Key Points
The anti-CD27 agonist antibody varlilumab was well tolerated in lymphoma patients with no maximum tolerated dose identified.
Varlilumab transiently increased pro-inflammatory cytokines and led to a durable complete response in a patient with refractory Hodgkin lymphoma.

Abstract
CD27, a costimulatory molecule on T cells, induces intracellular signals mediating cellular activation, proliferation, effector function, and cell survival on binding to its ligand, CD70. Varlilumab, a novel, first-in-class, agonist immunoglobulin G1 anti-CD27 antibody, mediates antitumor immunity and direct killing of CD27+ tumor cells in animal models. This first-in-human, dose-escalation, and expansion study evaluated varlilumab in patients with hematologic malignancies. Primary objectives were to assess safety and the maximum tolerated and optimal biologic doses of varlilumab. Secondary objectives were to evaluate pharmacokinetics, pharmacodynamics, immunogenicity, and antitumor activity. In a 3 + 3 dose-escalation design, 30 patients with B-cell (n = 25) or T-cell (n = 5) malignancies received varlilumab (0.1, 0.3, 1, 3, or 10 mg/kg IV) as a single dose with a 28-day observation period, followed by weekly dosing (4 doses per cycle, up to 5 cycles, depending on tumor response). In an expansion cohort, 4 additional patients with Hodgkin lymphoma received varlilumab at 0.3 mg/kg every 3 weeks (4 doses per cycle, up to 5 cycles). No dose-limiting toxicities were observed. Treatment-related adverse events, generally grade 1 to 2, included fatigue, decreased appetite, anemia, diarrhea, and headache. Exposure was linear and dose-proportional across dose groups and resulted in increases in proinflammatory cytokines and soluble CD27. One patient with stage IV Hodgkin lymphoma experienced a complete response and remained in remission at >33 months with no further anticancer therapy. These data support further investigation of varlilumab for hematologic malignancies, particularly in combination approaches targeting nonredundant immune regulating pathways. This trial was registered at www.clinicaltrials.gov as #NCT01460134.
Introduction
CD27, a member of the tumor necrosis factor receptor superfamily, acts as a potent costimulatory molecule that, unlike other related family members, is expressed constitutively on unstimulated T lymphocytes. CD27 is also expressed on B lymphocytes and is commonly expressed on nearly all subtypes of mature B-cell lymphomas.1,2 CD70, the ligand for CD27, is transiently expressed on antigen-presenting cells. CD27/CD70–mediated costimulation, concomitant with antigen-specific T-cell receptor (TCR) stimulation, results in T-cell activation, proliferation, survival, maturation of effector capacity, and T-cell memory (Figure 1).3,4 CD27-CD70 interactions also promote B-cell proliferation, generation of plasma cells, production of immunoglobulin, and B-cell memory,5-8 as well as the induction of natural killer cell cytolytic activity.9 CD27 is also expressed by T regulatory cells (Tregs) and may have a role in their expansion and activation.10
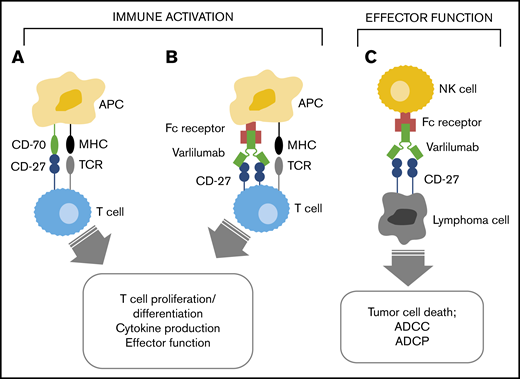
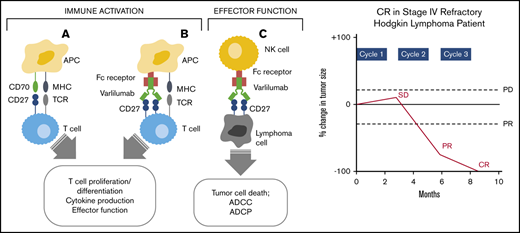
Mechanism of varlilumab antitumor activity. Interaction of CD27 and CD70 (A) and varlilumab and the Ag-specific TCR (B) in the immune activation of effector T cells. (C) Interaction of CD27-expressing tumor cells with varlilumab on natural killer (NK) cells for a cytolytic response. APC, antigen-presenting cell; MHC, major histocompatibility complex.
Mechanism of varlilumab antitumor activity. Interaction of CD27 and CD70 (A) and varlilumab and the Ag-specific TCR (B) in the immune activation of effector T cells. (C) Interaction of CD27-expressing tumor cells with varlilumab on natural killer (NK) cells for a cytolytic response. APC, antigen-presenting cell; MHC, major histocompatibility complex.
Varlilumab (CDX-1127) is a novel, first-in-class fully human immunoglobulin G1 kappa anti-CD27 monoclonal antibody that acts as an agonist of CD27 by interaction with the CD70-binding site.11 Varlilumab mimics CD70 to enhance the CD27-mediated T-cell costimulatory pathway when combined with TCR activation (Figure 1). Potent cytokine release and proliferation of T cells were observed when purified human T cells were cultured with varlilumab and OKT3 (TCR-stimulating antibody).12 Varlilumab has an unmodified Fc region allowing for Fc receptor–mediated crosslinking and Fc-dependent effector function such as antibody-dependent cellular cytotoxicity.
Varlilumab has potent antitumor activity in multiple animal models.13,14 For example, BCL1 B-lymphoma and CT26 (colon cancer) tumor challenge models using human CD27-transgenic mice showed that treatment with varlilumab resulted in substantially improved survival at high repeated dose levels (>150 μg × 5); a biologically effective response was observed for ≥0.5 mg/kg × 5 repeated doses. In addition, varlilumab exhibited potent antitumor activity against the EG7 mouse thymoma in syngeneic tumor models that rely on T cell–mediated immunity for response. In xenograft models in SCID mice, varlilumab showed significant antitumor effects against a variety of human tumor cell lines, including the lymphoblastic Burkitt’s lymphoma–derived cell lines (Raji, Daudi, and Namalwa) and an acute lymphocytic leukemia cell line (CCRF-CEM). In addition to the enhanced immune activation of cells, preclinical studies with varlilumab have shown direct therapeutic effects against CD27-expressing tumors. Together, these data provide support for targeting CD27 in hematologic malignancies as a mechanism to enhance antitumor immunity, as well as to mediate direct killing of CD27-expressing lymphomas.
The current first-in-human, phase 1, open-label, dose-escalating, and expansion study was conducted to assess the safety, pharmacokinetics, pharmacodynamics, and activity of varlilumab when administered as monotherapy to patients with advanced malignancies. Patients with solid tumors and hematologic malignancies were separately enrolled in parallel dose-escalation and expansion phases, given the potential for differing mechanisms of action, pharmacokinetics, and toxicity profile of anti-CD27–directed therapy in these populations. In the dose-escalation and expansion cohorts of patients with solid tumors (n = 56), varlilumab was well tolerated and showed clear evidence of biological and clinical activity.15 Durable clinical activity was observed in 2 patients with renal cell carcinoma. One patient achieved a partial response on study (78% shrinkage of target lesions and progression-free survival of >3.6 years). A second patient had stable disease for >3.9 years without additional therapy. This report provides results for the patients with hematologic malignancies enrolled in the phase 1 study.
Methods
Patients
The dose-escalation study portion included patients with a histologic diagnosis of any B-cell or T-cell malignancy, whereas the expansion cohort was restricted to patients with Hodgkin lymphoma. All patients were required to have disease that was progressive subsequent to previous therapies with no remaining alternative approved therapy options. Additional eligibility requirements included measurable or evaluable disease as per appropriate disease-specific staging criteria; ≥18 years of age; life expectancy ≥12 weeks; Eastern Cooperative Oncology Group performance status of 0 or 1; adequate renal, hepatic, and bone marrow function; and resolution of toxicity related to prior therapy (excluding alopecia) to grade 1 or lower. A washout of ≥4 weeks was required for chemotherapy, monoclonal-based therapies, systemic radiation therapy, and immunosuppressive medications, including systemic corticosteroids. Other immunotherapy, investigational drugs, and previous focal radiotherapy were prohibited within 2 weeks of study entry. Patients were excluded who were: pregnant; breastfeeding; or who had other prior malignancies within the last 5 years, active brain metastases, autoimmune disease, active infection, significant cardiovascular disease, or any other significant, active, concurrent medical illness that would have precluded study treatment.
The study was conducted at each of the participating institutions in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines, after approval by a local institutional review board and in accordance with an assurance filed with and approved by the Department of Health and Human Services, where appropriate. All patients signed written informed consent before initiation of any protocol-specific procedures. All authors had access to primary clinical trial data. This trial was registered at www.clinicaltrials.gov as #NCT01460134.
Study design and treatment
Study design and treatment schema are shown in Figure 2. Dose escalation followed a standard 3 + 3 design16 to evaluate the safety and tolerability of varlilumab, and, if possible, to determine the maximum tolerated dose and/or optimal biologic dose in patients with hematologic malignancies. The maximum tolerated dose was defined as the highest dose in which ≤1 of 6 patients experienced a dose-limiting toxicity (DLT). DLT was defined as any grade 3 or higher treatment-related toxicity, excluding grade 3 inflammation due to local therapeutic response persisting ≤7 days; grade 3 nonmalignant lymphocyte changes that improved to grade 2 or lower or within 20% of baseline within 28 days; and grade 3 nausea, vomiting, or diarrhea that resolved to grade 1 or lower within 48 hours.
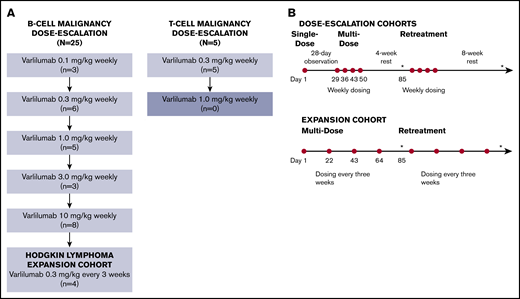
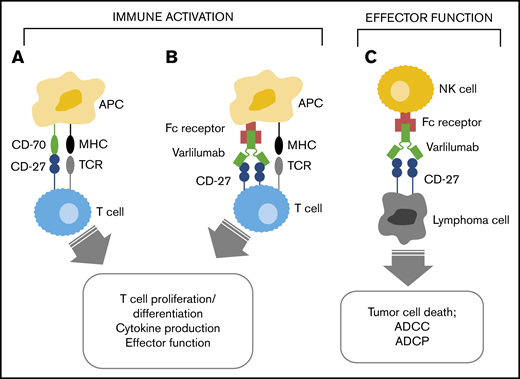
Study design/treatment schema. (A) Using a standard 3 + 3 design, the B-cell malignancy and T-cell malignancy dose escalations proceeded separately. Patients who did not complete the multidose phase for reasons other than DLT were replaced as necessary. The B-cell malignancy dose escalation completed through the maximum planned dose level (10 mg/kg). After the B-cell malignancy dose escalation component of the study, an expansion cohort was enrolled to further explore the clinical and biological activity of varlilumab (at 0.3 mg/kg every 3 weeks) in patients with Hodgkin lymphoma. The study was closed after treatment of 4 patients in the Hodgkin lymphoma expansion cohort and 5 patients at the 3 mg/kg dose level in the T-cell malignancy dose-escalation phase. (B) During the dose-escalation phase, varlilumab (represented by red circles) was initially administered as a single dose with 28-day observation. Additional multidose treatment (4 weekly doses with a 4-week observation) and retreatment (up to 4 additional cycles, each consisting of 4 weekly doses with an 8-week observation) were allowed for patients who had not experienced progressive disease or DLT. Patients in the Hodgkin lymphoma expansion cohort received varlilumab at 0.3 mg/kg, every 3 weeks (up to 5 cycles, each consisting of 4 doses). Diagnostic imaging and restaging were repeated every 12 weeks (as indicated by asterisks).
Study design/treatment schema. (A) Using a standard 3 + 3 design, the B-cell malignancy and T-cell malignancy dose escalations proceeded separately. Patients who did not complete the multidose phase for reasons other than DLT were replaced as necessary. The B-cell malignancy dose escalation completed through the maximum planned dose level (10 mg/kg). After the B-cell malignancy dose escalation component of the study, an expansion cohort was enrolled to further explore the clinical and biological activity of varlilumab (at 0.3 mg/kg every 3 weeks) in patients with Hodgkin lymphoma. The study was closed after treatment of 4 patients in the Hodgkin lymphoma expansion cohort and 5 patients at the 3 mg/kg dose level in the T-cell malignancy dose-escalation phase. (B) During the dose-escalation phase, varlilumab (represented by red circles) was initially administered as a single dose with 28-day observation. Additional multidose treatment (4 weekly doses with a 4-week observation) and retreatment (up to 4 additional cycles, each consisting of 4 weekly doses with an 8-week observation) were allowed for patients who had not experienced progressive disease or DLT. Patients in the Hodgkin lymphoma expansion cohort received varlilumab at 0.3 mg/kg, every 3 weeks (up to 5 cycles, each consisting of 4 doses). Diagnostic imaging and restaging were repeated every 12 weeks (as indicated by asterisks).
Varlilumab dose levels of 0.1, 0.3, 1, 3, and 10 mg/kg were selected for the initial dose escalation in patients with B-cell malignancies, based on the no observable effect level of 25 mg/kg identified during nonclinical toxicology and to bridge the dose ranges with expected clinical activity. Subsequently, an abbreviated dose escalation (3 and 10 mg/kg) was initiated for patients with T-cell malignancies. In the dose-escalation study portion, varlilumab was initially administered as a single dose followed by a 4-week evaluation period. Additional multidose treatment (4 weekly doses with a 4-week observation) and retreatment (up to 4 additional cycles, each consisting of 4 weekly doses with an 8-week observation period) were allowed for patients who had not experienced progressive disease or DLT.
After completion of the dose-escalation phase, an expansion cohort was initiated to further evaluate the clinical and biological activity of varlilumab in up to 15 patients with Hodgkin lymphoma at the 0.3 mg/kg dose given every 3 weeks.
All patients received varlilumab as a 90-minute IV infusion, with a 4- to 6-hour observation after the first 2 infusions and a 2-hour observation after each subsequent dose. For the first dose in the dose-escalation portion, patients received 10% of the total dose over 10 minutes followed by a 1-hour observation before administration of the remaining dose over 80 minutes.
Assessments
Safety assessments included vital signs, physical examinations, laboratory test results, electrocardiograms, and Eastern Cooperative Oncology Group performance status. Patients were monitored for toxicity through 70 days posttreatment. Given the expected mechanism of action of varlilumab, particular attention was given to adverse events such as diarrhea/colitis, rash, endocrinopathies, and hepatitis, which have been observed with other immune antibodies and may be related to immune activation.17 Patients were also closely monitored for infusion reactions or cytokine release syndrome.18 Adverse events were coded with the latest available version of the Medical Dictionary for Regulatory Activities and graded according to the National Cancer Institute–issued Common Terminology Criteria for Adverse Events version 4.0.
Diagnostic imaging for assessment of tumor response was performed every 12 weeks. Antitumor activity was assessed by the investigator using the International Working Group response criteria for non-Hodgkin lymphoma, the Cheson criteria for Hodgkin lymphoma, or other appropriate disease-specific criteria.
Pretreatment tumor samples cut serially at 5 μm were collected for CD27 staining. The tissue sections were deparaffinized in 3 changes of xylene and cleared through graded ethanol series. Endogenous peroxidase was quenched by incubation in 50% methanol/H2O2. After rinsing with tap water, all sections were pretreated for 30 minutes with 50 mM EDTA, pH 8.0 using a steamer and cooled for an additional 5 minutes. All immunohistochemical staining was performed automatically on a DAKO Autostainer Plus using the following antibodies and their corresponding detection systems: CD27 (1 mg/mL; Abcam, ab#131254, 1:1000) or mouse immunoglobulin G1 control (DAKO, #x0931, 1:100000). All sections were stained with hematoxylin and rinsed well in tap water. All slides were observed with light microscopy (Olympus AX70, 200×/aperture 0.46, 400×/aperture 0.75, 600×/aperture 0.80; Olympus America) with images captured with a SPOT RT camera and software (Diagnostic Instruments).
Serial serum samples were collected for pharmacokinetic analyses from 30 patients receiving from 0.1 to 10 mg/kg varlilumab through 28 days after the first varlilumab dose and the fourth weekly varlilumab dose. Trough (predose) and peak (30 minutes after completion of the infusion) samples were collected for all other infusions. Serum levels of varlilumab were determined by enzyme-linked immunosorbent assay (ELISA), using murine Fc-human CD27 fusion protein for capture and a goat F(ab′)2 specific anti-human IgG horseradish peroxidase conjugate for detection. The assay is validated with a sensitivity of 180 ng/mL.
Immunogenicity samples were collected prestudy, before the first dose, and at 1 week after the last dose in each multidose cycle, and at the time of study discontinuation. Anti-drug antibodies (ADAs) were determined by a bridging ELISA and a cut-point based on a 5% false-positive rate. Positive samples were confirmed in a secondary assay to determine specificity of response by immunodepletion with study drug and a cut-point based on a 1% false-positive rate, as previously described.15
Cytokines associated with T-cell function, immune regulation, and cell migration pretherapy and posttherapy were measured. Serum samples were analyzed by using a multiplex enzyme-linked immunosorbent assay (Thermo Fisher Scientific) to measure 30 serum cytokines. A Luminex 200 System, version 1.7, was used for reading plates, and MasterPlex QT 1.0 system (MiraiBio) was used to analyze data. Cytokines included the following: epidermal growth factor, eotaxin, basic fibroblast growth factor, granulocyte macrophage–colony stimulating factor, hepatocyte growth factor, interferon-α, interferon-γ, interleukin 1 receptor antagonist (IL-1RA), IL-1β, IL-2, IL-2R, IL-4 to IL-8, IL-10, IL-12, IL-13, IL-15, IL-17, inducible protein-10 (CXCL10), monocyte chemotactic protein 1, monokine induced by interferon-γ (CXCL9), macrophage inflammatory protein-1α (MIP-1α/CCL3), MIP-1β (CCL4), regulated on activation normal T-cell expressed and secreted protein, tumor necrosis factor-α, and vascular endothelial growth factor (VEGF). Internal control serum was included in all assays to control for interassay variation, and controls were randomized across plates.
Mean levels of soluble CD27 were assessed by using a sandwich ELISA that is not obstructed by the presence of bound varlilumab, as previously described.15 Whole blood was collected and processed into peripheral blood mononuclear cells with Ficoll and frozen for analysis by flow cytometry. Peripheral blood mononuclear cells were thawed and stained with the following antibodies: CD3 FITC, CD8 PerCP, CD4 PE, and FoxP3 Alexa 488. Cells were first gated on lymphocytes and then subsequently gated on the fluorescence of the cell of interest. Percentages of positive cells were calculated as a percentage of the lymphocytes.
Statistical considerations
The primary study objectives were to assess safety and the maximum tolerated and optimal biologic doses of varlilumab. Secondary study objectives were to evaluate pharmacokinetics, pharmacodynamics, immunogenicity, and antitumor activity.
Pharmacokinetic parameters were derived via noncompartmental analysis of individual patient serum drug levels using NCA Plasma Model 200-202 (Phoenix WinNonlin 8.0; Certara L.P. [Pharsight]). Briefly, elimination half-life and exposure parameters were estimated following visual inspection of dose 1 and 5 time vs concentration profiles to identify the terminal phase, and goodness-of-fit criteria were used to refine the terminal phase for each patient. Area under the curve was calculated by using the linear trapezoidal rule with linear interpolation with uniform weighting. All calculations were based on actual sampling times and infusion durations, including test infusion in dose 1.
For the pharmacodynamic end points, significant differences were calculated by using a 2-tailed Student t test using paired or unpaired data.
Results
Patient characteristics
Thirty-four patients were enrolled at 7 centers from April 2012 to June 2015 (Figure 2). Twenty-five patients with B-cell malignancies were treated with varlilumab in the initial dose escalation, which was completed through the maximum planned dose level (10 mg/kg). Subsequent to recruitment challenges in the T-cell dose-escalation and Hodgkin lymphoma expansion cohorts, which enrolled 5 patients and 4 patients, respectively, the study was closed.
Pretreatment demographic and disease characteristics for enrolled patients are shown in Table 1. Of the treated patients, 18 had B-cell non-Hodgkin lymphoma (10 diffuse large B-cell, 6 follicular, and 2 not specified), 11 had Hodgkin lymphoma, 3 had T-cell lymphoma, and 2 patients had mycosis fungoides. The majority (91%) had stage III or IV disease. Enrolled patients were heavily pretreated, with a median of 5 (range, 1-12) previous anticancer therapies. All patients had received at least 1 previous line of cytotoxic chemotherapy, 12% had received checkpoint inhibition, and 48% had undergone autologous transplant.
Study treatments and tolerability
Patients received a median of 4 (range, 1-21) varlilumab doses on study. Eighteen patients completed at least 1 multidose cycle of varlilumab. Four of these patients received multiple treatment cycles, ranging from 2 to 4.
Varlilumab, at doses up to 10 mg/kg in B-cell malignancies and 3 mg/kg in T-cell malignancies, was well tolerated without DLT. The most frequently reported treatment-related adverse events (supplemental Table 1) were fatigue (8 [24%]), decreased appetite (6 [18%]), anemia (4 [12%]), headache (3 [9%]), and diarrhea (3 [9%]). Except for one grade 3 event of transient increased blood alkaline phosphatase levels, treatment-related events were all grade 1 to 2 in severity. One patient discontinued study treatment after the first varlilumab dose (10 mg/kg) due to grade 1 vision changes, assessed as related to treatment by the investigator. The most common reason patients discontinued treatment was because of progressive disease (22 [65%]) or symptomatic deterioration (7 [21%]). Two patients (6%) requested to discontinue treatment; 1 patient each (3%) discontinued due to death, lost to follow-up, and a grade 1 adverse event of vision change (described earlier). There were no unexpected or high-grade toxicities in patients who previously received immune checkpoint blockade therapy.
Antitumor activity
Hodgkin lymphoma.
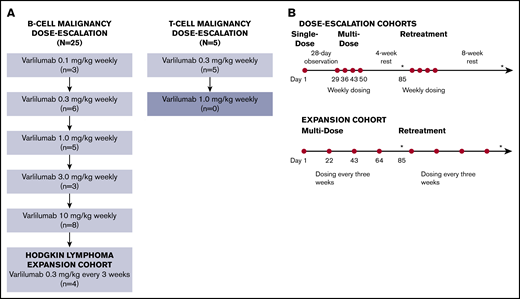
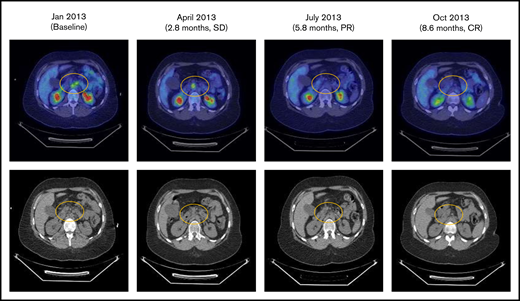
Eleven patients with Hodgkin lymphoma were enrolled. Of the 10 evaluable patients, there was 1 complete response, 1 patient with stable disease, and the remaining patients had progressive disease. The patient with a complete response was a 28-year-old woman with stage IV Hodgkin lymphoma who had previously experienced an inadequate response to induction therapy and progression after 4 subsequent regimens, including hematopoietic stem cell transplantation followed by brentuximab vedotin consolidation. The patient had not previously received treatment with anti–programmed cell death 1 (PD-1) or anti–programmed death-ligand 1 therapy. After 3 cycles of varlilumab therapy (0.3 mg/kg), the patient had a complete response (Figure 3). Although varlilumab was discontinued with achievement of the complete response, the patient remained in remission without further anticancer therapy at the last follow-up, >33 months after enrollment in the study.
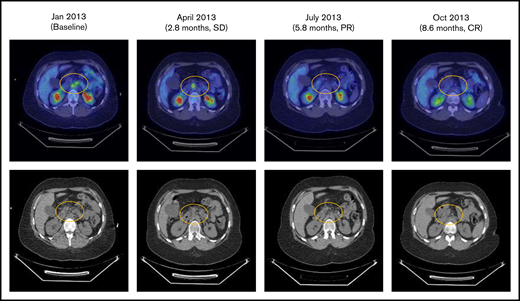
Patients with stage IV Hodgkin lymphoma with complete response (CR) to varlilumab. PR, partial response; SD, stable disease.
Patients with stage IV Hodgkin lymphoma with complete response (CR) to varlilumab. PR, partial response; SD, stable disease.
Non-Hodgkin lymphoma.
Of 18 patients with B-cell non-Hodgkin lymphoma, there was no objective response, and 3 patients experienced stable disease. A 67-year-old man with stage III follicular lymphoma who had previously received multiple courses of therapy, including combination chemotherapy, rituximab, ibritumomab tiuxetan, and traditional radiation therapy, experienced stable disease for 5.6 months with varlilumab (0.3 mg/kg). The tumor response included 36% shrinkage of measurable disease, with complete disappearance of disease in inguinal and iliac regions. A 52-year-old man with recurrent stage IV follicular lymphoma who received a single dose of varlilumab 0.3 mg/kg followed by 5 treatment cycles at 0.1 mg/kg experienced stable disease for 14 months. Finally, a 58-year-old man with stage IV follicular lymphoma, who had received 6 previous courses of therapy, including combination chemotherapy, rituximab, ibritumomab tiuxetan, and experimental therapy, experienced stable disease for 4.5 months while receiving varlilumab (0.1 mg/kg). One patient with T-cell non-Hodgkin lymphoma experienced stable disease, and the other 4 patients with T cell non-Hodgkin lymphoma experienced progressive disease.
CD27 expression.
Pretreatment tumor samples were sufficient for analysis of CD27 expression for 18 patients (supplemental Table 2). The 2 follicular lymphomas analyzed had the highest intensity of tumor expression, although apparently less lymphocyte infiltration. The remaining lymphomas (nine Hodgkin lymphoma, five diffuse B cell, and two T cell) had lower levels of tumor expression, including 5 Hodgkin lymphoma and 4 diffuse B-cell with no tumor staining. However, all of these showed CD27 staining of infiltrating lymphocytes.
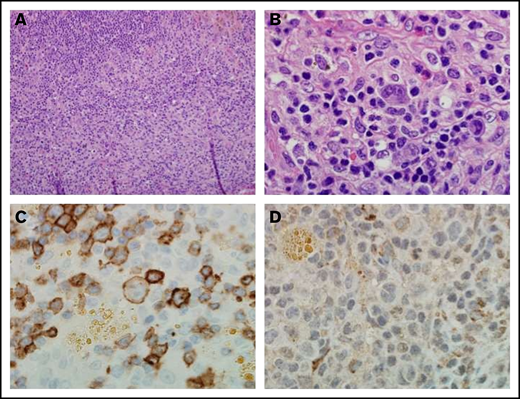
Notably, the pretreatment tumor sample for the patient with Hodgkin lymphoma who experienced a complete response exhibited the highest level of CD27 expression on infiltrating lymphocytes, relative to the other patients with Hodgkin lymphoma. Reed-Sternberg cells were CD27 positive (Figure 4).
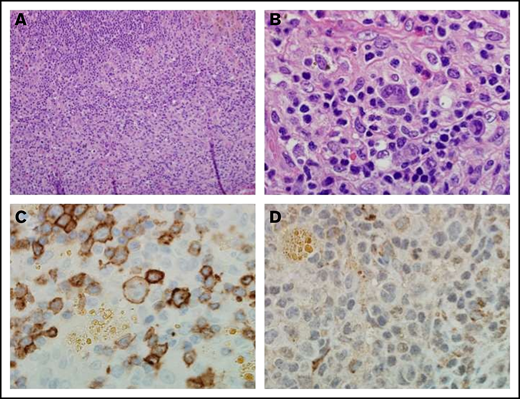
Immunohistochemistry of a pretreatment lymph node biopsy sample from a patient with complete response to varlilumab. (A) Hematoxylin and eosin stain (magnification ×20) from a patient with classical Hodgkin lymphoma. (B) Atypical lymphohistiocytic infiltrate within a background of normal reactive cells (magnification ×100). (C) CD27 expression on malignant cells, intratumoral lymphocytes, and histiocytes. Malignant cells and intratumoral T cells were positive for CD27 expression (magnification ×100). (D) CD70 staining was negative on both malignant cells and the intratumoral immune infiltrate (magnification ×100).
Immunohistochemistry of a pretreatment lymph node biopsy sample from a patient with complete response to varlilumab. (A) Hematoxylin and eosin stain (magnification ×20) from a patient with classical Hodgkin lymphoma. (B) Atypical lymphohistiocytic infiltrate within a background of normal reactive cells (magnification ×100). (C) CD27 expression on malignant cells, intratumoral lymphocytes, and histiocytes. Malignant cells and intratumoral T cells were positive for CD27 expression (magnification ×100). (D) CD70 staining was negative on both malignant cells and the intratumoral immune infiltrate (magnification ×100).
Pharmacokinetics, immunogenicity, and pharmacodynamics
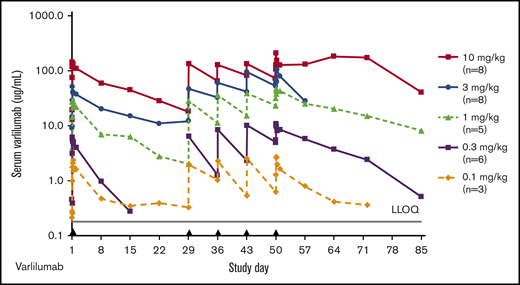
Varlilumab exposure was dose proportional, with a mean elimination half-life ranging from 2.9 days (0.3 mg/kg) to 12.5 days (3 mg/kg) after day 1 dosing (Figure 5; supplemental Table 3). Following steady-state dosing, the elimination half-life ranged from 4.3 days (0.3 mg/kg) to 14.5 days (10 mg/kg), which is consistent with the pharmacokinetic results from dose escalation of human monoclonal antibodies in patients with solid malignancies.15 There were no specific on-study anti-varlilumab antibodies detected.
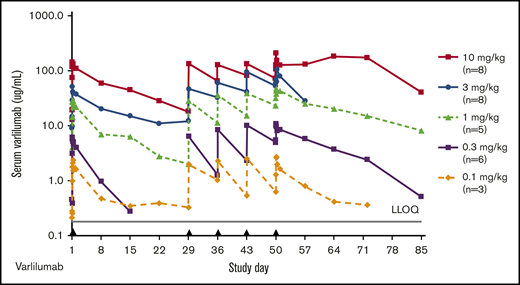
Varlilumab serum levels in patients with hematologic malignancies. Mean concentration–time curves for varlilumab for the dose-escalation cohorts. Black triangles represent varlilumab dosing. LLOQ, lower limit of quantification.
Varlilumab serum levels in patients with hematologic malignancies. Mean concentration–time curves for varlilumab for the dose-escalation cohorts. Black triangles represent varlilumab dosing. LLOQ, lower limit of quantification.
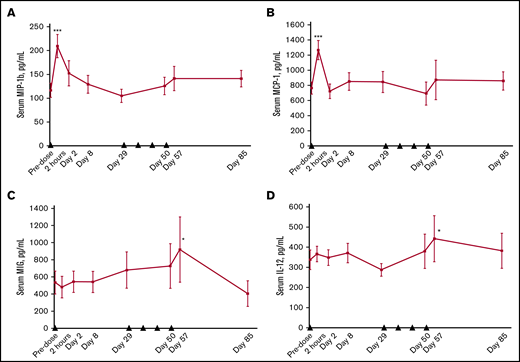
We observed significant changes in several soluble factors present in serum. The levels of soluble CD27 were markedly increased after varlilumab treatment and were generally dose dependent, with the most prominent effect at the 10 mg/kg dose (supplemental Figure 1). The level of chemokines, particularly MIP-1β (CCL4) and monocyte chemoattractant protein-1 (CCL2), showed a marked transient elevation after the initial dose of varlilumab, consistent with an inflammatory response (Figure 6A-B). Other soluble markers of inflammation, such as monokine induced by interferon-γ (CXCL9) and IL-12, were significantly increased compared with baseline values during the weekly dosing phase (Figure 6C-D). The changes in chemokines and cytokines were not dose dependent, as similar effects were observed in each cohort (not shown).
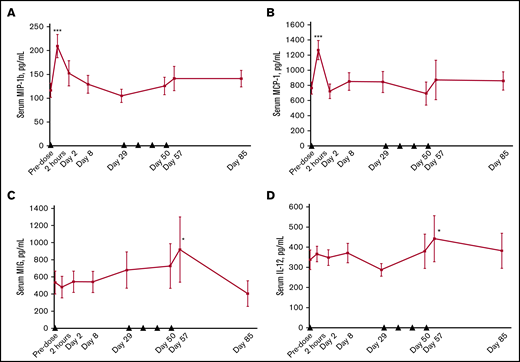
Changes in serum levels of soluble factors after varlilumab administration. MIP-1β (A), monocyte chemoattractant protein-1 (MCP-1) (B), monokine induced by gamma interferon (MIG) (C), and IL-12 (D). Serum cytokines were measured by using the Luminex 200 System at the indicated times. Data represent the mean and standard error of patients across different dose levels (n = 9 to 11). Black triangles represent varlilumab dosing. Statistics are shown for paired Student t test vs baseline samples. *P < .05; ***P < .001.
Changes in serum levels of soluble factors after varlilumab administration. MIP-1β (A), monocyte chemoattractant protein-1 (MCP-1) (B), monokine induced by gamma interferon (MIG) (C), and IL-12 (D). Serum cytokines were measured by using the Luminex 200 System at the indicated times. Data represent the mean and standard error of patients across different dose levels (n = 9 to 11). Black triangles represent varlilumab dosing. Statistics are shown for paired Student t test vs baseline samples. *P < .05; ***P < .001.
Varlilumab administration was associated with a decrease in the absolute lymphocyte count in some patients that was generally low grade, stable during continued treatment, and not associated with a clear dose effect. However, analysis of peripheral blood lymphocyte populations by using flow cytometry did not reveal consistent and significant changes in percentage of CD8+ T cells or CD4+ T cells across the various dose levels (supplemental Figure 2). A transient or maintained decrease in CD4+ Tregs was observed in some patients (supplemental Figure 3) but less consistently than reported for patients with solid tumors treated with varlilumab.15
Discussion
This first-in-human study in patients with hematologic malignancies evaluated the safety, pharmacokinetics, pharmacodynamics, and early activity of varlilumab, an agonist anti-CD27 monoclonal antibody. Varlilumab doses up to 10 mg/kg weekly were well tolerated with no DLT, and a maximum tolerated dose was not identified.
Evidence of single-agent clinical activity was seen in this heavily pretreated population of patients with advanced disease. One patient with heavily pretreated, stage IV Hodgkin lymphoma, including para-aortic involvement and B-symptoms, experienced a complete response to varlilumab (0.3 mg/kg) that persisted as of the last follow-up at nearly 3 years; 3 patients experienced stable disease ranging from 4.5 to 14 months. The kinetics of the complete response were consistent with an immune-mediated mechanism, with an initial increase in tumor that gradually disappeared after 2 subsequent cycles. Interestingly, this patient also had an abundance of CD27+ tumor-infiltrating lymphocytes and the Reed-Sternberg cells were CD27-positive; in contrast the Reed Sternberg cells were CD27-negative in the majority of Hodgkin lymphomas analyzed. This suggests the potential that varlilumab may have benefited this patient via dual mechanisms (ie, that the clinical response was mediated by an antitumor immune response and potentially by direct killing of the tumor cells).
It is interesting to note that among hematologic malignancies, the PD-1 inhibitors have shown the greatest activity in Hodgkin lymphoma. During the course of this study, data became available showing that nivolumab resulted in an 87% objective response rate, with 17% achieving a complete response in refractory classical Hodgkin lymphoma.19 However, activity of checkpoint blockade in non-Hodgkin lymphoma has been relatively modest, with primary mediastinal large B-cell lymphoma showing an overall response rate of 40% to 45% and other non-Hodgkin lymphoma subtypes being less responsive, with an overall response rate of 3% to 10%.
In this study, varlilumab exhibited clinical benefit in 1 patient with Hodgkin lymphoma (14%) who had a durable complete response among the 7 PD-1 blockade-naive patients with Hodgkin lymphoma. No responses were observed among the 3 patients with Hodgkin lymphoma who had previously received PD-1 blockade. Four patients with Hodgkin lymphoma were treated with a lower and less frequent varlilumab dosing regimen of 0.3 mg/kg on a 3-week schedule to potentially avoid overstimulation of T cells, but no clinical responses were observed. The only distinguishing factor identified in the 1 responding Hodgkin lymphoma patient relative to the nonresponders was the marked intensity of the CD27-expressing, tumor-infiltrating T cells in the responder, and possibly also the presence of CD27 expressing Reed Sternberg cells. However, CD27 expression did not correlate with clinical benefit in any of the other patients.
Varlilumab exposure was dose proportional from 0.1 to 10 mg/kg, with half-lives ranging from 3 to 12.5 days, which is consistent with the pharmacokinetic results from dose escalation in patients with solid malignancies.15 Accumulation was not calculated because the dosing intervals were not equivalent from the first to the fifth dose, but visual analysis of trough levels in dose escalation and retreatment patients indicate accumulation for all doses at a weekly interval (Figure 5). Screening ADA testing resulted in a 13% positivity rate, exceeding the 5% false-positive target, but specific ADAs were not detected posttreatment, even in patients with multiple cycles (up to 13 doses) of varlilumab. Because the varlilumab ADA assay is susceptible to interference from circulating levels of drug >100 ng/mL, ADAs in some patients may be masked. However, visual inspection of pharmacokinetic profiles did not reveal any evidence of attenuated exposure following repeat dosing, suggesting that ADA response is minimal.
We observed a significant dose-dependent increase in soluble CD27, which we believe reflects the stabilization of shed CD27 by circulating varlilumab, as has been reported for the anti-VEGF antibody (bevacizumab) stabilizing serum VEGF levels.20 However, this could also reflect an increase in the level of CD27 shedding from activated T cells, and increased soluble CD27 is consistent with an inflammatory effect that has been associated with improved response to immunotherapy.15
Other pharmacodynamic effects include transient upregulation of inflammatory chemokines and cytokines across all dose levels within hours of varlilumab administration. These effects are consistent with immune activation and with our observations of varlilumab in solid tumor patients.15 Notably, no cases of hyperprogression were seen. In contrast to the significant decrease of Tregs in patients with solid tumors, we observed a variable effect of varlilumab on Tregs in hematologic malignancies, with only a subset having pronounced decreases in circulating Tregs.
Combination strategies targeting multiple nonredundant pathways regulating tumor burden and immune responses may be synergistic and enhance antitumor immune responses as shown in preclinical studies. In particular, recent studies show that CD27 and PD-1 pathways synergize in their antitumor effects by directing complementary transcriptional programs, leading to enhanced survival and cytotoxicity of T cells.21 Mice inoculated with the aggressive BCL1 lymphoma and treated with varlilumab and PD-1 blockade showed statistically significant prolonged survival than mice that were not treated or given each of the 2 compounds separately. In addition, studies by Lim et al22 reported synergy when combining CD27 agonist monoclonal antibodies with anti-CD20 antibody therapy in lymphoma models. These studies pointed to a new mechanism of CD27 anti-tumor activity through the release of cytokines that attract and activate innate cells (natural killer and macrophage) which promote the antitumor effect of CD20 antibody. Synergistic activity23 has also been reported in animal models with varlilumab and the agonist anti-CD40 monoclonal antibody CDX-1140,24 which has recently entered clinical trials.
Collectively, these data support the safety and activity of varlilumab in hematologic malignancies. However, based on the modest single-agent activity of varlilumab, future studies will explore combination therapy to enhance overall outcomes for patients. Ongoing studies include varlilumab in combination with nivolumab in refractory non-Hodgkin lymphoma and varlilumab in combination with rituximab in CD20+ rituximab-refractory non-Hodgkin lymphoma.
This article is a continuation of a previous report.15
Presented in abstract form at the 2014 Society for Immunotherapy of Cancer Annual Meeting, National Harbor, MD, 7-9 November 2014 (Abstract 52514/Poster P115); the 2014 American Society of Clinical Oncology Annual Meeting, Chicago, IL, 30 May-3 June 2014 (Abstract ID #1127/Poster); and the 2013 Society for Immunotherapy of Cancer Annual Meeting, National Harbor, MD, 8-10 November 2013 (Abstract 1771910/Poster 146).
Deidentified individual participant data that underlie the reported results will be made available 6 months after publication for a period of up to 3 years following the publication date. Proposals for access should be sent to info@celldex.com. The study protocol is included as a data supplement available with the full-text version of this article.
Acknowledgments
Assistance with clinical trial management was provided by Elsa Paradise (Celldex Therapeutics, Inc.), and assistance with correlative data analysis was provided by Laura Vitale, Venky Ramakrishna, Pamela Morani, and Jason DelCarpini (Celldex Therapeutics, Inc.). An earlier version of the manuscript was prepared with medical writing assistance provided by Mary-Ann Zalman (M. A. Zalman & Associates, LLC).
This study was funded by Celldex Therapeutics, Inc. It was also supported by a US Department of Defense grant (W81XWH1810650) (S.M.A.).
Authorship
Contribution: S.M.A. designed research, performed research, analyzed and interpreted data, and reviewed/revised the manuscript; I.F. and J.N. performed research and reviewed/revised the manuscript; M.H.T. and J.B. performed research, collected data, analyzed and interpreted data, and reviewed/revised the manuscript; B.I.S. performed research, collection and assembly of data, data analysis, and interpretation; A.F. collected data, analyzed and interpreted data, and reviewed/revised the manuscript; T.R.H. analyzed and interpreted data, performed statistical analysis, and reviewed/revised the manuscript; T.R. analyzed and interpreted data, and wrote and revised the manuscript; T.K. analyzed and interpreted data, and reviewed/revised the manuscript; M.J.Y. designed research, analyzed and interpreted data, and reviewed/revised the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: S.M.A. reports research funding (to his institution) from Celldex, BMS, Merck, Pfizer, Seattle Genetics, Takeda, AI Therapeutics, Regeneron, and Affimed. I.F. reports research funding and consultancy from AbbVie, Seattle Genetics, and Verastem; research funding from Acerta, Agios, ArQule, BeiGene, Calithera, Celgene, Constellation, Curis, Forma, Forty-Seven, Genentech, Gilead, Incyte, Infinity, Janssen, Karyopharm, KITE, Merck, Novartis, Pfizer, Pharmacyclics, Portola, Roche, Takeda, Teva, TG Therapeutics, and Trillium; and consultancy for TG Therapeutics. M.H.T. reports honoraria and being an advisory board member and speaker (unbranded/nonpromotional talks) for BMS and Eisai Inc; and reports honoraria and being an advisory board member for Array Biopharma, Blueprint Medicines, LOXO Oncology, Arqule, Bayer, and Novartis. B.I.S. is a member of Data Monitoring Committees for Pfizer and Immune Design, Inc. J.B. has received research funding from Merck, BMS, Celldex, Acerta, Celgene, Seattle Genetics, and Pharmacyclics. T.R.H., T.R., T.K., and M.J.Y. are employed by and have ownership interest (including stock options, but excluding direct investments through mutual funds and the like) in Celldex Therapeutics, Inc. The remaining authors declare no competing financial interests.
The current affiliation for M.H.T. is Providence Portland Medical Center, Portland, OR.
Correspondence: Stephen M. Ansell, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: ansell.stephen@mayo.edu.