Key Points
Baseline mutations are associated with response (JAK2) or resistance (NRAS, PTPN11) to ivosidenib monotherapy in mIDH1 R/R AML.
2-HG restoration via novel IDH1 second-site and IDH2 mutations, and non-IDH–related pathways, are identified at relapse.

Abstract
Isocitrate dehydrogenase (IDH) 1 and 2 mutations result in overproduction of D-2-hydroxyglutarate (2-HG) and impaired cellular differentiation. Ivosidenib, a targeted mutant IDH1 (mIDH1) enzyme inhibitor, can restore normal differentiation and results in clinical responses in a subset of patients with mIDH1 relapsed/refractory (R/R) acute myeloid leukemia (AML). We explored mechanisms of ivosidenib resistance in 174 patients with confirmed mIDH1 R/R AML from a phase 1 trial. Receptor tyrosine kinase (RTK) pathway mutations were associated with primary resistance to ivosidenib. Multiple mechanisms contributed to acquired resistance, particularly outgrowth of RTK pathway mutations and 2-HG–restoring mutations (second-site IDH1 mutations, IDH2 mutations). Observation of multiple concurrent mechanisms in individual patients underscores the complex biology of resistance and has important implications for rational combination therapy design. This trial was registered at www.clinicaltrials.gov as #NCT02074839
Introduction
Somatic mutations in the genes encoding the metabolic enzymes isocitrate dehydrogenase (IDH) 1 and 2 are found in multiple solid and hematologic tumors, including acute myeloid leukemia (AML). The altered enzymes have gain-of-function activity, catalyzing the reduction of α-ketoglutarate to the oncometabolite D-2-hydroxyglutarate (2-HG).1,2 Accumulation of 2-HG results in metabolic dysregulation and inhibition of α-ketoglutarate-dependent enzymes, which drive oncogenesis via epigenetic dysregulation and a block in cellular differentiation.3-5 Mutations in IDH1 and IDH2 are present in 6% to 10%2,6-8 and 9% to 13%8,9 of patients with AML, respectively, and IDH1 mutations have been associated with poor clinical outcomes in patients with AML.10-12
The mutant IDH (mIDH) inhibitors ivosidenib (Tibsovo; Agios Pharmaceuticals, Inc.; formerly AG-120)13 and enasidenib (Idhifa; Celgene Corp.; formerly AG-221)14 are targeted, allosteric inhibitors of the mIDH1 and mIDH2 enzymes, respectively. Both are approved in the United States for the treatment of adults with relapsed or refractory (R/R) AML with an IDH1 or IDH2 mutation, respectively, as detected by a test approved by the US Food and Drug Administration, and ivosidenib is approved for patients with mIDH1 newly diagnosed AML who are at least 75 years of age or who have comorbidities that preclude the use of intensive chemotherapy.15,16 Pharmacologic engagement of the mIDH1/2 protein by the respective inhibitor results in lowering of 2-HG levels in most treated patients. However, with both mIDH inhibitors, the proportion of patients in whom 2-HG is reduced exceeds the proportion of patients achieving clinical response, indicating that 2-HG reduction alone is not sufficient for a response.17,18
Therapeutic resistance to mIDH1/2 inhibitors has implications for treatment and the development of rational combination regimens, but mechanisms of primary and secondary resistance to these inhibitors are not yet well understood. Preliminary analyses of co-occurring mutations at baseline, defined as the most recent measurement before the first administration of ivosidenib, have found that certain co-occurring mutations are associated with primary resistance. Baseline mutations in receptor tyrosine kinase (RTK) pathway genes were more common in patients with mIDH1 R/R AML who did not achieve complete remission (CR) or CR with partial hematologic response (CRh) with ivosidenib treatment.17 In mIDH2 R/R AML treated with enasidenib, significantly fewer patients with NRAS mutations achieved CR.18 Secondary resistance after a clinical response to enasidenib was associated with the emergence of AML-related mutations, such as RUNX1 and FLT3,19 and IDH-related mutations, including outgrowth of mIDH1 in patients who initially had mIDH2 (isoform switching),19,20 and the emergence of second-site IDH2 mutations.21 However, these reports were based on a limited number of patients, and the frequency and breadth of resistance mechanisms have not been comprehensively characterized.
To fully characterize the mechanisms of response and relapse to ivosidenib monotherapy, we conducted a comprehensive genomic analysis of samples from a large cohort of patients with mIDH1 R/R AML treated in a phase 1 study17 whose starting dose of ivosidenib was the approved dose of 500 mg once daily (QD). Here, we confirm that RTK pathway mutations are associated with primary resistance to ivosidenib. Importantly, we found that multiple mechanisms contribute to relapse after ivosidenib therapy, including emergence or outgrowth of AML-related mutations, such as RTK pathway genes, and IDH-related mutations (comprising second-site mutations in IDH1 and mutations in IDH2), which were associated with increased 2-HG. These various mechanisms of resistance occurred in isolation or in combination, underscoring the complex biology of treatment resistance.
Methods
Study design
The design of this phase 1, multicenter, open-label, dose-escalation and dose-expansion study of ivosidenib in patients with mIDH1 advanced hematologic malignancies (ClinicalTrials.gov number: NCT02074839) has been reported elsewhere, along with eligibility criteria.17 Patients received ivosidenib orally QD in 28-day cycles (supplemental Methods).
Biomarker analysis methods are reported in the supplemental Methods.
Results
Patient cohort
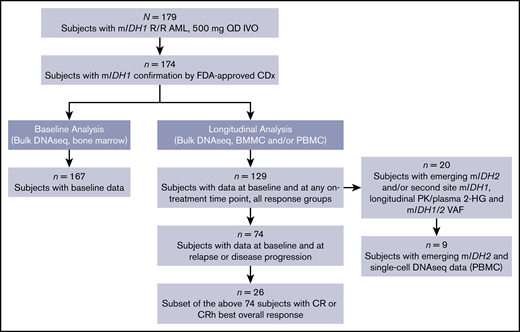
A total of 179 patients with R/R AML were enrolled and treated with a starting dose of 500 mg QD in the first-in-human, open-label, dose-escalation and dose-expansion trial of ivosidenib in patients with mIDH1 advanced hematologic malignancies; follow-up is ongoing.17 The current analysis focused on the subset of 174 patients with R/R AML with mIDH1 confirmation, using the Abbott RealTime IDH1 assay. The baseline demographic characteristics are summarized in supplemental Table 1. The number of patients included in each of the different molecular analyses reported here is shown in Figure 1.

Patient flow diagram summarizing analysis sets. Analyses did not include all patients treated in this population because of several factors, including lack of complete sample availability for all protocol-designated time points and/or suboptimal quantity and/or quality of some samples, resulting in failure to obtain valid data. BMMC, bone marrow mononuclear cell; CDx, companion diagnostic test; DNAseq, DNA sequencing; FDA, US Food and Drug Administration; IDH, isocitrate dehydrogenase; IVO, ivosidenib; PBMC, peripheral blood mononuclear cell; PK, pharmacokinetics; VAF, variant allele frequency.
Patient flow diagram summarizing analysis sets. Analyses did not include all patients treated in this population because of several factors, including lack of complete sample availability for all protocol-designated time points and/or suboptimal quantity and/or quality of some samples, resulting in failure to obtain valid data. BMMC, bone marrow mononuclear cell; CDx, companion diagnostic test; DNAseq, DNA sequencing; FDA, US Food and Drug Administration; IDH, isocitrate dehydrogenase; IVO, ivosidenib; PBMC, peripheral blood mononuclear cell; PK, pharmacokinetics; VAF, variant allele frequency.
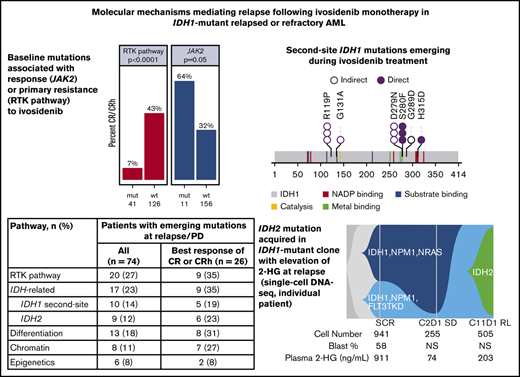
Baseline comutations in RTK pathway genes are associated with primary treatment resistance
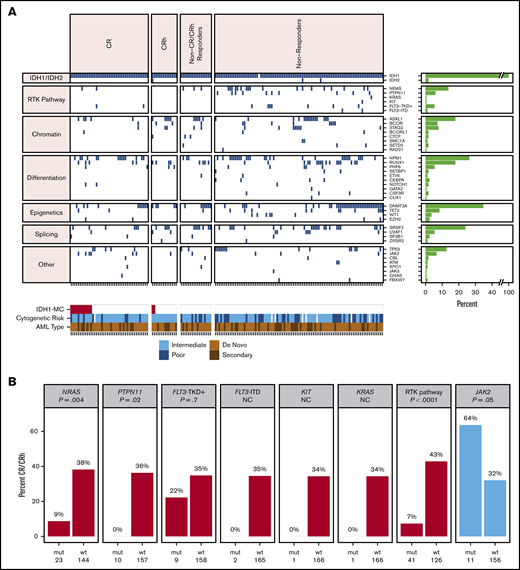
Co-occurring mutation profiling was performed on whole bone marrow by next-generation sequencing (NGS; detection sensitivity, 1%-5%). Per patient co-occurring mutations at baseline (n = 167), organized by best overall response category, are shown in Figure 2A. The subset of 101 patients whose baseline mutational profiles were reported previously17 are all included in this analysis. The most frequent oncogenic co-occurring mutations detected at baseline were DNMT3A (35%), NPM1 (26%), SRSF2 (24%), ASXL1 (18%), RUNX1 (18%), NRAS (14%), and TP53 (13%; see supplemental Table 2 for full list of genes tested). Among 11 patients with mutations in FLT3, 2 were internal tandem duplications, and 9 were in the tyrosine kinase domain (TKD) or juxtamembrane domain point mutations. There were 14 patients with mutations in TET2 at baseline. The mean number of mutations (including mIDH1) as an estimate of genomic complexity was lower in patients achieving CR or CRh responses (2.8) than in patients with non-CR/CRh responses and nonresponders (3.7), with P < .001 (Student t test; supplemental Figure 2). RTK pathway mutations, along with an increased number of mutations, are similarly associated with primary treatment resistance to enasidenib in mIDH2 R/R AML.18
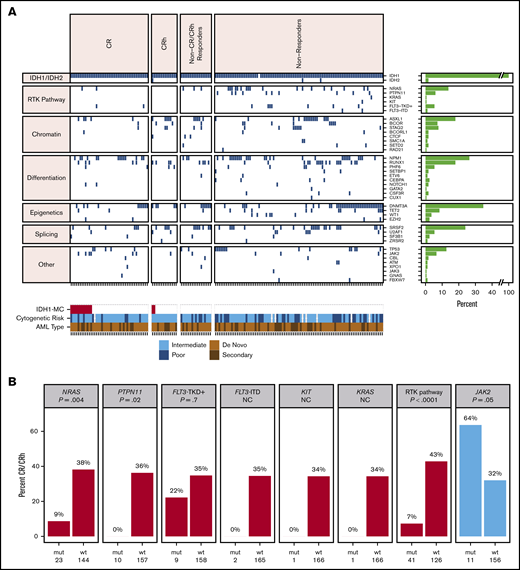
Baseline co-occurring mutation analysis. (A) Heat map showing co-occurring mutations at baseline by best overall response category (NGS; whole bone marrow; n = 167 patients). Merge of dose-escalation and dose-expansion data (VAF cutoff, 1%-5%). Cytogenetic risk was classified according to the National Comprehensive Cancer Network Clinical Practice Guidelines for AML, version 1.2015. The bar graph depicts the frequency of genes with co-occurring mutations. A similar heat map with detected mutations by VAF level is provided in supplemental Figure 1. (B) Association between mutation status and best response. P value is based on Fisher’s exact test examining the association between specific pathway or gene mutations and best overall response of CR/CRh vs non-CR/CRh responders and nonresponders. NC denotes P value not calculated because of small number of patients with mutation. FLT3-TKD+ denotes FLT3-TKD or juxtamembrane domain point mutations. Data source: 167 patients with baseline NGS data from whole bone marrow. IDH1-MC, IDH1 mutation clearance; ITD, internal tandem duplication; mut, mutant; wt, wild-type.
Baseline co-occurring mutation analysis. (A) Heat map showing co-occurring mutations at baseline by best overall response category (NGS; whole bone marrow; n = 167 patients). Merge of dose-escalation and dose-expansion data (VAF cutoff, 1%-5%). Cytogenetic risk was classified according to the National Comprehensive Cancer Network Clinical Practice Guidelines for AML, version 1.2015. The bar graph depicts the frequency of genes with co-occurring mutations. A similar heat map with detected mutations by VAF level is provided in supplemental Figure 1. (B) Association between mutation status and best response. P value is based on Fisher’s exact test examining the association between specific pathway or gene mutations and best overall response of CR/CRh vs non-CR/CRh responders and nonresponders. NC denotes P value not calculated because of small number of patients with mutation. FLT3-TKD+ denotes FLT3-TKD or juxtamembrane domain point mutations. Data source: 167 patients with baseline NGS data from whole bone marrow. IDH1-MC, IDH1 mutation clearance; ITD, internal tandem duplication; mut, mutant; wt, wild-type.
Baseline mutations in the individual RTK pathway genes of NRAS and PTPN11, and in the RTK pathway genes as a group (defined in this analysis as NRAS, KRAS, PTPN11, KIT, and FLT3), were associated with a significantly lower likelihood of achieving a best response of CR or CRh with ivosidenib (P = .004, P = .016, and P < .001, respectively; Fisher’s exact test; Figure 2B), suggesting a role in primary treatment resistance. Nevertheless, CR or CRh was observed in 2 (9%) of 23 patients with baseline NRAS mutations (see single-cell DNA sequencing analyses for further details). Responses were also observed in patients with mutations in genes associated with adverse prognosis,22 including ASXL1 (10 of 30; 33%), RUNX1 (9 of 30; 30%), and TP53 (5 of 21; 24%). In addition, 7 (64%) of 11 patients with baseline JAK2 V617 mutations, which are commonly observed in patients with myeloproliferative neoplasms and secondary AML with a history of myeloproliferative neoplasms, achieved CR (n = 6) or CRh (n = 1; P = .05; Figure 2B). Two patients with baseline FLT3-TKD mutations achieved CR. No significant association was seen between response and mutations in other genes tested (see supplemental Table 2 for full list).
Clinical response is not predicted by the position of mIDH1 within the clonal hierarchy
We previously demonstrated that in 101 patients with mIDH1 R/R AML who received ivosidenib in the phase 1 study, there was no significant difference in baseline mIDH1 variant allele frequency (VAF) between CR or CRh responders and those with other responses or no response.17 The lack of significant correlation was also observed in this larger data set of 167 patients (P = .087; Fisher’s exact test), with mIDH1 VAF in whole bone marrow ranging from 3% to 58%, as measured by NGS (supplemental Figure 3). Response was observed in patients with lower mIDH1 VAF. For example, among 18 patients with mIDH1 VAF of 10% or less, 7 achieved CR or CRh.
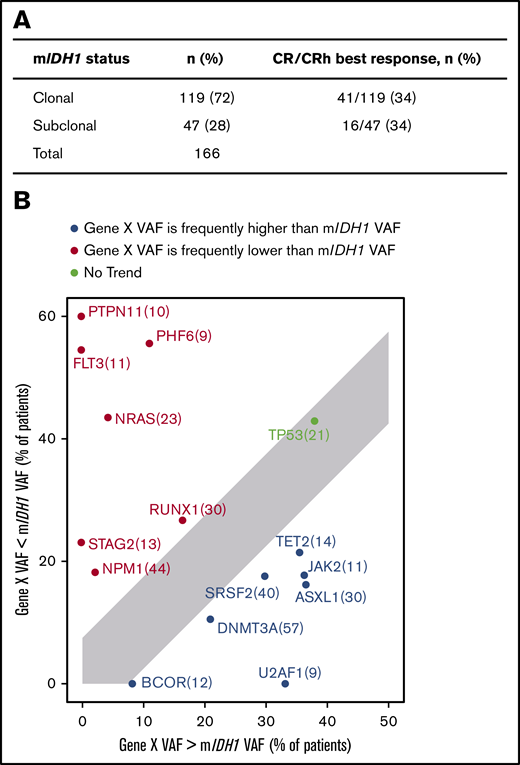
In the present analysis, we investigated the position of mIDH1 in the clonal hierarchy by examining the baseline VAF of co-occurring gene mutations in relation to mIDH1 in bone marrow samples, as well as the association with clinical response. mIDH1 was defined as subclonal in a sample if any comutation VAF was greater than mIDH1 VAF +5%; otherwise, it was defined as clonal. The 5% cutoff was selected based on the distribution of VAFs in replicate samples on this NGS platform,23 and this cutoff was also used by Molenaar et al.24 Details on the manual curation steps that were taken to refine this analysis are provided in the supplemental Methods.
In this data set, mIDH1 was subclonal in 28% of patients and clonal in 72% (Figure 3A). There was no association between clonal or subclonal mIDH1 status and achieving a best response of CR or CRh (P = 1.000; Fisher’s exact test). Mutations in SRSF2, ASXL1, JAK2, TET2, U2AF1, BCOR, and DNMT3A occurred more frequently with higher VAF than mIDH1, whereas mutant NRAS, NPM1, PTPN11, PHF6, RUNX1, STAG2, and FLT3 VAFs were more frequently lower than mIDH1 VAF (Figure 3B). Other groups have reported that mutations in RTK pathway genes generally occur later in clonal evolution than mutations in IDH1/2.25 Baseline mIDH1 VAF in relation to other gene mutations is shown in supplemental Figure 4.
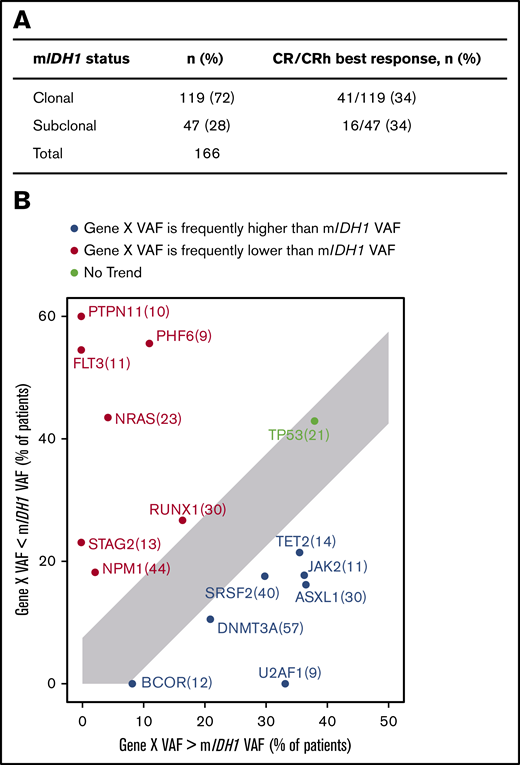
Clonal hierarchy of mIDH1 and co-occurring mutations. (A) Association of clinical response and mIDH1 clonal/subclonal status. mIDH1 was defined as subclonal in a sample if any comutation VAF was greater than mIDH1 VAF +5%; otherwise, it was defined as clonal. See details in the supplemental Methods. (B) Plot showing the tally of how often a co-occurring gene VAF is observed to be lower (red) or higher (blue) than mIDH1 VAF by NGS. Only genes that were mutated in at least 5% of patients are shown. Per-gene VAFs are detailed in supplemental Figure 4. For each gene X, the subset of patients with a mutation in gene X is selected (n in parentheses) and plotted, with the x-axis showing the percentage of patients where gene X VAF > mIDH1 VAF +5% and the y-axis showing the percentage of patients where gene X VAF < mIDH1 VAF −5%.
Clonal hierarchy of mIDH1 and co-occurring mutations. (A) Association of clinical response and mIDH1 clonal/subclonal status. mIDH1 was defined as subclonal in a sample if any comutation VAF was greater than mIDH1 VAF +5%; otherwise, it was defined as clonal. See details in the supplemental Methods. (B) Plot showing the tally of how often a co-occurring gene VAF is observed to be lower (red) or higher (blue) than mIDH1 VAF by NGS. Only genes that were mutated in at least 5% of patients are shown. Per-gene VAFs are detailed in supplemental Figure 4. For each gene X, the subset of patients with a mutation in gene X is selected (n in parentheses) and plotted, with the x-axis showing the percentage of patients where gene X VAF > mIDH1 VAF +5% and the y-axis showing the percentage of patients where gene X VAF < mIDH1 VAF −5%.
Relapse is characterized by multiple mechanisms, including emergence of RTK pathway mutations and 2-HG–restoring mutations
We performed longitudinal bulk NGS on bone marrow mononuclear cells and/or peripheral blood mononuclear cells to characterize the mutational profile both at baseline and at the time of relapse or disease progression. Longitudinal data were available for 129 patients with samples at baseline and at least 1 on-treatment point irrespective of response (see supplemental Table 3 for all mutations newly identified during treatment). Of these patients, data were available for 74 patients at relapse (after CR, CRh, CR with incomplete hematologic recovery, CR with incomplete platelet recovery, or morphologic leukemia-free state) or at disease progression (for patients with best overall response of stable disease or progressive disease), and the mutational profile of these 74 patients at relapse or progression was compared with the mutational profile at baseline (Figure 4A).
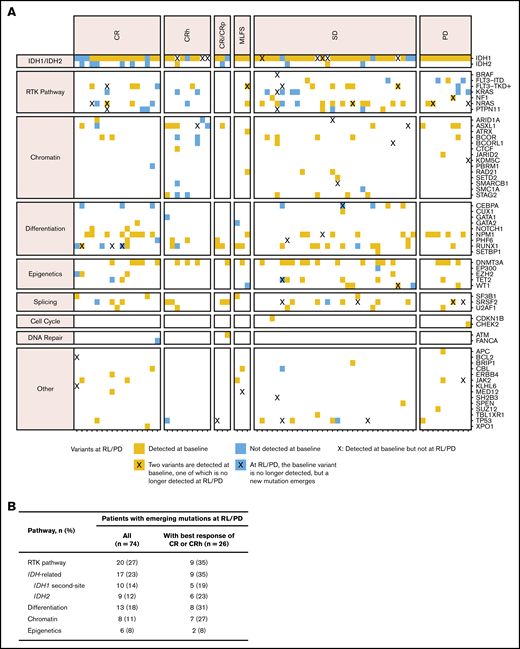
Mutations in genes and pathways detected at relapse and progression. (A) Gene mutations detected at relapse/progressive disaease (RL/PD) in patients with paired baseline and RL/PD data (n = 74), with genes organized by pathway and patients sorted by best overall response. Blue squares indicate new mutations at RL/PD that were not detected at baseline. Blue squares in the IDH1 mutation row indicate emergent second-site IDH1 mutations at time of RL/PD. Orange squares indicate mutations identified at RL/PD that were also detected at baseline. An X indicates that a variant in that gene that was detected at baseline is no longer detected at relapse. (B) Frequency of emergence of mutations by pathway in patients with data at baseline and at RL/PD (n = 74). CRi, complete remission with incomplete hematologic recovery; CRp, complete remission with incomplete platelet recovery; MLFS, morphologic leukemia-free state; SD, stable disease.
Mutations in genes and pathways detected at relapse and progression. (A) Gene mutations detected at relapse/progressive disaease (RL/PD) in patients with paired baseline and RL/PD data (n = 74), with genes organized by pathway and patients sorted by best overall response. Blue squares indicate new mutations at RL/PD that were not detected at baseline. Blue squares in the IDH1 mutation row indicate emergent second-site IDH1 mutations at time of RL/PD. Orange squares indicate mutations identified at RL/PD that were also detected at baseline. An X indicates that a variant in that gene that was detected at baseline is no longer detected at relapse. (B) Frequency of emergence of mutations by pathway in patients with data at baseline and at RL/PD (n = 74). CRi, complete remission with incomplete hematologic recovery; CRp, complete remission with incomplete platelet recovery; MLFS, morphologic leukemia-free state; SD, stable disease.
Mutations identified by bulk NGS at the time of relapse/progression, but not detected at baseline, were categorized as either AML related or IDH related (2-HG restoring). Mutations in RTK pathway genes (NRAS, KRAS, PTPN11, KIT, NF1, BRAF, or FLT3) were identified in 20 (27%) of 74 patients; IDH-related mutations occurred at a similar frequency (17 of 74; 23%) and included second-site mutations in IDH1 (10 of 74; 14%) and canonical mutations in IDH2 (9 of 74; 12%; Figure 4B). IDH2 mutations and second-site IDH1 mutations were not mutually exclusive (ie, both were observed in 2 patients). In the 26 patients with best response of CR/CRh who relapsed, IDH-related mutations emerged at a similar frequency (35%) as RTK pathway genes. Notably, 5 patients had newly identified mutations in both RTK pathway genes and IDH1 and/or IDH2 at relapse/progression.
Bulk NGS data showed that second-site IDH1 mutations and IDH2 mutations were rare at baseline, occurring in 4 (2.4%) of 167 patients with baseline bone marrow DNA profiling. Of these, 1 patient had 2 second-site mutations in IDH1 (R119P and G131A) and had previously received treatment with the mIDH1 inhibitor IDH305. Three (1.8%) patients had mIDH2-R140 at screening. In contrast, 41 (25%) of 167 patients had RTK pathway mutations at screening.
The second-site IDH1 mutations that emerged during treatment with ivosidenib monotherapy included the previously described mIDH1-S280F in addition to 5 novel, previously unidentified mutations (Figure 5A; supplemental Table 4). Although no dominant second-site IDH1 mutation was detected in this data set, the second-site IDH1 mutations clustered into 2 subregions of the IDH1 protein. Notably, all IDH2 mutations that emerged during treatment were R140Q in this R/R AML data set (supplemental Table 4). However, emergence of IDH2-R172K was observed in 1 patient with newly diagnosed AML in the phase 1 study (data on file).
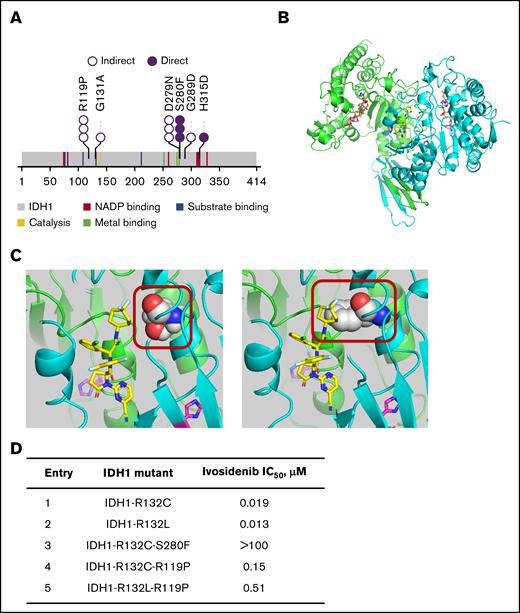
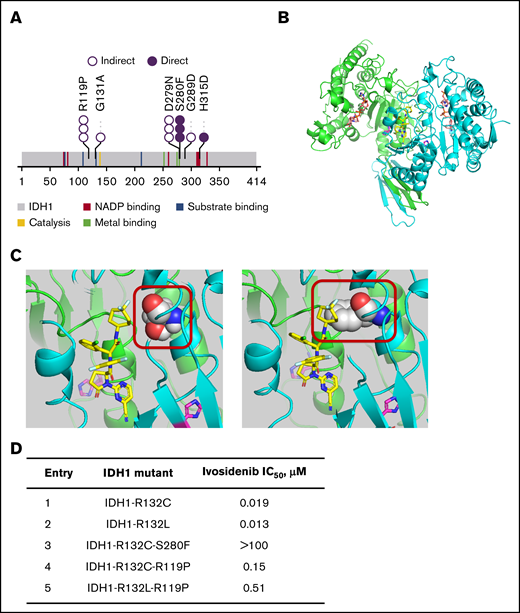
2-HG–restoring mutations emerging at relapse or disease progression. (A) Second-site IDH1 mutations emerging during ivosidenib treatment (n = 20). Each circle represents a single patient with detection of the mutation. Direct denotes that the mutation makes contact with the ivosidenib or cofactor (NADPH) binding pocket by 3-dimensional modeling. Indirect denotes that it induces structural changes in vicinity of ivosidenib or cofactor binding pockets (hypothesized). Novel mutations (not previously published) are mostly the indirect type. (B) Crystal structure of IDH1-R132H in the inactive, open conformation. The cocrystal structure of IDH1-R132H with inhibitor 20a (PBD accession no. 5L57)40 was obtained at 2.7 Ao resolution and used to develop the model for the ivosidenib analog AGI-14686 (supplemental Methods). The homodimeric protein structure is shown with the inhibitor (AGI-14686) in yellow, occupying the middle of the tetra-helical domain. The protein monomers are shown in green and cyan, the mutated 132H residue in magenta and cofactor NADPH in orange. (C) The mIDH1-AGI-14686 binding model for the IDH1-R132H-S280F mutation places the di-F cyclopentane of AGI-14686 near Ser280 (left). Mutation of Ser280 to Phe (right) will create a steric interference with ivosidenib (or its analogs), and thus will no longer bind to mIDH1. The mIDH1 protein is depicted in cartoon (green and blue for each monomer), the ivosidenib analog AGI-14686 in yellow sticks, and residue 280 in spheres (Ser in the left panel, Phe in the right panel). R132H is shown in magenta. (D) Biochemical 50% inhibitory concentration (IC50) for second-site IDH1 mutations. We expressed various combinations of IDH1 mutants with second-site mutations and examined biochemically whether they were still inhibited by ivosidenib.
2-HG–restoring mutations emerging at relapse or disease progression. (A) Second-site IDH1 mutations emerging during ivosidenib treatment (n = 20). Each circle represents a single patient with detection of the mutation. Direct denotes that the mutation makes contact with the ivosidenib or cofactor (NADPH) binding pocket by 3-dimensional modeling. Indirect denotes that it induces structural changes in vicinity of ivosidenib or cofactor binding pockets (hypothesized). Novel mutations (not previously published) are mostly the indirect type. (B) Crystal structure of IDH1-R132H in the inactive, open conformation. The cocrystal structure of IDH1-R132H with inhibitor 20a (PBD accession no. 5L57)40 was obtained at 2.7 Ao resolution and used to develop the model for the ivosidenib analog AGI-14686 (supplemental Methods). The homodimeric protein structure is shown with the inhibitor (AGI-14686) in yellow, occupying the middle of the tetra-helical domain. The protein monomers are shown in green and cyan, the mutated 132H residue in magenta and cofactor NADPH in orange. (C) The mIDH1-AGI-14686 binding model for the IDH1-R132H-S280F mutation places the di-F cyclopentane of AGI-14686 near Ser280 (left). Mutation of Ser280 to Phe (right) will create a steric interference with ivosidenib (or its analogs), and thus will no longer bind to mIDH1. The mIDH1 protein is depicted in cartoon (green and blue for each monomer), the ivosidenib analog AGI-14686 in yellow sticks, and residue 280 in spheres (Ser in the left panel, Phe in the right panel). R132H is shown in magenta. (D) Biochemical 50% inhibitory concentration (IC50) for second-site IDH1 mutations. We expressed various combinations of IDH1 mutants with second-site mutations and examined biochemically whether they were still inhibited by ivosidenib.
The majority of the newly identified second-site IDH1 and IDH2 mutations (15 of 16 with available 2-HG data at relapse) were associated with a concurrent increase in 2-HG levels, and are thus termed 2-HG–restoring mutations (see details in supplemental Figure 5).
Structural and biochemical characterization of second-site IDH1 mutations
IDH1/2 mutations in AML that lead to the generation of 2-HG localize to the active sites of the enzymes, on residues R132 in IDH1 and R140 or R172 in IDH2.1,2 Ivosidenib and enasidenib bind to allosteric sites, thus stabilizing the open, inactive conformation of the enzymes.14 The crystal structure model of IDH1-R132H in open conformation (when ivosidenib or an analog is bound) is shown in Figure 5B, indicating the active site with the cofactor NADPH and the location of the inhibitor in the allosteric, tetra-helical binding site. We performed 3-dimensional modeling of second-site IDH1 mutations using an analog of ivosidenib (AGI-14686; supplemental Figure 6A) to determine whether ivosidenib binding would be affected by these mutations. The model (see supplemental Methods for development) placed AGI-14686 in the tetra-helical center of the dimeric IDH1-R132H protein and allowed us to observe the second-site IDH1 mutations, all of which could be predicted to affect ivosidenib and/or cofactor binding. Two groups of mutations were postulated: those directly affecting the drug-binding pocket or NADPH cofactor/active sites and those that indirectly affect either the ivosidenib binding site or the cofactor/substrate site. The binding model for the IDH1-R132H-S280F mutation (Figure 5C) places the di-F cyclopentane group of the inhibitor near Ser280 within the binding pocket of the protein. Mutation of Ser280 to Phe creates a steric interference with ivosidenib (and its analogs), and thus prevents binding. The binding model for the IDH1-R119P mutation is shown in supplemental Figure 6B. This mutation is located on the protein loops covering the ivosidenib binding site; the proline-induced changes in backbone conformation and rigidity of the mIDH1 protein are predicted to alter the kinetics of ivosidenib binding. Modeling of additional second-site IDH1 mutations predicts that they would affect ivosidenib or NADPH binding either directly or indirectly (supplemental Figure 6C-F). Biochemical characterization of both IDH1-S280F and IDH1-R119P second-site mutations confirms loss of ivosidenib inhibitory potency (Figure 5D). At steady state, the free maximum and average plasma concentrations of ivosidenib are found to be 0.94 and 0.71 μM, respectively, given the mean percentage plasma protein binding of 91.6%.15,26 This level exceeds the IC50 of pathogenic IDH1-R132 mutants, but is below that of the IDH1-R132C-S280F double mutant, and is within close range of that of the IDH1-R132C/L-R119P mutant in the in vitro setting, suggesting that the level of ivosidenib in plasma is not sufficient to inhibit the second-site mutation. There were no differences in plasma 2-HG elevations at relapse or progression, irrespective of the nature of the second-site mutations (supplemental Figure 5).
Single-cell DNA sequencing analyses reveal complex polyclonal resistance mechanisms
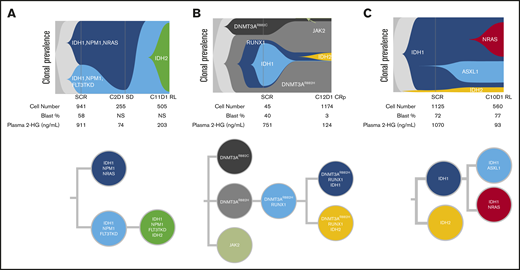
To assess clonal architecture heterogeneity and dynamics in the relapse setting at single-cell resolution with greater precision than is afforded by NGS, we conducted single-cell DNA sequencing analyses (Tapestri Platform; Mission Bio; 19-gene AML panel) on matched baseline and relapse peripheral blood mononuclear cell sample pairs from 9 patients who had a response to ivosidenib followed by emergence of mIDH2. The polyclonal relapse mechanisms were grouped into the following 3 categories: 6 of 9 patients had no detectable mIDH2 at baseline, but mIDH2 was identified in the same clone as mIDH1 at relapse (Figure 6A; elevated 2-HG observed at relapse); 1 of 9 patients had no detectable mIDH2 at baseline, but mIDH2 was identified in a separate clone than mIDH1 at relapse (isoform switching; Figure 6B); and 2 of 9 patients had detectable mIDH2 at baseline in a separate clone than mIDH1 (Figure 6C). Figure 6A represents the clonal architecture of AML in a patient with 2 distinct mIDH1 clones at baseline: 1 harboring NPM1/NRAS comutations and the other harboring NPM1/FLT3-TKD comutations. After ivosidenib treatment, the IDH1/NPM1/NRAS clone was no longer detected. Reduction in the IDH1/NPM1/FLT3-TKD clone was observed at cycle 2 day 1, but it ultimately expanded at relapse with the acquisition of mIDH2. Figure 6B demonstrates the clonal architecture in a patient without mIDH2 at baseline. Emergence of mIDH2 in a separate clone than mIDH1 was observed at cycle 12 day 1 with elevated 2-HG. Figure 6C shows a case in which mIDH2 was present at baseline by single-cell DNA sequencing, but not detected by bulk NGS, with relapse most likely a result of acquisition of NRAS mutation in the mIDH1 clone and/or expansion of the IDH1/ASXL1 clone, as 2-HG remained low at relapse. These findings show that relapse occurs as a result of expansion of multiple parallel clones spanning different pathways. Furthermore, phylogenetic tree reconstruction from clonotypes indicated patterns of both branching and linear clonal evolution. Additional cases are provided in supplemental Figure 7.
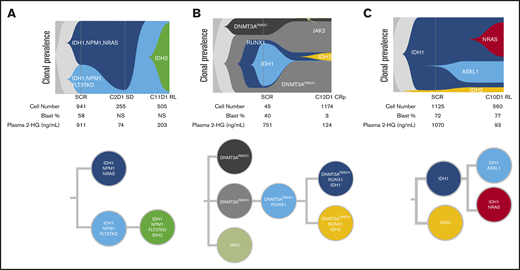
Polyclonal relapse mechanisms. Single-cell analysis showing clonal evolution in individual patients. (A) mIDH2 acquired in the same clone as mIDH1, with elevation of 2-HG at relapse. Two distinct mIDH1 clones were present at baseline: 1 harboring NPM1/NRAS and the other harboring NPM1/FLT3-TKD comutations. After ivosidenib treatment, the IDH1/NPM1/NRAS clone was no longer detected. Reduction in the IDH1/NPM1/FLT3-TKD clone was observed at cycle 2 day 1, but it ultimately expanded at relapse with the acquisition of mIDH2. (B) mIDH2 was not detected at baseline, but emergence of mIDH2 in a separate clone than mIDH1 was observed at cycle 12 day 1, with elevation of 2-HG. (C) mIDH2 was present at baseline (though not detected by bulk NGS) in a distinct clone to mIDH1, with clinical relapse associated with detection of an NRAS clone. In this case of NRAS-driven clinical relapse, the 2-HG levels remained low at relapse. Additional details on these cases shown in supplemental Figure 7. NS, not specified.
Polyclonal relapse mechanisms. Single-cell analysis showing clonal evolution in individual patients. (A) mIDH2 acquired in the same clone as mIDH1, with elevation of 2-HG at relapse. Two distinct mIDH1 clones were present at baseline: 1 harboring NPM1/NRAS and the other harboring NPM1/FLT3-TKD comutations. After ivosidenib treatment, the IDH1/NPM1/NRAS clone was no longer detected. Reduction in the IDH1/NPM1/FLT3-TKD clone was observed at cycle 2 day 1, but it ultimately expanded at relapse with the acquisition of mIDH2. (B) mIDH2 was not detected at baseline, but emergence of mIDH2 in a separate clone than mIDH1 was observed at cycle 12 day 1, with elevation of 2-HG. (C) mIDH2 was present at baseline (though not detected by bulk NGS) in a distinct clone to mIDH1, with clinical relapse associated with detection of an NRAS clone. In this case of NRAS-driven clinical relapse, the 2-HG levels remained low at relapse. Additional details on these cases shown in supplemental Figure 7. NS, not specified.
Discussion
We report a comprehensive analysis of primary and secondary mechanisms of resistance to ivosidenib monotherapy in patients with mIDH1 R/R AML. The bulk NGS and single-cell sequencing results highlight the polyclonal nature of AML and illustrate the complex cellular and molecular mechanisms that drive clinical relapse in the setting of ivosidenib treatment. Mutations in RTK pathway genes were associated with primary treatment resistance in this population and were a frequent secondary resistance mechanism. Interestingly, KRAS mutations were more frequent at relapse/progression than at baseline. In addition, 2-HG restoration as a result of mutations preventing drug/cofactor binding and/or emergence of an IDH2 mutation represents a mechanism of secondary resistance, emerging at a similar frequency as RTK pathway mutations.
We previously reported an association between baseline mutations in RTK pathway genes and primary resistance to ivosidenib.17 With a larger data set, we now observe significant associations between single RTK pathway genes (NRAS, PTPN11) and best overall response. This finding is consistent with previous work showing an association between NRAS mutation and a lower likelihood of response to enasidenib in patients with mIDH2 R/R AML.18 However, in the present study, CR was observed in 2 of 23 patients with NRAS mutations, indicating that these mutations at baseline do not confer universal resistance. This is supported by our single-cell DNA sequencing analyses, which revealed an example of a clone harboring an NRAS mutation (along with IDH1 and NPM1 comutations) that was no longer detected after ivosidenib treatment.
The results from this large data set also extend the findings of earlier isolated case studies, which revealed 2-HG–restoring mutations (including isoform switching and the emergence of mIDH1-S280F) to be mechanisms involved in secondary resistance to ivosidenib.20,21 In our data set, the frequent observation of 2-HG–restoring mutations at relapse after CR or CRh by virtue of mutations in IDH2 and second-site mutations in IDH1 (9 of 26 patients; 35%) highlights the key role of 2-HG production in mIDH1 AML. Further, single-cell DNA sequencing analyses revealed multiple instances in which mIDH2 was not detectable at baseline, but emerged within the same clone as mIDH1, suggesting the existence of a selective pressure to reestablish 2-HG even in the presence of other AML driver mutations. In other single-cell DNA sequencing profiles, mIDH2 was observed in an independent subclone, with an unknown effect on coexisting mIDH1 clones or clinical disease features. It is possible that 2-HG production and release by these mIDH2 subclones can affect all leukemic and/or stromal cells in a paracrine manner. Reports indicate that 2-HG can enter specific cell types (eg, tumor-associated immune cells), potentially leading to immunosuppression.27-30 The emergence of multiple second-site IDH1 mutations with no dominant mutation supports the use of mIDH1 inhibitors in combination with other therapies to reduce the likelihood of resistance.
Although 2-HG–restoring mutations are a major pathway of secondary resistance, other 2-HG-independent pathways are important and may be dominant over 2-HG restoration in some cases. McMahon et al.31 observed that reactivation of RTK pathway signaling was a major mechanism of secondary resistance to the FLT3 inhibitor gilteritinib, including the emergence of RAS mutations in FLT3-mutated subclones. We show that in addition to being associated with primary resistance, RTK pathway mutations are also associated with secondary resistance to ivosidenib monotherapy in this study population. Among patients who relapsed after achieving CR or CRh, RTK pathway mutations emerged at relapse in 9 (35%) of 26 patients. The biological processes explaining the association between RTK pathway mutations and both primary and secondary resistance to ivosidenib are not currently understood. Several hypotheses are possible. First, it may be that the proliferative and prosurvival effects of RTK pathway activation are sufficiently strong oncogenic signals to reduce dependency on 2-HG. Second, it is possible that RTK pathway-activating mutations contribute to a differentiation block that remains enforced after initiation of ivosidenib treatment. Some reports have shown that FLT3 inhibitors induce granulocytic differentiation and differentiation syndrome symptoms in some patients with FLT3-mutated AML.32,33 Thus, the combination of mIDH inhibitors with RTK pathway inhibitors, including FLT3 inhibitors, may present a rational treatment strategy. A third hypothesis is that IDH1/2 mutations result in activation of some components of RTK signaling, which would not be reversed by ivosidenib in cases with co-occurring RTK pathway mutations. In glioma, IDH1 mutations were reported to increase platelet-derived growth factor receptor α expression levels by methylation and inactivation of a nearby CCCTC-binding factor insulator site.34 Others have observed increased AKT/mTOR signaling in mIDH1 cells.35-37
An interesting observation is that RTK pathway mutations are associated with ivosidenib resistance despite being predominantly subclonal to mIDH1. Our longitudinal data show that RTK pathway mutation VAFs usually increase at relapse, but they do not generally become the dominant clone. Analogously, the FLT3 inhibitor midostaurin has shown efficacy in patients with a low mFLT3 allelic ratio.38 It is possible that RTK pathway signaling in subclones can influence all leukemia cells, including neighboring cells lacking RTK pathway mutations, via paracrine effects that may include cytokines and/or stromal cells. Alternatively, RTK pathway mutations may be correlated with another factor that is causally related to resistance (eg, comutational burden), and the observed association is an epiphenomenon. In this data set, the number of comutations is associated with clinical response, but not as strongly as RTK pathway mutations.
JAK2 mutations were associated with a high CR/CRh rate (64%), with the caveat of the limited number of patients (n = 11). Although JAK2 mutations are often classed together with other mutations affecting MAPK pathway signaling, their different pattern of response to ivosidenib treatment points to the distinct biology of JAK2 mutations, such as STAT pathway activation, frequent ancestral status during clonal evolution, and association with prior myeloproliferative neoplasm. The number of patients is insufficient to determine whether de novo or secondary disease has a prognostic role in the context of JAK2 mutation. Data on additional patients with JAK2 mutations are needed to gain a more robust picture of this patient subset.
We found no significant association between clonal hierarchy and response to ivosidenib therapy, as assessed by the VAF of mIDH1 in relation to the VAF of other co-occurring mutations. This demonstrates that mIDH1 inhibition can be efficacious even in patients with a subclonal IDH1 mutation.
Single-cell DNA sequencing analyses uncovered bona fide co-occurring mutations at single-cell resolution, including genes of the RTK pathway (eg, NRAS, KRAS, PTPN11, FLT3), transcription factors (RUNX1), chromatin/epigenetic regulators (DNMT3A, ASXL1), and splicing factors (U2AF, SF3B1). These co-occurring mutations indicate functional interplay between these genes and mIDH1, and reflect a more complex role of mIDH1 during leukemogenesis or maintenance of mIDH1 AML, such as cooperation with the constitutively activated RTK pathway to promote cell proliferation, and/or cooperation with chromatin/epigenetic regulators and transcription regulators to block cell differentiation. In the majority of cases, emerging mIDH2 is found within the same cell as mIDH1, indicating that mIDH1 AML is highly “addicted” to mIDH and tends toward reactivating the capability to produce 2-HG. Moreover, patterns of linear and branching clonal evolution were observed with single-cell DNA sequencing, highlighting additional complexity. Single-cell DNA sequencing proved to be a powerful tool in delineating molecular outcomes for patients with mIDH1 R/R AML.
Our findings highlight the interplay among baseline mutation profiles, response, and clonal evolution during ivosidenib therapy. The finding that the mechanism of resistance to ivosidenib is complex and polyclonal has implications for mIDH1/2 inhibitor treatment strategies, and supports the use of combination therapies or sequential treatment modifications at early relapse before overt clinical progression, rather than monotherapy with mIDH1/2 inhibitors. It will also be important to understand whether or not these patterns of resistance are replicated with combination therapies. Thorough cataloging of resistance mechanisms to targeted therapies has proven invaluable in the development of next-generation therapies, such as second- and third-generation inhibitors of BCR-ABL, EGFR, and ALK, and in the development of efficacious combination therapies such as BRAF-MEK dual inhibition in melanoma.39 In patients with mIDH1 AML with FLT3, RAS, or other signaling mutations, dual treatment with kinase inhibitors (FLT3 or MEK) may improve responses. In addition, dual pharmacologic inhibition of both mIDH1 and mIDH2 could potentially alleviate 2-HG restoration caused by isoform switching. Because individual patients often show multiple resistance mechanisms at relapse, combination of ivosidenib with nontargeted agents, such as intensive chemotherapy/cytotoxic therapies, hypomethylating agents, and venetoclax (BCL-2 inhibitor), may improve responses and decrease the likelihood of relapse.
Investigators interested in data sharing and collaboration should contact the corresponding author, Bin Wu, at bin.wu@agios.com.
Acknowledgments
The authors thank Pedro Mendez and Robert Durruthy-Durruthy (Mission Bio) for technical support of single-cell DNA sequencing analyses.
Medical writing assistance was provided by Helen Varley of Excel Medical Affairs, Horsham, United Kingdom, and funded by Agios.
Authorship
Contribution: S.C., H.W., C.D.D., E.M.S., S.d.B., G.J.R., J.K.A., A.S.M., J.W., D.A.P., A.T.F., M.S.T., H.M.K., R.M.S., L.Q., B.N., M.G., W.L., K.M., C.B., S.A.B., E.C.A., and B.W. participated in the interpretation of the translational data; S.C., H.W., E.C.A., and B.W. designed the translational studies, participated in the collection and analysis of the translational data, and drafted the manuscript; C.D.D., E.M.S., S.d.B., G.J.R., J.K.A., A.S.M., J.W., D.A.P., A.T.F., M.S.T., H.M.K., and R.M.S. participated in recruitment of patients and collection of data; Z.K. and L.D. provided data and interpretation of biochemical and structural characterization of second-site IDH1 mutations; P.N. participated in clinical sample management; G.L. oversaw the generation and collection of 2-HG data; V.Z. and H.L. participated in the statistical analyses of the translational data; H.L., M.G., and E.C.A. designed the clinical study and developed the protocol; B.W. developed and oversaw the translational science plan; and all authors contributed to the review and revision of the manuscript for important intellectual content and approved the final version for submission.
Conflict-of-interest disclosure: S.C., H.W., Z.K., L.D., B.N., P.N., G.L., V.Z., H.L., M.G., W.L., K.M., C.B., S.A.B., and B.W. are employees of and have equity ownership in Agios. C.D.D. has received honoraria and research funding from AbbVie, Agios, Celgene, and Daiichi Sankyo, and honoraria from MedImmune. E.M.S. has membership on the board of directors or advisory committee for Agios, Astellas, Celgene, Daiichi Sankyo, Genentech, Novartis, PTC Therapeutics, and Syros, and has acted as consultant for Agios. S.d.B. has acted as consultant for AbbVie, Agios, Astellas, Bayer, Celgene, Daiichi Sankyo, Forma, Janssen, Novartis, Pfizer, Pierre Fabre, Servier, and Syros; has received research funding from Agios and Forma; and is on a speaker bureau for Celgene. G.J.R. has acted as consultant or member of a data and safety monitoring committee for AbbVie, Actinium, Agios, Amphivena, Argenx, Astellas, Astex, Bayer, Celgene, Celltrion, Daiichi Sankyo, Eisai, Janssen, Jazz, MEI Pharma, Novartis, Orsenix, Otsuka, Pfizer, Roche/Genentech, Sandoz, Takeda, and Trovagene, and has received research funding from Cellectis. J.K.A. has acted as a consultant to AbbVie, Agios, Cancer Expert Now, Daiichi Sankyo, Glycomimetics, Novartis, and Theradex; is on a speaker bureau for France Foundation, PeerView, and prIME Oncology; and has received research funding to their institution from Agios, Astellas, Boehringer Ingelheim, Celgene, Fujifilm, and Genentech. A.S.M. has acted as a consultant to AbbVie, Agios, Astellas, Jazz, and PTC Therapeutics. J.W. has membership on the board of directors or advisory committee for Celgene and Pfizer, has received research funding from Takeda, and has acted as consultant and is on a speaker bureau for Jazz. D.A.P. has acted as a consultant to AbbVie, Agios, Argenx, Celgene, Celyad, Curis, Pfizer, and Servier, and has received research funding from Agios and Pfizer. A.T.F. has acted as a consultant for AbbVie, Agios, Amphivena, Astellas, Celgene, Daiichi Sankyo, Forty Seven, Jazz, Kite, NewLink Genetics, Novartis, PTC Therapeutics, Takeda, and Trovagene. M.S.T. has received research funding from AbbVie, ADC Therapeutics, BioSight, Cellerant, and Orsenix; has acted as a consultant and has membership on the board of directors or advisory committee for AbbVie, BioLineRx, Daiichi Sankyo, Delta Fly Pharma, Jazz, KAHR, Nohla, Oncolyze, Orsenix, Rigel, and Tetraphase; and holds patents or royalties with UpToDate. H.M.K. has received research funding from Ariad, Astex, Bristol Myers Squibb, Cyclacel, Daiichi Sankyo, Immunogen, Jazz, Novartis, and Pfizer and honorarium from Actinium, Immunogen, Pfizer, and Takeda. R.M.S. has acted as a consultant for AbbVie, Actinium, Agios, Amgen, Argenx, Arog, Astellas, AstraZeneca, BioLineRx, Celgene, Cornerstone Biopharma, Fujifilm, Jazz, Merck, Novartis, Ono, Orsenix, Otsuka, Pfizer, Sumitomo, and Trovagene; has received research funding from Agios, Arog, and Novartis; and is a member of a data and safety monitoring board for Argenx, Celgene, and Takeda Oncology. L.Q. has received research funding from Agios and Celgene and is on a speaker bureau for Celgene. E.C.A. was an employee of and had equity ownership in Agios at the time of the study and is currently an employee of Aprea Therapeutics.
Correspondence: Bin Wu, Agios, Clinical Biomarkers, 88 Sidney St, Cambridge, MA 02139; e-mail: bin.wu@agios.com.