

Key Points
This is the largest study on the genotype of patients with VWD3.
The VWD3 genotype was more heterogeneous in the European population than in the Iranian population.

Abstract
Type 3 von Willebrand disease (VWD3) is a rare and severe bleeding disorder characterized by often undetectable von Willebrand factor (VWF) plasma levels, a recessive inheritance pattern, and heterogeneous genotype. The objective of this study was to identify the VWF defects in 265 European and Iranian patients with VWD3 enrolled in 3WINTERS-IPS (Type 3 Von Willebrand International Registries Inhibitor Prospective Study). All analyses were performed in centralized laboratories. The VWF genotype was studied in 231 patients with available DNA (121 [115 families] from Europe [EU], and 110 [91 families] from Iran [IR]). Among 206 unrelated patients, 134 were homozygous (EU/IR = 57/77) and 50 were compound heterozygous (EU/IR = 43/7) for VWF variants. In 22 patients, no or only one variant was found. A total of 154 different VWF variants (EU/IR = 101/58 [5 shared]) were identified among the 379 affected alleles (EU/IR = 210/169), of which 48 (EU/IR = 18/30) were novel. The variants p.Arg1659*, p.Arg1853*, p.Arg2535*, p.Cys275Ser, and delEx1_Ex5 were found in both European and Iranian VWD3 patients. Sixty variants were identified only in a single allele (EU/IR = 50/10), whereas 18 were recurrent (≥3 patients) within 144 affected alleles. Nine large deletions and one large insertion were found. Although most variants predicted null alleles, 21% of patients carried at least 1 missense variant. VWD3 genotype was more heterogeneous in the European population than in the Iranian population, with nearly twice as many different variants. A higher number of novel variants were found in the Iranian VWD3 patients.
Introduction
von Willebrand factor (VWF) is a large plasma glycoprotein that plays a key role in primary hemostasis by mediating platelet interaction with the exposed collagen of the subendothelial matrix under high shear stress conditions.1 VWF also has a role in secondary hemostasis by acting as a carrier for coagulation factor VIII (FVIII), protecting it from premature cleavage and thus prolonging its half-life.2 A deficiency or dysfunction of VWF causes von Willebrand disease (VWD), a clinically heterogeneous hemorrhagic disorder.3 VWD is classified as type 1 and 3 in the presence of quantitative defects of VWF, whereas qualitative defects are classified as type 2 (2A, 2B, 2M, and 2N).4 VWD type 1 presents with mildly reduced plasma VWF levels, with a definitive diagnosis when levels are <30 IU/dL,5 whereas VWD type 3 (VWD3) presents with virtually undetectable VWF antigen (VWF:Ag) levels (<5 IU/dL) and markedly reduced FVIII. Patients with VWD3 have moderate to severe mucocutaneous bleeding symptoms but also muscle hematomas and hemarthrosis.6 VWD3 is inherited in an autosomal recessive pattern, patients being either homozygous for a gene variant or compound heterozygous for 2 different variants. VWD3 is a rare disorder (1 per million), but, as a recessive condition, its prevalence is higher in countries with a high rate of consanguinity.
VWD3 has been investigated in several studies, and >300 different gene variants have been reported.7-10 The majority of these studies involved small groups of patients, and the authors have either focused on clinical symptoms and biochemical phenotype or on the patients’ genotype.6,7 The 3WINTERS-IPS (Type 3 Von Willebrand International Registries Inhibitor Prospective Study) is an investigator-initiated, multicenter, European/Iranian observational study that aims to comprehensively evaluate the clinical symptoms of VWD3 patients,11 their phenotypic laboratory findings, and the VWF variants, as well as the safety and efficacy of replacement therapy for the treatment and prevention of bleeding manifestations (registered at www.clinicaltrials.gov as #NCT02460458).
We report here the spectrum of VWF variants in 2 large groups of VWD3 patients from Europe and the Islamic Republic of Iran (Iran). The objectives of the current study were to identify the VWF defects in the largest cohort of VWD3 patients investigated thus far and to compare the VWF variants between the European VWD3 (EU-VWD3) and the Iranian VWD3 (IR-VWD3) patients. Our findings highlight the distinct features regarding the heterogeneity and the localization of the VWF variants identified in these 2 populations.
Methods
Patients
A total of 265 patients previously diagnosed with VWD3 were enrolled in the 3WINTERS-IPS study from 9 European countries (Italy, Spain, France, Germany, The Netherlands, United Kingdom, Hungary, Finland, and Sweden) and Iran (Figure 1). Patients were enrolled if they had a history of bleeding, VWF levels <5 IU/dL, and an autosomal recessive pattern of inheritance.4 Written informed consent was obtained from patients in accordance with the Declaration of Helsinki. Of the 265 patients enrolled (146 from Europe and 119 from Iran), 231 were evaluated at the molecular level (121 European patients from 115 families and 110 Iranian patients from 91 families). The members of each of the 206 families investigated are reported in supplemental Tables 1 and 2. The remaining 34 patients were not investigated due to insufficient plasma and/or buffy coat being available. We confirm that the institutional review board submissions were performed and approved in all participating centers by their local institutional review boards.The study was conducted in accordance with the Declaration of Helsinki.
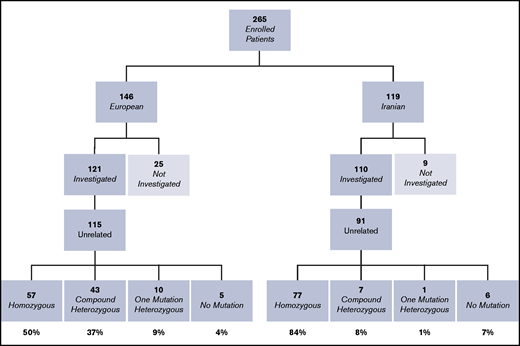
Flowchart and summary of the basic results of the molecular analysis of the cohort of VWD3 patients. Patients not investigated were excluded from the study because of insufficient plasma and/or buffy coat aliquots.
Flowchart and summary of the basic results of the molecular analysis of the cohort of VWD3 patients. Patients not investigated were excluded from the study because of insufficient plasma and/or buffy coat aliquots.
Clinical and laboratory characteristics of the patients investigated at the molecular level
| Total patients fully characterized | Total patients no or only partially characterized | P | EU patients fully characterized | EU patients no or only partially characterized | P | IR patients fully characterized | IR patients no or only partially characterized | P | |
|---|---|---|---|---|---|---|---|---|---|
| N | 207 | 24 | 105 | 16 | 102 | 8 | |||
| Sex | |||||||||
| Male | 87 (42%) | 8 (33%) | 37 (35.2%) | 6 (37.5%) | 50 (49%) | 2 (25%) | |||
| Female | 120 (58%) | 16 (67%) | 68 (64.8%) | 10 (62.5%) | 52 (51%) | 6 (75%) | |||
| Age at study inclusion, y | 28 (16-43) | 38 (20-51) | .08 | 40 (25-49) | 42 (33-54) | .40 | 22 (8-30) | 21 (14-34) | .39 |
| Bleeding score | 15 (8-21) | 11 (7-18) | .30 | 18 (14-25) | 15 (11-19) | .07 | 10 (4-16) | 8 (5-10) | .42 |
| N* | 157 | 20 | 69 | 12 | 88 | 8 | |||
| VWF:Ag* | 0.5 (0.5-0.5) | 2.2 (0.8-3.9) | <.001 | 0.5 (0.5-1.3) | 3.9 (0.8-4.6) | .003 | 0.5 (0.5-0.5) | 1.8 (1.1-2.3) | <.001 |
| FVIII:C* | 2.4 (1.9-3.2) | 2.7 (1.8-8.4) | .29 | 2.7 (2.2-3.7) | 5.5 (2.5-16.9) | .07 | 2.2 (1.7-2.6) | 1.8 (1.6-2.3) | .42 |
| Total patients fully characterized | Total patients no or only partially characterized | P | EU patients fully characterized | EU patients no or only partially characterized | P | IR patients fully characterized | IR patients no or only partially characterized | P | |
|---|---|---|---|---|---|---|---|---|---|
| N | 207 | 24 | 105 | 16 | 102 | 8 | |||
| Sex | |||||||||
| Male | 87 (42%) | 8 (33%) | 37 (35.2%) | 6 (37.5%) | 50 (49%) | 2 (25%) | |||
| Female | 120 (58%) | 16 (67%) | 68 (64.8%) | 10 (62.5%) | 52 (51%) | 6 (75%) | |||
| Age at study inclusion, y | 28 (16-43) | 38 (20-51) | .08 | 40 (25-49) | 42 (33-54) | .40 | 22 (8-30) | 21 (14-34) | .39 |
| Bleeding score | 15 (8-21) | 11 (7-18) | .30 | 18 (14-25) | 15 (11-19) | .07 | 10 (4-16) | 8 (5-10) | .42 |
| N* | 157 | 20 | 69 | 12 | 88 | 8 | |||
| VWF:Ag* | 0.5 (0.5-0.5) | 2.2 (0.8-3.9) | <.001 | 0.5 (0.5-1.3) | 3.9 (0.8-4.6) | .003 | 0.5 (0.5-0.5) | 1.8 (1.1-2.3) | <.001 |
| FVIII:C* | 2.4 (1.9-3.2) | 2.7 (1.8-8.4) | .29 | 2.7 (2.2-3.7) | 5.5 (2.5-16.9) | .07 | 2.2 (1.7-2.6) | 1.8 (1.6-2.3) | .42 |
For quantitative variables, median and interquartile range are reported; descriptive variables are reported as numbers with percentages. Mann-Whitney U test was used to evaluate the group comparisons.
Includes only the patients without prophylaxis.
Candidate missense mutations identified in the VWD3 3WINTERS-IPS cohort
| EU/IR | VWF Gene Defect | Domain (exon) | SIFT | Poly Phen-2 | Mutation Taster | CADD PHRED score | ClinGen | dbSNP(re) –HGMD (CM) | First reported | In vitro study |
|---|---|---|---|---|---|---|---|---|---|---|
| IR | NM_000552.3: c.220G>A; p.Gly74Arg | D1 (3) | T | D | D | 33 | VUS | – | ||
| EU | NM_000552.3:c.449T>C; p.Leu150Pro | D1 (5) | D | D | D | 28 | LP† | rs61753994 | 63 * | |
| IR | NM_000552.3:c.498C>A; p.Asn166Lys | D1 (5) | D | D | D | 22.7 | VUS | – | ||
| EU/IR | NM_000552.3:c.823T>A; p.Cys275Ser | D1 (7) | D | D | D | 28.1 | LP | rs61753998 | 31 | 65 |
| IR | NM_000552.3:c.1574G>A; p.Gly525Glu | D2 (14) | D | D | D | 26.6 | VUS | – | ||
| EU | NM_000552.3:c.2311A>G; p.Met771Val | D' (18) | D | P | D | 23 | LP | rs1212894308 - CM061227 | 44 | |
| IR | NM_000552.3:c.2376C>G; p.Cys792Trp | D' (18) | D | D | D | 26.4 | LP | – | ||
| IR | NM_000552.3:c.2560C>T; p.Arg854Trp | D' (20) | D | D | D | 31 | LP | rs61748482 | 64 | 64 |
| EU | NM_000552.3:c.2771G>A; p.Arg924Gln | D3 (21) | T | B | N | 14.2 | LB† | rs33978901 | 47 | 53,57 |
| EU | NM_000552.3:c.3467C>T; p.Thr1156Met | D3 (26) | D | D | D | 26.2 | LP† | rs267607328 | 65 | 66 |
| EU | NM_000552.3:c.3568T>C; p.Cys1190Arg | D3 (27) | D | D | D | 27 | LP† | rs61749364 | 50 | 67 |
| EU | NM_000552.3:c.3679T>C; p.Cys1227Arg | D3 (28) | D | D | D | 28.7 | LP | rs61749366 | 68 | 67 |
| IR | NM_000552.3:c.4007G>A; p.Arg1336Gln‡ | A1 (28) | T | B | N | 21.8 | VUS† | rs886049741 | ||
| EU | NM_000552.3:c.5096C>T; p.Ser1699Phe | A3 (29) | D | D | D | 24.4 | VUS | – | 46 | |
| EU | NM_000552.3:c.5146G>A; p.Ala1716Thr | A3 (29) | T | B | D | 20.8 | VUS | CM115145 rs61750623 | ||
| IR | NM_000552.3:c.5429C>A; p.Ala1810Glu | A3 (31) | T | B | N | 7.8 | VUS | – | ||
| EU | NM_000552.3:c.5779T>C; p.Cys1927Arg | D4 (34) | D | D | D | 26.8 | VUS | CM930738 | 69 * | |
| IR | NM_000552.3:c.6520T>G; p.Cys2174Gly | D4 (37) | D | D | D | 27.1 | VUS | rs61750619 | 31 | |
| EU | NM_000552.3:c.6551G>C; p.Cys2184Ser | D4 (37) | D | D | D | 25.8 | LP | CM125355 | 36 * | 36 |
| EU | NM_000552.3:c.6634T>C; p.Cys2212Arg | D4 (38) | D | D | D | 29 | LP | CM125353 | 36 * | 36 |
| EU | NM_000552.3:c.6697G>A; p.Glu2233Lys | D4 (38) | D | D | D | 27.9 | VUS | rs61750623 | 46 | |
| EU | NM_000552.3:c.6847T>C; p.Cys2283Arg | C1 (39) | D | D | D | 28 | LP | CM137380 | 70 * | 70 |
| EU | NM_000552.3:c.6911G>A; p.Cys2304Tyr | C1 (40) | D | D | D | 24.7 | LP | rs61750626 | 49 | 71 |
| EU | NM_000552.3:c.6973T>A; p.Cys2325Ser | C1 (40) | D | D | D | 25.8 | LP | rs125608270 CM125358 | 36 * | 36 |
| EU | NM_000552.3:c.7085G>T; p.Cys2362Phe | C2 (42) | D | D | A | 27.6 | P† | rs61750630 | 72 * | 73 |
| EU | NM_000552.3:c.7182T>G; p.Cys2394Trp§ | C2 (42) | D | D | D | 21.9 | VUS | CM095418 | 32 * | |
| EU | NM_000552.3:c.7405T>C; p.Ser2469Pro | C3 (43) | T | P | N | 22.7 | VUS | rs61751287 | 49 * | |
| IR | NM_000552.3:c.7619T>A; p.Val2540Asp | C4 (45) | D | D | D | 27.9 | VUS | – | ||
| EU | NM_000552.3:c.7636A>T; p.Asn2546Tyr | C4 (45) | D | D | D | 32 | VUS | rs61751298 | 74 * | |
| EU | NM_000552.3:c.8012G>A; p.Cys2671Tyr§ | C6 (49) | D | D | D | 26.3 | LP | rs61751303 | 72 * | 75 |
| EU | NM_000552.3:c.8171G>A; p.Cys2724Tyr | CK (51) | D | D | D | 24.9 | VUS | – | ||
| EU | NM_000552.3:c.8216G>A; p.Cys2739Tyr | CK (51) | D | D | D | 26.6 | LP | rs61751305 | 76 | 75 |
| IR | NM_000552.3:c.8323T>C; p.Ser2775Pro | CK (52) | D | P | D | 23.1 | VUS | – |
| EU/IR | VWF Gene Defect | Domain (exon) | SIFT | Poly Phen-2 | Mutation Taster | CADD PHRED score | ClinGen | dbSNP(re) –HGMD (CM) | First reported | In vitro study |
|---|---|---|---|---|---|---|---|---|---|---|
| IR | NM_000552.3: c.220G>A; p.Gly74Arg | D1 (3) | T | D | D | 33 | VUS | – | ||
| EU | NM_000552.3:c.449T>C; p.Leu150Pro | D1 (5) | D | D | D | 28 | LP† | rs61753994 | 63 * | |
| IR | NM_000552.3:c.498C>A; p.Asn166Lys | D1 (5) | D | D | D | 22.7 | VUS | – | ||
| EU/IR | NM_000552.3:c.823T>A; p.Cys275Ser | D1 (7) | D | D | D | 28.1 | LP | rs61753998 | 31 | 65 |
| IR | NM_000552.3:c.1574G>A; p.Gly525Glu | D2 (14) | D | D | D | 26.6 | VUS | – | ||
| EU | NM_000552.3:c.2311A>G; p.Met771Val | D' (18) | D | P | D | 23 | LP | rs1212894308 - CM061227 | 44 | |
| IR | NM_000552.3:c.2376C>G; p.Cys792Trp | D' (18) | D | D | D | 26.4 | LP | – | ||
| IR | NM_000552.3:c.2560C>T; p.Arg854Trp | D' (20) | D | D | D | 31 | LP | rs61748482 | 64 | 64 |
| EU | NM_000552.3:c.2771G>A; p.Arg924Gln | D3 (21) | T | B | N | 14.2 | LB† | rs33978901 | 47 | 53,57 |
| EU | NM_000552.3:c.3467C>T; p.Thr1156Met | D3 (26) | D | D | D | 26.2 | LP† | rs267607328 | 65 | 66 |
| EU | NM_000552.3:c.3568T>C; p.Cys1190Arg | D3 (27) | D | D | D | 27 | LP† | rs61749364 | 50 | 67 |
| EU | NM_000552.3:c.3679T>C; p.Cys1227Arg | D3 (28) | D | D | D | 28.7 | LP | rs61749366 | 68 | 67 |
| IR | NM_000552.3:c.4007G>A; p.Arg1336Gln‡ | A1 (28) | T | B | N | 21.8 | VUS† | rs886049741 | ||
| EU | NM_000552.3:c.5096C>T; p.Ser1699Phe | A3 (29) | D | D | D | 24.4 | VUS | – | 46 | |
| EU | NM_000552.3:c.5146G>A; p.Ala1716Thr | A3 (29) | T | B | D | 20.8 | VUS | CM115145 rs61750623 | ||
| IR | NM_000552.3:c.5429C>A; p.Ala1810Glu | A3 (31) | T | B | N | 7.8 | VUS | – | ||
| EU | NM_000552.3:c.5779T>C; p.Cys1927Arg | D4 (34) | D | D | D | 26.8 | VUS | CM930738 | 69 * | |
| IR | NM_000552.3:c.6520T>G; p.Cys2174Gly | D4 (37) | D | D | D | 27.1 | VUS | rs61750619 | 31 | |
| EU | NM_000552.3:c.6551G>C; p.Cys2184Ser | D4 (37) | D | D | D | 25.8 | LP | CM125355 | 36 * | 36 |
| EU | NM_000552.3:c.6634T>C; p.Cys2212Arg | D4 (38) | D | D | D | 29 | LP | CM125353 | 36 * | 36 |
| EU | NM_000552.3:c.6697G>A; p.Glu2233Lys | D4 (38) | D | D | D | 27.9 | VUS | rs61750623 | 46 | |
| EU | NM_000552.3:c.6847T>C; p.Cys2283Arg | C1 (39) | D | D | D | 28 | LP | CM137380 | 70 * | 70 |
| EU | NM_000552.3:c.6911G>A; p.Cys2304Tyr | C1 (40) | D | D | D | 24.7 | LP | rs61750626 | 49 | 71 |
| EU | NM_000552.3:c.6973T>A; p.Cys2325Ser | C1 (40) | D | D | D | 25.8 | LP | rs125608270 CM125358 | 36 * | 36 |
| EU | NM_000552.3:c.7085G>T; p.Cys2362Phe | C2 (42) | D | D | A | 27.6 | P† | rs61750630 | 72 * | 73 |
| EU | NM_000552.3:c.7182T>G; p.Cys2394Trp§ | C2 (42) | D | D | D | 21.9 | VUS | CM095418 | 32 * | |
| EU | NM_000552.3:c.7405T>C; p.Ser2469Pro | C3 (43) | T | P | N | 22.7 | VUS | rs61751287 | 49 * | |
| IR | NM_000552.3:c.7619T>A; p.Val2540Asp | C4 (45) | D | D | D | 27.9 | VUS | – | ||
| EU | NM_000552.3:c.7636A>T; p.Asn2546Tyr | C4 (45) | D | D | D | 32 | VUS | rs61751298 | 74 * | |
| EU | NM_000552.3:c.8012G>A; p.Cys2671Tyr§ | C6 (49) | D | D | D | 26.3 | LP | rs61751303 | 72 * | 75 |
| EU | NM_000552.3:c.8171G>A; p.Cys2724Tyr | CK (51) | D | D | D | 24.9 | VUS | – | ||
| EU | NM_000552.3:c.8216G>A; p.Cys2739Tyr | CK (51) | D | D | D | 26.6 | LP | rs61751305 | 76 | 75 |
| IR | NM_000552.3:c.8323T>C; p.Ser2775Pro | CK (52) | D | P | D | 23.1 | VUS | – |
Variants reported in bold are not listed in the EAHAD,17 HGMD,18 or Ensembl19 databases. Variants reported in italics have been identified in both European and Iranian populations. Underlined variants might affect normal splice site.
SIFT (Sorting Intolerant From Tolerant): T = tolerated; D = deleterious. PolyPhen-2: D = probably damaging; P = possibly damaging; B = benign. MutationTaster: D = disease causing; N = polymorphism; A = disease-causing automatic. ClinGen: P = pathogenic; LP = likely pathogenic; LB = likely benign; VUS = uncertain significance. In Combined Annotation-Dependent Depletion (CADD), a cutoff of 20 was used so that variants with score ≥20 were considered harmful.21
Indicates that the previous publication was concerning the same patient evaluated in this study.
Prediction already reported in ClinVar.
The pathogenicity of p.Arg1336Gln is unclear as it was found in the same allele together with c.2397_2400dupCATG, which implies that in this patient the p.Arg1336Gln does not play a role.
The second variants identified in this patient do not match the variant previously reported in the original publication of the patient.
Phenotypic evaluation
Assessment of the patients’ bleeding score (BS) was performed in each center by using a validated questionnaire.12 Patients’ plasma VWF:Ag level was measured by using a sensitive enzyme-linked assay.11 Factor VIII coagulant activities (FVIII:C) were evaluated with a one-stage clotting assay using TriniCLOT aPTT reagent (Tcoag Ireland Ltd., Wicklow, Ireland) and coagulation FVIII-deficient plasma (Siemens Healthcare GmbH, Erlangen, Germany).
Molecular analysis
The genetic evaluation was performed in 3 nationally and internationally recognized European laboratories under the technical supervision of the study authors (A.G. and I.P., Sheffield, United Kingdom; R.S., Hamburg, Germany; and L.B. and F.P., Milan, Italy). EU-VWD3 samples were assessed in Sheffield and Hamburg, and those from IR-VWD3 were all tested in Milan. Genomic DNA was isolated from the buffy coat aliquots collected at the time of patient enrollment using the following: QIAsymphony DNA Kit (QIAGEN Lake Constance GmbH, Stockach, Germany) in Sheffield; QIAamp DNA Blood Mini Kit (QIAGEN Lake Constance GmbH) in Hamburg; and a salting out method in Milan.13
Sequence analysis.
The molecular analysis was performed at the 3 centers using next-generation sequencing (NGS)14 and/or Sanger sequencing to evaluate the VWF coding regions, including intron–exon boundaries and a portion of the 5' and 3' untranslated (UTR) coding regions (up to: 5'-UTR c.-650 and 3'-UTR c.-150). In Sheffield and Milan, genetic evaluation was initially performed by using NGS analysis. At Sheffield, the custom oligonucleotide baits were designed by using Agilent Technologies SureDesign (https://earray.chem.agilent.com/suredesign/index.htm). The enriched libraries were prepared by using a SureSelectXT reagent kit (Agilent Technologies Inc., Palo Alto, CA), and the pooled library underwent paired-end sequencing on the HiSeq 2500 system (Illumina, Inc., San Diego, CA). In Milan, the custom panel was designed by using Illumina Design Studio on Human (hg19) reference genome (https://emea.support.illumina.com/sequencing/sequencing_software/designstudio.html). The VWF was amplified by using TruSeq Custom Amplicon strategy (Illumina, Inc.), and the paired-end sequencing was performed on a MiSeq sequencing platform (Illumina, Inc.). NGS data were analyzed according to the guidelines reported by the Broad Institute (https://software.broadinstitute.org/gatk/best-practices/), and the variant call format files were annotated by ANNOVAR.15 Further details of the NGS analysis are reported in the supplemental Materials. All variants identified in Sheffield and Milan were confirmed by Sanger sequencing using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Warrington, UK) on a 3730 and 3130 DNA Analyzer (Applied Biosystems), respectively. In Hamburg, genetic analysis was performed by using direct Sanger sequencing. Exons 2 to 52, including intron–exon boundaries, were amplified by polymerase chain reaction (PCR), and PCR products were analyzed on an ABI Prism 3130 Genetic Analyzers (Applied Biosystems) using the genomic kit ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction (Applied Biosystems). The variants identified were confirmed on a second PCR amplicon using direct sequencing. PCR conditions and oligonucleotides used for Sanger sequencing analysis in all 3 centers are available on demand. Nucleotide numbering is reported as per the Human Genome Variation Society nomenclature (http://varnomen.hgvs.org/). The full Human Genome Variation Society nomenclature is used in the tables and supplemental tables but for brevity not in text and figures. All websites cited here were accessed January 2021.
Multiplex ligation-dependent probe amplification analysis.
Copy number variations within the VWF were detected by multiplex ligation-dependent probe amplification (MLPA) using VWF-specific kits (SALSA MLPA Probemix P011/P012 VWF, MRC Holland; http://www.mlpa.com/, last accessed January 2021) to confirm large homozygous gene deletions identified by PCR amplification or further investigate patients in whom no variants were found by using NGS or Sanger sequencing.16 Reactions were set up as described by MRC Holland (MLPA General Protocol), and fragment size analysis was performed by using either the 3730 DNA Analyzer (Sheffield) or the 3130 Genetic Analyzer (Hamburg and Milan) (Applied Biosystems).
Screening of VWF variants.
The variants identified were prioritized on the basis of allelic frequency and type of mutation. Missense, splicing, nonsense, insertion, duplication, and deletion variants with minor allele frequency <0.01 were considered as candidate pathogenic variants. Any detected VWF sequence variation was checked to determine whether they had been earlier recorded in online databases. They were sought in the Coagulation Factor Variant Databases portal supported by the European Association for Haemophilia and Allied Disorders (EAHAD) 17 (https://grenada.lumc.nl/LOVD2/VWF/home.php?select_db=VWF), the Human Gene Mutation Database (HGMD) 18 (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=VWF), and the Ensembl database19 (www.ensembl.org), all accessed January 2021.
In silico analysis.
The impact of the missense changes on VWF protein structure and function was evaluated by using the following tools: Combined Annotation-Dependent Depletion PHRED score20 (applying a threshold of 20),21 Sorting Intolerant From Tolerant22 (https://sift.bii.a-star.edu.sg/), Polymorphism Phenotyping v2,23 PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster24 (http://www.mutationtaster.org/StartQueryEngine.html), all accessed November 2020. Pathogenicity of potential splice site variants was evaluated by using splice site prediction tools such as varSEAK (https://varseak.bio/index.php), Alternative Splice Site Predictor25 (http://wangcomputing.com/assp/index.html), Berkeley drosophila genome project26 (https://www.fruitfly.org/seq_tools/splice.html), and NatGen227 (https://services.healthtech.dtu.dk/service.php?NetGene2-2.42), all accessed January 2021. Furthermore, variants classification was performed following the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology28 by ClinGen Pathogenicity Calculator.29
Statistical methods
Medians and interquartile range are used to report quantitative values, and descriptive variables are reported as numbers with percentages. All patients investigated in this study were divided into 2 groups: those fully characterized at the molecular level (ie, variants identified in both alleles) vs those with no variant or only partially characterized (ie, variants identified in only 1 allele). This comparison was conducted for the whole cohort of VWD3 and for the EU-VWD3 and the IR-VWD3. Patients’ age (at study inclusion), BS, VWF:Ag, and FVIII:C of the different groups were analyzed by using the Mann-Whitney U test. The χ2 test was used to evaluate the different proportion of the various unique variants found in the two VWD3 populations.
Results
Patients
The median age of the fully characterized EU-VWD3 patients was almost twice the median age of the IR-VWD3 patients, and the BS in the first group was almost twice the score of the second group (Table 1). The percentage of the two sexes was nearly equal in the IR-VWD3, whereas almost two-thirds of the patients were female in the EU-VWD3. In both populations, the median VWF:Ag values of the fully characterized patients were statistically significantly lower than those of the patients with no mutations or only partially characterized.
Number of VWF variants in the 3WINTERS-IPS cohort
A total of 231 patients (Europe/Iran [EU/IR] = 121/110), previously diagnosed with VWD3, were investigated at the molecular level. The variants identified in these 231 patients are reported in supplemental Tables 1 and 2; the data reported hereafter represent only the unrelated patients from 206 families (EU/IR = 115/91). The majority (134 [EU/IR = 57/77]) were homozygous for the variants identified, but no variant was identified in five EU-VWD3 patients and in six IR-VWD3 patients (Figure 1). A total of 154 different unique variants were identified in the 2 populations, 101 in European patients and 58 in Iranian patients (Figure 2). Only 5 variants were found in both populations. Three of these are among the 11 “hotspot” arginine codons present in the VWF,30 p.Arg1659* (EU/IR = 9/8), p.Arg1853* (EU/IR = 2/3), and p.Arg2535* (EU/IR = 2/2). One is a missense variant, p.Cys275Ser31 (EU/IR = 1/2), and one is a large deletion, delEx1_Ex532 (EU/IR = 1/2). Figure 3 shows the allele distributions among the different unique variants found in the 2 populations. For comparison, the alleles (EU/IR = 210/169) of the different unique variants identified in each population (EU/IR = 101/58) were divided into 3 groups: a first group of different variants found only in a single patient, either in the heterozygous or homozygous state (1 or 2 alleles); a second group of different variants found in 2 or 3 patients (up to 4 alleles), and a third group of different variants found in ≥3 patients (5 or more alleles). The first group included 50.5% of the alleles in the EU-VWD3 (78 patients) vs 45% of the alleles in the IR-VWD3 (43 patients). The second group in the EU-VWD3 is characterized by 15 different variants that account for 18.5% of the alleles, whereas in the IR-VWD3, four different variants represent 8.3% of the alleles. The third group consists of the most common variants found in the 2 populations (8 in EU-VWD3 and 10 in the IR-VWD3) and accounts for 31% and 46.7%, respectively, of the alleles identified in each VWD3 population. The distribution of the different type of variants in the 2 populations is shown in Figure 4. Variant data have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/; SUB9482148).
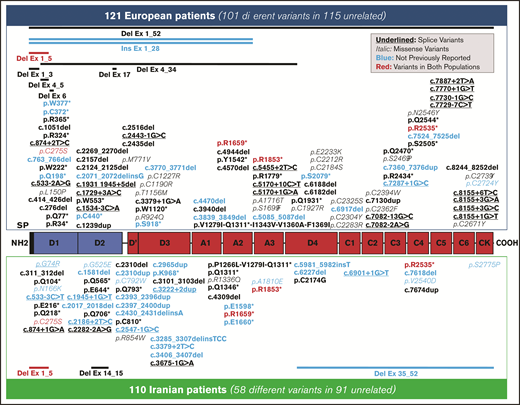
Distribution of the 154 different unique variants identified in the 3WINTERS-IPS cohort. A schematic representation of the pro-VWF polypeptide with the homologous repeated domains structure. Variants identified in the European population are reported at the top; those found in the Iranian population are reported at the bottom. The candidate missense mutations are reported in italics, and the potential splice site mutations are underlined. The 5 variants identified in both populations are reported in red. The variants not previously reported in the EAHAD, HGMD, or Ensembl databases are reported in blue.
Distribution of the 154 different unique variants identified in the 3WINTERS-IPS cohort. A schematic representation of the pro-VWF polypeptide with the homologous repeated domains structure. Variants identified in the European population are reported at the top; those found in the Iranian population are reported at the bottom. The candidate missense mutations are reported in italics, and the potential splice site mutations are underlined. The 5 variants identified in both populations are reported in red. The variants not previously reported in the EAHAD, HGMD, or Ensembl databases are reported in blue.

Distribution of alleles among the 154 different unique variants found in the 3WINTERS-IPS cohort. (A) European population. (B) Iranian population. Group 1 reports the alleles of different variants identified only in 1 patient, in either the heterozygous or homozygous state. Group 2 reports the alleles of different variants found in 2 or 3 patients (2 up to 4 alleles). Group 3 reports the alleles of the variants identified in ≥3 patients (>4 alleles). The variants identified in both populations are reported in red. The potential splice site mutations are underlined. The variants not previously reported in the EAHAD, HGMD, or Ensembl databases are reported in blue. The p.Arg1336Gln has been counted together with c.2397_2400dupCATG because these 2 variants are on the same allele.
Distribution of alleles among the 154 different unique variants found in the 3WINTERS-IPS cohort. (A) European population. (B) Iranian population. Group 1 reports the alleles of different variants identified only in 1 patient, in either the heterozygous or homozygous state. Group 2 reports the alleles of different variants found in 2 or 3 patients (2 up to 4 alleles). Group 3 reports the alleles of the variants identified in ≥3 patients (>4 alleles). The variants identified in both populations are reported in red. The potential splice site mutations are underlined. The variants not previously reported in the EAHAD, HGMD, or Ensembl databases are reported in blue. The p.Arg1336Gln has been counted together with c.2397_2400dupCATG because these 2 variants are on the same allele.
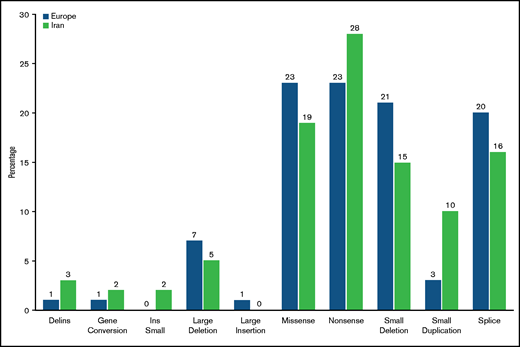
Comparison of the distribution of the different type of variants identified. The different types of variants identified were reported in percentages of the different unique variants identified in each population: 101 European patients and 58 Iranian patients.
Comparison of the distribution of the different type of variants identified. The different types of variants identified were reported in percentages of the different unique variants identified in each population: 101 European patients and 58 Iranian patients.
Type of VWF variants in the 3WINTERS-IPS cohort
Nonsense variants.
Thirty-six different nonsense variants were identified, 23 in the EU-VWD3 and 16 in the IR-VWD3 population. Eight of the 11 “hotspot” arginine codons were found in EU-VWD3 patients and 3 of them were also found in IR-VWD3 patients. Of the 36 different nonsense variants, 9 (EU/IR = 6/3) were not reported in the EAHAD, HGMD, or Ensembl databases. An additional nonsense variant, p.Gln1311*, was found in 2 patients (EU/IR = 1/1) in the homozygous state as a consequence of 2 different gene conversions (supplemental Tables 1 and 2).
Missense variants.
Table 2 reports the 33 different missense variants identified in this study (EU/IR = 23/11). Eight of them (EU/IR = 1/7) were not reported in the EAHAD, HGMD, or Ensembl databases, whereas the p.Cys275Ser was found in both populations. The pathogenicity of p.Arg1336Gln is unclear, as it was found, in 2 related patients, in the same allele together with the c.2397_2400dupCATG (supplemental Table 2), which implies that, in these patients, the p.Arg1336Gln does not play a role. The classification of these variants following the recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines is reported in Table 2. Only one variant (p.Arg924Gln) was classified as “likely benign,” whereas almost one-half of the variants were classified as “uncertain significance.”28
In most of the missense variants, all the prediction bioinformatics tools agreed in suggesting their possible deleterious role (Table 2). Conversely, the p.Arg924Gln and p.Ala1810Glu were recognized as not harmful according to all prediction tools. Two variants (p.Ala1716Thr and p.Ser2469Pro) were evaluated as harmful only by 2 tools. The variant p.Gly74Arg is due to the c.220G>A at the last nucleotide of exon 3 and might affect the normal splicing process, as predicted by the splicing prediction tools (Table 3). Sixteen of the missense variants identified involve the loss of cysteine residues, and 9 of these (EU/IR = 8/1) cluster in the D4-C2 domains. Twenty-one percent of patients (41 of 195) carry missense variants, in which at least 1 variant was identified (in 3 of these 41 patients, only 1 variant allele was found). Of the 379 mutated alleles, 65 are missense (17.2%). In EU-VWD3, the percentage of patients carrying missense variants is 26.4% (29 of 110 patients), compared with 14.1% in IR-VWD3 (12 of 85 patients). The percentage of the missense alleles in EU-VWD3 is 20.0% (42 alleles of 210); in IR-VWD3, it is 13.6% (23 alleles of 169).
Potential splice site variants identified in the VWD3 3WINTERS-IPS cohort
| EU/IR | VWF Gene Defect | Domain (IVS) | varSEAK | ASSP score (WT; MUT) threshold 4.5 (donor)/2.2 (acceptor) | BDGP score (WT; MUT) threshold 0.40 | NatGen2 (WT; MUT) | ClinGen | dbSNP(rs)– HGMD (CS) | First reported | mRNA study |
|---|---|---|---|---|---|---|---|---|---|---|
| IR | NM_000552.3: c.220G>A (p.Gly74Arg) | D1(3) | Splicing effect | 13.65; <4.5 | 0.96; 0.52 | 0.99; 0.70 | VUS | – | ||
| IR | NM_000552.3: c.533-3C>G | D1 (5) | Splicing effect | 9.38; 4.95 | 0.90; <0.40 | 0.71; 0.43 | VUS | – | ||
| EU | NM_000552.3: c.533-2A>G | D1 (5) | – | – | – | – | LP | rs267607301 | 77 | 33 p.Thr179Profs*30 |
| EU | NM_000552.3: c.874 + 2T>C | D1 (7) | Splicing effect | 9.85; <4.5 | 0.80; <0.40 | 0.99; not detected | VUS | CS1310896 | 78 * | |
| IR | NM_000552.3: c.874+ 1G>A | D1 (7) | Splicing effect | 9.85; <4.5 | 0.80; <0.40 | 0.99; not detected | VUS | rs267607302 | 79 | |
| EU | NM_000552.3: c.1534-3C>A | D2 (13) | – | – | – | – | LP | rs61754009 | 72 | 34 p.Leu512Profs*11 |
| EU | NM_000552.3: c.1729 + 3A>C | D2 (14) | Likely splicing effect | 6.60; <4.5 | 0.17; <0.01 | 0.70; not detected | VUS | CS095425 | 32 * | |
| EU | NM_000552.3: c.1931_1945 + 5del | D2(15) | Splicing effect | 11.34; <4.5 | 0.48; <0.40 | 0.65; not detected | LP | CD942301 | 76 | |
| IR | NM_000552.3: c.1945 + 1G>T | D2 (15) | Splicing effect | 11.34; <4.5 | 0.48; <0.40 | 0.36; not detected | VUS | – | ||
| IR | NM_000552.3: c.2186 + 2T>C | D2 (16) | Splicing effect | 13.30; <4.5 | 1.00; <0.40 | 1.00; 0.70 | VUS | – | ||
| IR | NM_000552.3: c.2282-2A>G | D' (17) | Splicing effect | 3.58; <2.2 | 0.32; < 0.01 | 0.24; not detected | VUS | CS143311 | 55 | |
| EU | NM_000552.3: c.2443-1G>C | D' (18) | – | – | – | – | LP† | rs61748480 | 43 | 35 p.Val815Cysfs*15 |
| IR | NM_000552.3: c.2547-1G>C | D' (19) | Splicing effect | n.a. | 0.05; <0.01 | 0.55; not detected | VUS | . | ||
| IR | NM_000552.3: c.3222 + 2dupT | D3 (24) | Splicing effect | n.a. | 0.97; <0.40 | 0.95; not detected | VUS | |||
| IR | NM_000552.3: c.3379 + 2T>C | D3 (25) | Splicing effect | 10.05; <4.5 | 0.90; <0.40 | 0.95; not detected | VUS | . | ||
| EU | NM_000552.3: c.3379 + 1G>A | D3 (25) | Splicing effect | 10.05;<4.5 | 0.90; <0.40 | 0.95; not detected | P† | rs2363337 | 81 | |
| IR | NM_000552.3: c.3675-1G>A | D3 (27) | Splicing effect | 9.27; <2.2 | 0.97; <0.40 | 0.95; not detected | LP† | rs746457842 | 46 | |
| EU | NM_000552.3: c.5170 + 1G>A | A3 (29) | Splicing effect | 12.33; <4.5 | 0.84; <0.40 | 0.83; not detected | LP | rs764543553 | ||
| EU | NM_000552.3: c.5170 + 10C>T | A3 (29) | – | – | – | – | LB† VUS† | rs61750601 | 31,42 | 33 (No splicing alteration) |
| EU | NM_000552.3: c.5455 + 2T>C | A3 (31) | Splicing effect | 12.02; <4.5 | 0.89; <0.40 | 0.71; 0.06 | VUS | CS1310562 | 59 | |
| IR | NM_000552.3: c.6901 + 1G>T | C1 (39) | Splicing effect | 13.93; <4.5 | 0.98; <0.40 | 0.99; not detected | VUS | – | ||
| EU | NM_000552.3: c.7082-2A>G | C2 (41) | – | – | – | – | LP | rs267607358 | 63 * | 33 (Nonsense mediated decay) |
| EU | NM_000552.3: c.7082-13G>C | C2 (41) | No splicing effect | 7.48; 8.79 | 0.72; 0.77 | 0.00; 0.00 | VUS† | rs71581025 | ||
| EU | NM_000552.3: c.7287 + 1G>C | C2 (42) | Splicing effect | 11.26; <4.5 | 1.00; <0.40 | 0.93; not detected | VUS | – | ||
| EU | NM_000552.3: c.7729 + 7C>T | C4 (45) | – | – | – | – | P | rs61751301 | 42 * | 36 p.Glu2577Glyfs*21 |
| EU | NM_000552.3: c.7730-1G>C | C4 (45) | – | – | – | – | LP | rs267607366 | 63 | 33 p.Glu2577Alafs*58 |
| EU | NM_000552.3: c.7770 + 1G>T | C4 (46) | – | – | – | – | LP | – | 37 * | 37 (Three alternative splicing) |
| EU | NM_000552.3: c.7887 + 2T>A | C4 (47) | Splicing effect | 13.80; <4.5 | 0.99; <0.40 | 0.93; not detected | VUS | rs113814258 | ||
| EU | NM_000552.3: c.8155 + 6T>C | C6 (50) | – | – | – | – | LP | rs1223422347 - CS100200 | 38 * | 38 p.Gly2706Valfs*25 |
| EU | NM_000552.3: c.8155 + 3G>C | C6 (50) | – | – | – | – | LP | rs61751304 | 63 * | 33 p.Gly2706Valfs*25 |
| EU | NM_000552.3: c.8155 + 3G>A | C6 (50) | No splicing effect | 11.67; 12.35 | 0.94; 0.99 | 0.60; 0.63 | VUS | CS1211912 | 81 * | |
| EU | NM_000552.3: c.8155 + 1G>T | C6 (50) | – | – | – | – | LP | rs746457842 | 36 | 36 p.G2706_C2719delfs*25 |
| EU/IR | VWF Gene Defect | Domain (IVS) | varSEAK | ASSP score (WT; MUT) threshold 4.5 (donor)/2.2 (acceptor) | BDGP score (WT; MUT) threshold 0.40 | NatGen2 (WT; MUT) | ClinGen | dbSNP(rs)– HGMD (CS) | First reported | mRNA study |
|---|---|---|---|---|---|---|---|---|---|---|
| IR | NM_000552.3: c.220G>A (p.Gly74Arg) | D1(3) | Splicing effect | 13.65; <4.5 | 0.96; 0.52 | 0.99; 0.70 | VUS | – | ||
| IR | NM_000552.3: c.533-3C>G | D1 (5) | Splicing effect | 9.38; 4.95 | 0.90; <0.40 | 0.71; 0.43 | VUS | – | ||
| EU | NM_000552.3: c.533-2A>G | D1 (5) | – | – | – | – | LP | rs267607301 | 77 | 33 p.Thr179Profs*30 |
| EU | NM_000552.3: c.874 + 2T>C | D1 (7) | Splicing effect | 9.85; <4.5 | 0.80; <0.40 | 0.99; not detected | VUS | CS1310896 | 78 * | |
| IR | NM_000552.3: c.874+ 1G>A | D1 (7) | Splicing effect | 9.85; <4.5 | 0.80; <0.40 | 0.99; not detected | VUS | rs267607302 | 79 | |
| EU | NM_000552.3: c.1534-3C>A | D2 (13) | – | – | – | – | LP | rs61754009 | 72 | 34 p.Leu512Profs*11 |
| EU | NM_000552.3: c.1729 + 3A>C | D2 (14) | Likely splicing effect | 6.60; <4.5 | 0.17; <0.01 | 0.70; not detected | VUS | CS095425 | 32 * | |
| EU | NM_000552.3: c.1931_1945 + 5del | D2(15) | Splicing effect | 11.34; <4.5 | 0.48; <0.40 | 0.65; not detected | LP | CD942301 | 76 | |
| IR | NM_000552.3: c.1945 + 1G>T | D2 (15) | Splicing effect | 11.34; <4.5 | 0.48; <0.40 | 0.36; not detected | VUS | – | ||
| IR | NM_000552.3: c.2186 + 2T>C | D2 (16) | Splicing effect | 13.30; <4.5 | 1.00; <0.40 | 1.00; 0.70 | VUS | – | ||
| IR | NM_000552.3: c.2282-2A>G | D' (17) | Splicing effect | 3.58; <2.2 | 0.32; < 0.01 | 0.24; not detected | VUS | CS143311 | 55 | |
| EU | NM_000552.3: c.2443-1G>C | D' (18) | – | – | – | – | LP† | rs61748480 | 43 | 35 p.Val815Cysfs*15 |
| IR | NM_000552.3: c.2547-1G>C | D' (19) | Splicing effect | n.a. | 0.05; <0.01 | 0.55; not detected | VUS | . | ||
| IR | NM_000552.3: c.3222 + 2dupT | D3 (24) | Splicing effect | n.a. | 0.97; <0.40 | 0.95; not detected | VUS | |||
| IR | NM_000552.3: c.3379 + 2T>C | D3 (25) | Splicing effect | 10.05; <4.5 | 0.90; <0.40 | 0.95; not detected | VUS | . | ||
| EU | NM_000552.3: c.3379 + 1G>A | D3 (25) | Splicing effect | 10.05;<4.5 | 0.90; <0.40 | 0.95; not detected | P† | rs2363337 | 81 | |
| IR | NM_000552.3: c.3675-1G>A | D3 (27) | Splicing effect | 9.27; <2.2 | 0.97; <0.40 | 0.95; not detected | LP† | rs746457842 | 46 | |
| EU | NM_000552.3: c.5170 + 1G>A | A3 (29) | Splicing effect | 12.33; <4.5 | 0.84; <0.40 | 0.83; not detected | LP | rs764543553 | ||
| EU | NM_000552.3: c.5170 + 10C>T | A3 (29) | – | – | – | – | LB† VUS† | rs61750601 | 31,42 | 33 (No splicing alteration) |
| EU | NM_000552.3: c.5455 + 2T>C | A3 (31) | Splicing effect | 12.02; <4.5 | 0.89; <0.40 | 0.71; 0.06 | VUS | CS1310562 | 59 | |
| IR | NM_000552.3: c.6901 + 1G>T | C1 (39) | Splicing effect | 13.93; <4.5 | 0.98; <0.40 | 0.99; not detected | VUS | – | ||
| EU | NM_000552.3: c.7082-2A>G | C2 (41) | – | – | – | – | LP | rs267607358 | 63 * | 33 (Nonsense mediated decay) |
| EU | NM_000552.3: c.7082-13G>C | C2 (41) | No splicing effect | 7.48; 8.79 | 0.72; 0.77 | 0.00; 0.00 | VUS† | rs71581025 | ||
| EU | NM_000552.3: c.7287 + 1G>C | C2 (42) | Splicing effect | 11.26; <4.5 | 1.00; <0.40 | 0.93; not detected | VUS | – | ||
| EU | NM_000552.3: c.7729 + 7C>T | C4 (45) | – | – | – | – | P | rs61751301 | 42 * | 36 p.Glu2577Glyfs*21 |
| EU | NM_000552.3: c.7730-1G>C | C4 (45) | – | – | – | – | LP | rs267607366 | 63 | 33 p.Glu2577Alafs*58 |
| EU | NM_000552.3: c.7770 + 1G>T | C4 (46) | – | – | – | – | LP | – | 37 * | 37 (Three alternative splicing) |
| EU | NM_000552.3: c.7887 + 2T>A | C4 (47) | Splicing effect | 13.80; <4.5 | 0.99; <0.40 | 0.93; not detected | VUS | rs113814258 | ||
| EU | NM_000552.3: c.8155 + 6T>C | C6 (50) | – | – | – | – | LP | rs1223422347 - CS100200 | 38 * | 38 p.Gly2706Valfs*25 |
| EU | NM_000552.3: c.8155 + 3G>C | C6 (50) | – | – | – | – | LP | rs61751304 | 63 * | 33 p.Gly2706Valfs*25 |
| EU | NM_000552.3: c.8155 + 3G>A | C6 (50) | No splicing effect | 11.67; 12.35 | 0.94; 0.99 | 0.60; 0.63 | VUS | CS1211912 | 81 * | |
| EU | NM_000552.3: c.8155 + 1G>T | C6 (50) | – | – | – | – | LP | rs746457842 | 36 | 36 p.G2706_C2719delfs*25 |
Variants reported in bold are not listed in the EAHAD,17 HGMD,18 or Ensembl19 databases. Splice prediction tools: varSEAK, Alternative Splice Site Predictor (ASSP),25 Berkeley drosophila genome project (BDGP),26 NatGen2.27 ClinGen: P = Pathogenic; LP = Likely pathogenic; LB = likely benign; VUS = uncertain significance.
IVS, intervening sequence; n.a., native splice site not identified by the prediction tool.
Indicates that the previous publication was concerning the same patient evaluated in this study.
Prediction already reported in ClinVar.
Splicing variants.
Table 3 reports the 29 splice site variants identified in this study (EU/IR = 20/9). Seven of these (EU/IR = 1/6) were not reported in the EAHAD, HGMD, or Ensembl databases. Eleven splicing variants have been previously evaluated at the messenger RNA (mRNA) levels,33-38 and the rest of them were tested by using 4 splicing prediction tools. Most variants were predicted to alter the normal splicing process with at least 3 tools. However, in the cases of c.7082-13G>C and c.8155 + 3G>A, all 4 tools predicted no splicing alteration.
Deletions, insertions, and duplications.
Thirty-one different small deletions were identified (EU/IR = 22/9); 12 of them (EU/IR = 7/5) were novel in the EAHAD, HGMD, or Ensembl databases. All the small deletions cause frameshifts predicting premature termination codons, with the exception of 4. Three of these, c.3101_3103del (p.Thr1034del), c.5085_5087delCCT (p.Leu1696del) and c.8244_8252del (p.His2748_Cys2750del), result in small in-frame deletions (1 up to 3 amino acids). The fourth deletion, located at the last portion of exon 16 and the first 5 nucleotides of intron 16 (c.1931_1945 + 5del), obviously affects the normal splicing process (Table 3).
Nine distinct small duplications were identified (EU/IR = 3/6); six of them (EU/IR = 1/5) were not reported in the EAHAD, HGMD or Ensembl databases. All the small duplications cause frameshifts predicting premature termination codons, with the exception of c.3222 + 2dupT located in intron 24, which might affect the normal splicing process, as suggested by 3 splicing prediction tools (Table 3).
Nine different large deletions were identified (EU/IR = 7/3), with delEx1_Ex532 found in both populations. All the large deletions were found in a single patient, with the exception of delEx1_Ex5 found in 2 patients, delEx1_Ex3 in 3 patients (all of Hungarian descent)36,39,40 and delEx1_Ex5241 in 4 patients. The delEx35_Ex52 was novel according to the EAHAD and HGMD databases. Only 3 distinct del-ins variants (c.2071_2072delCCinsG, c.2430_2431delCCinsA, and c.3285_3307delinsTCC) were identified (EU/IR = 1/2), and all of them were novel. Only 2 insertions were identified: a small one (c.5981_5982insT) in a case of IR-VWD3 and a large one (insEx1_Ex28) in a case of EU-VWD3 (both were novel).
Discussion
In this study, the genetic background of VWD3 was very heterogeneous, with 154 different unique VWF variants among 379 defective alleles, which is a very high rate considering the recessive nature of VWD3 and thus the inclusion of many homozygous patients. The main findings are that the VWF genotypes were completely different in EU-VWD3 and IR-VWD3. Indeed, among the 154 different variants identified, only 5 were shared by both populations, whereas all other variants were unique for each population. Based on these data, no specific genetic hotspots for mutation seem to play a major role in VWD3 pathogenesis, or at least not in the IR-VWD3, where, to date, only 4 of the 11 “hotspot” arginine codons mutations have been reported.42,43 Furthermore, the heterogeneity of the gene defects was more pronounced among EU-VWD3 (101 different variants among 210 alleles, 48.1%) than among IR-VWD3 (58 different variants among 169 alleles, 34.3%; P = .0094), reflecting the higher rate of consanguinity and thus of homozygosity in the IR-VWD3. Despite the large heterogeneity between the 2 populations, the spectrum of variant types is rather similar (Figure 4).
Pathogenicity of the variants
In the majority of patients (79%), VWD3 is caused by null alleles or putative null alleles, as expected for a quantitative VWF defect and in accordance with other VWD3 cohorts.7 However, in 21% of the patients, missense mutations were found, and their VWD3 pathogenicity is not self-evident. The large majority of the previously reported missense variants were found in patients with VWD3 or type 1 VWD (Table 2). However, in a few cases, some of these variants were associated with qualitative VWF defects, such as type 2N/1 (p.Met771Val),44 2N (p.Arg854Trp),45 and 2M with collagen-binding defects (2MCB) (p.Ser1699Phe)46 or even with a variant of questionable significance (p.Arg924Gln).47-53 However, some of these variants also affect VWF levels. Indeed, p.Met771Val and p.Arg854Trp have also been reported in VWD3 patients from India, confirming their pathogenic role also in this study.54,55 The role of p.Ser1699Phe in VWD3 is less certain. It was reported in a type 2MCB patient, but no VWF plasma level was mentioned.46 Other type 2MCB patients have VWF:Ag levels of ∼30 IU/dL56; if this is also the case for p.Ser1699Phe, its finding along with the p.Arg2535* in our patient, might explain the VWD3 phenotype. The role of p.Arg924Gln is the most controversial.47-53 However, expression studies of p.Arg924Gln showed no alteration of VWF synthesis and secretion.53,57 Thus, its finding in our patient is unlikely to justify the phenotype. About one-half of the previously reported missense variants were found in patients assessed in this study. Hence, their previous finding in VWD3 does not confirm their pathogenic role. In addition, not all of them have been evaluated by expression studies. However, in most missense variants, all bioinformatics tools agreed in predicting their possible deleterious role (Table 2). This was not the case of p.Arg924Gln and p.Arg1810Glu, both predicted to be benign by all tools and therefore unlikely to cause the patients’ phenotype. The p.Ala1716Thr and p.Ser2469Pro were predicted to be deleterious by only 2 tools. Expression studies of these 2 variants should be done to confirm their role in VWD3.
One-half of the 33 different missense variants identified involve the loss of cysteine residues (Table 2). The important role that this amino acid plays in the posttranslational modification of pro-VWF58 supports the deleterious nature of these variants. A higher percentage of EU-VWD3 (26.4%) was found to carry missense variants compared with the IR-VWD3 (14.1%). However, the median VWF:Ag values of the fully characterized patients were the same in both EU-VWD3 and IR-VWD3 (Table 1). The higher percentage of missense variants in EU-VWD3 vs those of IR-VWD3 does not correspond to a lower median BS value in this population. The BS of the EU-VWD3 was higher than the BS of IR-VWD3, probably because the median age of EU-VWD3 was almost twice the median age of IR-VWD3.
The pattern of variants observed in this study, with 17% missense variant alleles contrasting with null alleles, are concordant with other VWD3 studies,6,8,36,46,55,59,-61 reporting that roughly 80% of mutations were null alleles.
All splice site variants evaluated at the mRNA level (Table 3) affect normal splicing, with the exception of c.5170 + 10C>T, which plays no role in VWD3.33 Most of the remaining variants, using prediction splicing tools, were considered able to alter the normal splicing process. However, in the cases of c.7082-13G>C and c.8155 + 3G>A, all 4 tools predicted their benign role; therefore, these variants also are likely to play no role in VWD3.
Recurrent variants
Of the 101 different variants found in the EU-VWD3 patients, only 23 were identified in more than one patient (groups 2 and 3 of Figure 3A). Of these, 12 were found in patients from more than one country: 10 in two countries, one (p.Arg324*) in three countries (Germany, Spain, and United Kingdom) and one (c.2435delC) in 5 countries (Finland, Sweden, Hungary, Germany, and Italy). Indeed, the c.2435delC in exon 1862 was the most common variant in Northern Europe.
The 10 most common IR-VWD3 variants represented 46.7% of the alleles found in patients enrolled at all 7 Iranian centers. Only 3 variants were found in patients from >2 centers (c.311_312delAG, c.2547-1G>C, and c.7618delG), whereas the remaining variants were mostly found in patients from single centers. Also, some of the “hotspot” variants30 were recurrent but mainly in EU-VWD3 (Figure 2).
Variant distribution along the VWF
In the EU-VWD3, the variants were scattered throughout the pro-VWF coding region (Figure 2), whereas in the Iranian population, they tended to concentrate at the NH2 terminal of the pro-VWF. In particularly, 18 of the 58 different variants identified are located in the D′-D3 domains. This uneven variant distribution suggests that these defects have been positively selected, probably due to consanguinity, in a relatively closed population. A similar distribution, although to a lesser extent, was described in the context of Indian8 and French46 VWD3 studies. The patients’ geographic origin probably plays a relevant role in this regard, as suggested by the more even distribution of EU-VWD3 variants that have been found in patients from 9 different countries.
VWD3 patients without variants
No mutations were identified in five EU-VWD3 and six IR-VWD3. Moreover, in ten EU-VWD3 and one IR-VWD3, only one mutated allele was found. Alleles without identified variants were reported from all 3 genetic centers (11 from Sheffield, 10 from Hamburg, and 13 from Milan), refuting any methodologic bias. There are several possible reasons for the missing variants. Mutations outside the sequences analyzed in the study, including deep intronic sequence regions, are a possibility; large gene conversions with the VWF pseudogene might also play a role, and further studies are therefore suggested. It is important to recognize that these VWD3 patients exist and that further VWD3 genotypes are possible.
In conclusion, the fruitful collaboration among many European and Iranian investigators participating in the 3WINTERS-IPS project has allowed collection of detailed information about VWF defects in the largest cohort of VWD3 patients available thus far, and it has helped to elucidate several differences between the EU-VWD3 and IR-VWD3. There was a remarkable diversity in the degree of heterogeneity of these VWF variants. In the IR-VWD3, the number of different unique variants was basically one-half the number of those identified in EU-VWD3. With only 5 exceptions, no variants were shared by the 2 populations, underlying their different genetic background. Furthermore, in the IR-VWD3, a cluster of variants was in the D′-D3 domains, whereas the EU-VWD3 variants were spread throughout the VWF. Notwithstanding these discrepancies, the spectrum of the variants identified in the two VWD3 populations was similar.
Acknowledgments
The authors thank the Members and current Chair of the Sub-Committee on VWF of the Scientific Standardization Committee of the International Society on Thrombosis and Haemostasis who promoted the endorsement of the 3WINTERS-IPS project among the scientific activities of this Sub-Committee. The authors also thank Javier Battle, Erik Berntorp, Cosimo Ettorre, Charles R.M. Hay, Massimo Morfini, Maria Gabriella Mazzucconi, Johannes Oldenburg, Rafael Parra Lòpez, and Omidreza Zekavat for patients’ enrollment and collection of clinical data. The 3WINTERS-IPS project received unconditional research grants from several pharmaceutical companies, and the authors therefore acknowledge the representatives of Baxter-Shire-Takeda, Grifols, CSL Behring, LFB, and Octapharma. The authors thank Paolo De Simoni, Luca Maravigna, and Elisabetta Musazzi of the CRO Sintesi Research for the study coordination and L.F. Ghilardini for the illustration work.
This work was partially supported by the Hungarian National Research Development and Innovation Office (NFKI) grant OTKA-K19_131945 (I.B.). This work was also partially supported by the Italian Ministry of Health–Bando Ricerca Corrente (F.P.).
Authorship
Contribution: F.P., A.B.F., I.P., and J.E. conceived, designed, and supervised the study; L.B., A.C., A.G., N.N., R.S., F.O., and W.H. performed laboratory measurements; L.B., A.G., and R.S. performed data analysis; L.B., J.E., A.G., R.S., F.P., A.B.F., I.P., P.M.M., and U.B. wrote and revised the manuscript; M.A., Z.B., M.-R.B., O.B., I.B., G.C., J.E., P.E., J.G., H.H., M.K., B.K., R.L., F.W.G.L., M.F.L.F., R.M., F.P., C.S., R.S., A. Tiede, G.T., A. Tosetto, M.T., and E.M.K.Z. enrolled patients and collected patient data; M.A. supervised the laboratory samples collected at the Iranian centers; and all authors contributed to the editing of the manuscript and approved the final version.
Conflict-of-interest disclosure: O.B. reports advisory board participation for Sobi, CSL Behring, and Pfizer. F.W.G.L. received unrestricted research grants from CSL Behring, Shire/Takeda, and uniQure; is a consultant for CSL Behring, Shire/Takeda, BioMarin, and uniQure, the fees of which go to the university; has received travel support from Sobi; and is a data and safety monitoring board member of a study sponsored by Roche. R.L. reports board or advisory committee membership for Abbott, Roche, BioMarin, and Takeda, and employment (Chief Scientific Officer) for Aplagon Ltd. P.M.M. received speaker fees from Bayer, Grifols, Kedrion, Novo Nordisk, and Octapharma; and reports advisory board membership for Bayer and Kedrion. R.M. reports advisory board participation for Bayer, Sobi, Novo Nordisk, Shire/Takeda, Roche, and CSL Behring. C.S. reports advisory board participation for Bayer, Novo Nordisk, Roche, Shire/Takeda, and Sobi; and is on the speaker bureau for CSL Behring, Roche, and Shire/Takeda. A. Tiede received grants and personal fees for lectures and consultancy from Alnylam, Bayer, Biogen Idec, BioMarin, Biotest, Boehringer Ingelheim, Chugai, CSL Behring, Daiichi Sankyo, Leo Pharma, Novo Nordisk, Octapharma, Pfizer, Portola, Roche, Shire/Takeda, and Sobi. J.E. received research support from CSL Behring outside the scope of this project and fees for educational activities from Roche and Celgene (fees go to the institution). A.B.F. reports advisory board participation of CSL Behring, Grifols, Takeda, Octapharma, LFB, and Kedrion. F.P. has received honoraria for participating as a speaker at satellite symposia organized by Bioverativ, Grifols, Roche, Sanofi, Sobi, Spark, and Takeda; and advisory board participation of Roche, Sanofi, and Sobi. The remaining authors declare no competing financial interests.
Hamid Hoorfar died on 2 August 2020.
Correspondence: Luciano Baronciani, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Via Pace 9, 20122 Milan, Italy; e-mail: luciano.baronciani@policlinico.mi.it.

