL and by 40% at 25 μg/mL of CRP (Figure 6D). The data show that the absence of JAK2-mediated signals did not affect the early signaling intermediates Lyn and Syk downstream of GPVI, but affected other downstream signaling intermediates, leading to impaired PLC-γ2 phosphorylation and activation.
Discussion
The cellular mechanisms by which somatic mutations in the tyrosine kinase JAK2 increase the risk of developing arterial and venous thrombosis in patients with MPNs remain unclear. In this study, we examined the role of platelet JAK2 in primary hemostasis using Jak2Plt−/− mice lacking JAK2 in platelets and MKs and described a novel mechanism by which platelet JAK2 contributes to thrombosis in mice.
Jak2Plt−/− mice had severe MK hyperplasia and thrombocytosis.17 Previous studies have linked the endocytosis of the TPO receptor Mpl and regulation of plasma TPO by platelets to JAK2 expression and activity and Mpl and JAK2 protein levels to MK proliferation.33-35 Jak2Plt−/− mice have elevated plasma TPO levels caused by defective endocytosis of the cytokine by platelets and MKs.17 Mouse models lacking Mpl, the endocytic GTPase dynamin 2, or the ubiquitin ligase Cbl in platelets and MKs, where blunted TPO endocytosis results in hemapoietic stem and progenitor cell and MK hyperplasia related to increased TPO stimulation, support the role of Mpl-mediated endocytosis regulating TPO plasma levels.26,36,37
Thrombocytosis is typically associated with thrombosis and thrombocytopenia with bleeding. Thus, the most confounding observation was the severe tail-bleeding diathesis and hemostatic defects of Jak2Plt−/− mice. By 2 independent in vivo mouse models of thrombosis, we demonstrated that JAK2 expression in platelets contributes to primary hemostasis. Occlusion failed in Jak2Plt−/− mice in a FeCl3-induced arterial injury model, which was associated with development of unstable thrombus, and no stable thrombi were created in a cremaster laser–induced injury system, in which Jak2Plt−/− platelets did not successfully stabilize at the site of vascular injury. The genetic approach of deleting JAK2 in platelets is consistent with earlier reports of a pharmacological approach, where treatment with the specific JAK2 inhibitor AG490 also led to prolonged microvessel occlusion time in mice.38 Hemostatic defects have been observed in knockin mouse models expressing the hyperactive JAK2 V617F in platelets.15,16 Why loss of JAK2 function, related to genetic deletion or pharmacological inhibition, and expression of hyperactive JAK2 V617F in platelets may lead to similar in vivo and in vitro hemostatic defects remains unexplained.
Under arterial shear rates, the initial platelet adhesion to subendothelial collagen is mediated by collagen-induced VWF binding to the platelet GPIb-IX complex, and subsequent activation of platelet collagen receptor GPVI.28,29Jak2Plt−/− platelets had thrombus formation defects after normal adhesion to immobilized type 1 collagen under arterial shear rates. Jak2Plt−/− mice had normal plasma ppVWF and VWF plasma levels, and Jak2Plt−/− platelets expressed the GPIb-IX complex and the collagen receptor GPVI normally. Furthermore, plasma VWF binding to Jak2Plt−/− platelets in response to botrocetin was indistinguishable from controls. The data show that the interaction between plasma VWF and the platelet GPIb-IX complex was not disrupted by JAK2 deletion in platelets. By contrast, Jak2Plt−/− platelet spreading was impaired on immobilized collagen or fibrinogen after the addition of low doses of the collagen receptor GPVI agonist CRP, but not thrombin. Together, our data suggest that in vivo hemostatic defects and consequent bleeding of Jak2Plt−/− mice is not caused by defective VWF and GPIbα-VWF interactions, but is the result of defective platelet activation in response to collagen exposure, a critical initial step in thrombus development.
Supporting a role for JAK2 in intrinsic platelet GPVI signaling and function, we identified defects in the ability of Jak2Plt−/− platelets to secrete α-granule, activate integrin αIIbβ3, and aggregate in response to exposure collagen and CRP, but not in response to the GPCR agonists, thrombin, ADP, and the thromboxane A2 analogue U46619. The GPVI-specific impairments of Jak2Plt−/− platelets observed are consistent with previous observations showing that pharmacological JAK2 inhibition results in impaired platelet aggregation after stimulation by collagen, but not by thrombin.38 The contribution of ADP release from dense granules and thromboxane A2 formation in the responses of Jak2Plt−/− platelets to GPVI agonists was not evaluated in our study and cannot be ruled out, as ADP scavengers and aspirin affect, to various degrees, platelet responses to collagen, CRP, and convulxin.39-41 Elegant studies using an in vivo platelet depletion model have shown that newly formed young platelets display GPVI signaling defects.42 Although a minimal increase in immature platelet fraction was observed in Jak2Plt−/− mice, impaired GPVI signaling in this fraction alone is unlikely to be responsible for the hemostatic defects, as most Jak2Plt−/− platelets are mature and circulate normally.17
Additional support for the role of GPVI in the hemostatic defect of in Jak2Plt−/− platelets came from the reduced intracellular protein tyrosine phosphorylation in response to CRP. The absence of JAK2-mediated signals did not affect the early downstream GPVI signaling kinases Lyn and Syk but showed impaired phosphorylation of PLC-γ2. Thus, our data posit a role for JAK2 on unidentified signaling intermediates downstream of Syk and upstream of PLC-γ2. The phenotype of Jak2Plt−/− mice resembles that of mice lacking PLC-γ2,43 which is activated downstream of both GPVI and the hemITAM receptor CLEC-2. Because GPVI and the CLEC-2 share many signaling molecules, it is likely that JAK2 also affects CLEC-2 signaling. Consistent with the hypothesis, α-granule secretion, and integrin αIIbβ3 activation were mildly attenuated in Jak2Plt−/− platelets stimulated with the CLEC-2 agonist rhodocytin. Phosphoproteomics-based studies have identified JAK2 tyrosine phosphorylation downstream of both GPVI and CLEC-2.44,45 In addition, Mpl/JAK2 signaling may also contribute to the regulation of platelet hemostatic function in vivo, as TPO potentiates GPVI signaling through a phosphatidylinositol 3-kinase–dependent pathway in human platelets.46,47 Potentiation of platelet GPVI signaling by TPO is lost in platelets of JAK2 V617F patients.48 Mice expressing the activating JAK2 V617F mutation also develop platelet hemostatic defects, further supporting this notion.15,16 Thus, combined GPVI, CLEC-2, and Mpl signaling defects likely explain the bleeding diathesis of Jak2Plt−/− mice.
In conclusion, although our study identified a role for the tyrosine kinase JAK2 in platelet GPVI and CLEC-2 signaling that is necessary for successful in vivo hemostasis in mice, the contribution of other JAK2 signaling-dependent receptors should be investigated further.
Acknowledgments
The authors thank Kay-Uwe Wagner for providing the Jak2fl/fl mice, Johannes Eble for providing the rhodocytin, François Maignen for statistical expertise, and Karin Hoffmeister for helpful discussions.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute R01 grants HL136430 (S.L.H.), HL133348 (H.W.), and HL126743 (H.F.).
Authorship
Contribution: N.E. designed and performed experiments, collected, analyzed, and interpreted data, and wrote and revised the manuscript; S.S., M.L.S., C.D., and D.J. designed and performed the experiments; collected, analyzed, and interpreted the data; and revised the manuscript; S.L.H. and H.W. analyzed and interpreted data and revised the manuscript; and H.F. conceived and designed the study, designed and performed experiments, collected, analyzed, and interpreted the data and wrote and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for S.S. is Department of Medicine, Boston University School of Medicine, Boston, MA.
Correspondence: Hervé Falet, Versiti Blood Research Institute, 8733 W Watertown Plank Rd, Milwaukee, WI 53226; e-mail: hfalet@versiti.org.