Abstract
Central nervous system (CNS) complications are among the most common, devastating sequelae of sickle cell disease (SCD) occurring throughout the lifespan.
These evidence-based guidelines of the American Society of Hematology are intended to support the SCD community in decisions about prevention, diagnosis, and treatment of the most common neurological morbidities in SCD.
The Mayo Evidence-Based Practice Research Program supported the guideline development process, including updating or performing systematic evidence reviews. The panel used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, including GRADE evidence-to-decision frameworks, to assess evidence and make recommendations.
The panel placed a higher value on maintaining cognitive function than on being alive with significantly less than baseline cognitive function. The panel developed 19 recommendations with evidence-based strategies to prevent, diagnose, and treat CNS complications of SCD in low-middle– and high-income settings.
Three of 19 recommendations immediately impact clinical care. These recommendations include: use of transcranial Doppler ultrasound screening and hydroxyurea for primary stroke prevention in children with hemoglobin SS (HbSS) and hemoglobin Sβ0 (HbSβ0) thalassemia living in low-middle–income settings; surveillance for developmental delay, cognitive impairments, and neurodevelopmental disorders in children; and use of magnetic resonance imaging of the brain without sedation to detect silent cerebral infarcts at least once in early-school-age children and once in adults with HbSS or HbSβ0 thalassemia. Individuals with SCD, their family members, and clinicians should become aware of and implement these recommendations to reduce the burden of CNS complications in children and adults with SCD.
Summary of recommendations
Background
Stroke, silent cerebral infarcts (silent strokes), and cognitive morbidity are the most common permanent sequelae of sickle cell disease (SCD) in children and adults. Prior to 1990 in the United States, a large prospective cohort study demonstrated that by 40 years of age, ∼20% and ∼10% of adults with phenotype hemoglobin SS (HbSS) or hemoglobin SC (HbSC) had a cerebrovascular accident, respectively (Figure 1).1 Over the last decades, screening with transcranial Doppler ultrasound (TCD) and treatment with regular blood transfusion in children with abnormal TCD velocities may result in a 10-fold decrease in the prevalence of strokes in children with HbSS and hemoglobin Sβ0 (HbSβ0) thalassemia living in high-income settings.2

High incidence of cerebrovascular accidents in children and adults with SCD prior to the onset of primary stroke prevention with transcranial Doppler (TCD) and regular blood transfusion or hydroxyurea. Data from the 3647 children and adults with SCD followed prospectively from 1978 to 1988 in the Cooperative Study for Sickle Cell Disease cohort. The incidence rates of cerebrovascular accidents (CVA) were used to determine CVA-free survival curves. The estimated age at first CVA was significantly different for individuals with HbSS (SS) and HbSC (SC; P < .001). Chances of having a first CVA by 20 years of age, 30 years of age, and 45 years of age were estimated at 11%, 15%, and 24%, respectively, for HbSS patients and 2%, 4%, and 10%, respectively, for those with HbSC.1 Reprinted from Ohene-Frempong et al with permission.1
High incidence of cerebrovascular accidents in children and adults with SCD prior to the onset of primary stroke prevention with transcranial Doppler (TCD) and regular blood transfusion or hydroxyurea. Data from the 3647 children and adults with SCD followed prospectively from 1978 to 1988 in the Cooperative Study for Sickle Cell Disease cohort. The incidence rates of cerebrovascular accidents (CVA) were used to determine CVA-free survival curves. The estimated age at first CVA was significantly different for individuals with HbSS (SS) and HbSC (SC; P < .001). Chances of having a first CVA by 20 years of age, 30 years of age, and 45 years of age were estimated at 11%, 15%, and 24%, respectively, for HbSS patients and 2%, 4%, and 10%, respectively, for those with HbSC.1 Reprinted from Ohene-Frempong et al with permission.1
The most common cause of permanent neurological injury in children and adults with HbSS and HbSβ0 thalassemia is a silent cerebral infarct, occurring in ∼39% of children by 18 years3 of age and >50% of adults by 30 years of age (Figure 2).4 Silent cerebral infarcts require magnetic resonance imaging (MRI) to detect and a formal neurological examination to exclude the presence of an overt stroke.5 Both stroke and silent cerebral infarcts are associated with significant cognitive impairments6 that may significantly alter educational attainment, employment status, and quality of life.

Prevalence of SCIs in unselected children and adults with HbSS or HbSβ0thalassemia. Silent cerebral infarcts (SCI) were detected with MRI of the brain in children and adults with HbSS or HbSβ0 thalassemia and no history of focal neurological deficits or strokes. Each point represents distinct cross-sectional studies in children and adults with HbSS or HbSβ0 thalassemia.4 Reprinted from Kassim et al with permission.4
Prevalence of SCIs in unselected children and adults with HbSS or HbSβ0thalassemia. Silent cerebral infarcts (SCI) were detected with MRI of the brain in children and adults with HbSS or HbSβ0 thalassemia and no history of focal neurological deficits or strokes. Each point represents distinct cross-sectional studies in children and adults with HbSS or HbSβ0 thalassemia.4 Reprinted from Kassim et al with permission.4
One of the panel’s chief objectives was to establish guidelines applicable to the >95% of children born with HbSS and HbSβ0 thalassemia in low-middle–income countries. Conservatively, <5% of all children born in the world with HbSS or HbSβ0 thalassemia live in the United States and Europe.7 This estimate is based on the consensus that there are ∼300 000 children annually born with HbSS or HbSβ0 thalassemia in the world,7 coupled with the evidence that there are a total of 1971, 334, and 353 infants born with SCD per year in the United States, United Kingdom, and France, respectively.8-10 Children and adults with HbSS living in low-middle–income settings without resources to implement evidence-based strategies for primary and secondary stroke prevention have high lifetime stroke risk,11 a risk similar to that documented in the 1990s among children with HbSS in high-income settings prior to the implementation of TCD screening and regular blood transfusion therapy.1
The panel recognized that most of the recommendations would be difficult to implement in low-middle–income settings where the majority of children and adults with SCD live; however, when applicable, the panel provided recommendations for these regions based on the best available evidence. Major barriers to transferring knowledge about neurological injury prevention from high-income to low-middle–income settings include the low number of TCD machines and MRI scanners to detect central nervous system (CNS) pathology; the lack of sufficiently trained health care providers to perform TCD; the low number of physicians with expertise in hematology, neurology, and neuroradiology; and access to therapy for primary and secondary stroke prevention.
Given the high prevalence of neurological morbidity in children and adults with SCD, a critical component of the recommendations includes involving individuals with SCD and their families in medical decision-making. Families must be informed of (1) the presence of neurological morbidity as diagnosed through imaging, cognitive testing, or both; (2) the increased risk for future neurological morbidity; and (3) plausible disease-modifying therapies that may attenuate or abate risks of further neurological injury without data from phase 3 randomized controlled trials. The panel developed 19 recommendations with evidence-based strategies to prevent, screen, and treat CNS complications of SCD in low-middle– and high-income settings.
These guidelines are based on original and updated systematic reviews of evidence conducted under the direction of the Mayo Evidence-Based Practice Research Program. The panel followed best practices for guideline development recommended by the Institute of Medicine and the Guidelines International Network (GIN).12-15 The panel used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach16-21 to assess the certainty in the evidence and formulate recommendations.
Interpretation of strong and conditional recommendations
The strength of a recommendation is expressed as either strong (“the guideline panel recommends…”) or conditional (“the guideline panel suggests…”) and has the following interpretation:
Strong recommendation
For patients: Most individuals in this situation would want the recommended course of action, and only a small proportion would not.
For clinicians: Most individuals should follow the recommended course of action. Formal decision aids are not likely to be needed to help individual patients make decisions consistent with their values and preferences.
For policy makers: The recommendation can be adopted as policy in most situations. Adherence to this recommendation according to the guideline could be used as a quality criterion or performance indicator.
For researchers: The recommendation is supported by credible research or other convincing judgments that make additional research unlikely to alter the recommendation. On occasion, a strong recommendation is based on low or very low certainty in the evidence. In such instances, further research may provide important information that alters the recommendations.
Conditional recommendation
For patients: The majority of individuals in this situation would want the suggested course of action, but many would not. Decision aids may be useful in helping patients to make decisions consistent with their individual risks, values, and preferences.
For clinicians: Different choices will be appropriate for individual patients, and clinicians must help each patient arrive at a management decision consistent with the patient's values and preferences. Decision aids may be useful in helping individuals make decisions consistent with their individual risks, values, and preferences.
For policy makers: Policy-making will require substantial debate and involvement of various stakeholders. Performance measures about the suggested course of action should focus on whether an appropriate decision-making process is duly documented.
For researchers: This recommendation is likely to be strengthened (for future updates or adaptation) by additional research. An evaluation of the conditions and criteria (and the related judgments, research evidence, and additional considerations) that determined the conditional (rather than strong) recommendation will help identify possible research gaps.
Interpretation of good practice statements
As described by the GRADE Guidance Group, good practice statements endorse interventions or practices that the guideline panel agreed have unequivocal net benefit yet may not be widely recognized or used.22 Good practice statements in these guidelines are not based on a systematic review of available evidence. Nevertheless, they may be interpreted as strong recommendations.
Recommendations
Primary stroke prevention for children with SCD living in low-middle– and high-income settings
Recommendation 1.1.
For children with HbSS or HbSβ0 thalassemia (ages 2-16 years), the American Society of Hematology (ASH) guideline panel recommends annual TCD screening (strong recommendation based on moderate certainty in the evidence about effects ⊕⊕⊕◯).
Recommendation 1.2.
For children who have compound heterozygous SCD other than HbSC and have evidence of hemolysis in the same range as those with HbSS, the ASH guideline panel suggests TCD screening (conditional recommendation based on very low certainty in the evidence about effects ⊕◯◯◯).
Recommendation 2.1.
For children with HbSS or HbSβ0 thalassemia (ages 2-16 years) who have abnormal TCD velocities and live in a high-income setting (where regular blood transfusion therapy, typically every 3-4 weeks, is feasible to maintain the maximum HbS level <30% and maintain the hemoglobin level >9.0 g/dL), the ASH guideline panel recommends regular blood transfusion for at least a year (vs no transfusion) with the goal of keeping maximum HbS levels below 30% and maintaining hemoglobin levels >9.0 g/dL to reduce the risk of stroke (strong recommendation based on moderate certainty in the evidence about effects ⊕⊕⊕◯).
Recommendation 2.2.
For children who have compound heterozygous SCD other than HbSC, who have evidence of hemolysis in the same range as those with HbSS, an abnormal TCD velocity, and live in a high-income setting (where regular blood transfusion therapy is feasible), the ASH guideline panel suggests regular blood transfusion for at least a year (vs no transfusion) with the goal of keeping maximum HbS levels below 30% to reduce the risk of stroke (conditional recommendation based on very low certainty in the evidence about effects ⊕◯◯◯).
Recommendation 2.3.
For children with SCD (ages 2-16 years) and abnormal TCD results who have been receiving transfusion therapy for at least 1 year and are interested in stopping transfusion, according to the clinical trial risk stratification with an MRI and magnetic resonance angiography (MRA) of the brain (see Technical remarks in supplemental File 5), the ASH guideline panel suggests that hydroxyurea treatment at the maximum tolerated dose can be considered to substitute for regular blood transfusions (conditional recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Recommendation 3.
For children (ages 2-16 years) with HbSS, HbSβ0 thalassemia, or compound heterozygous SCD who have abnormal TCD screening and live in low-middle-income settings (where regular blood transfusion therapy and chelation therapy are not available or affordable), the ASH guideline panel suggests hydroxyurea therapy with at least 20 mg/kg per day at a fixed dose or the maximum tolerated dose (conditional recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Acute and timely treatment of suspected or confirmed ischemic stroke or TIA
Recommendation 4.1.
For children or adults with SCD and acute neurological deficits, including transient ischemic attack (TIA), the ASH guideline panel recommends prompt blood transfusion. The transfusion should be given immediately upon recognition of symptoms without delay beyond 2 hours of acute neurological symptom presentation. The type of transfusion (simple, modified exchange, or apheresis) is dependent on individual patient factors and local transfusion resources (strong recommendation based on high certainty in the evidence about effects ⊕⊕⊕⊕).
Recommendation 4.2.
For children or adults with SCD and acute neurological deficits including TIA, the ASH guideline panel suggests exchange transfusion vs simple transfusion. When exchange transfusion is not available within 2 hours of presentation for medical care and hemoglobin is ≤8.5 g/dL, simple transfusion can be performed to avoid delays in treatment while a manual exchange transfusion or an automated apheresis is planned (conditional recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Secondary prevention of ischemic strokes in children and adults with HbSS or HbSβ0 thalassemia
Recommendation 5.
For children with HbSS or HbSβ0 thalassemia and a history of prior ischemic stroke, the ASH guideline panel recommends blood transfusion goals for secondary stroke prevention of increasing the hemoglobin above 9 g/dL at all times and maintaining the HbS level at <30% of total hemoglobin until the time of the next transfusion (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Recommendation 6.
For adults and children with SCD, moyamoya syndrome, and a history of stroke or TIA, the ASH guideline panel suggests evaluation for revascularization surgery in addition to regular blood transfusion (conditional recommendation based on very low certainty in the evidence about effects ⊕⊕⊕◯).
Acute management of ischemic strokes and the use of tPA for adults with SCD presenting with stroke symptoms
Recommendation 7.
For adults with SCD presenting with symptoms of acute ischemic stroke who are being evaluated for IV tissue plasminogen activator (tPA; age ≥18 years, no hemorrhage on computed tomography [CT] scan, within 4.5 hours of onset of symptoms/signs and without contraindications for thrombolysis), the ASH guideline panel suggests management using a shared decision-making approach that follows these principles:
For all patients, the administration of tPA should not delay prompt simple or exchange blood transfusion therapy.
Patients may be considered for IV tPA based on its established inclusion and exclusion criteria detailed in stroke management algorithms.
The following factors suggest likely benefit from IV tPA: older age, atrial fibrillation, diabetes, hypertension, and hyperlipidemia. Management of younger patients without these risk factors should emphasize early transfusion.
There are no validated risk stratification or reliable age cutoff criteria to guide the choice of initial therapy. IV tPA is not recommended for children with SCD (<18 years of age).
IV tPA is not recommended for children with SCD (<18 years of age).
Screening for developmental delay or cognitive impairment in children and adults with SCD
Recommendation 8.1.
Given the high prevalence of developmental delay and cognitive impairments in children with SCD, coupled with the guidelines set by the American Academy of Pediatrics, the ASH guideline panel recommends that clinicians supervising care of children with SCD conduct surveillance using simplified signaling questions for the following:
Concerns about developmental delays in preschool-age children.
Concerns about neurodevelopmental disorders in school-age children with SCD, such as academic or behavioral problems or symptoms of inattention, hyperactivity, or impulsivity.
Recommendation 8.2.
For children with SCD who have abnormal surveillance results suggesting increased risk for developmental delay or cognitive impairment, the ASH guideline panel recommends screening or referral for formal screening by a psychologist or a pediatrician able to perform screening with the available validated tools (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Recommendation 8.3.
Given the high prevalence of cognitive impairment in adults with SCD, coupled with the guidelines set by the American Academy of Neurology, the ASH guideline panel recommends that clinicians supervising care of adults with SCD conduct surveillance for cognitive impairment using simplified signaling questions (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Recommendation 8.4.
For adults who have abnormal surveillance results suggesting cognitive impairment, the ASH guideline panel recommends formal referral to a psychologist or a primary care physician able to perform more in-depth cognitive evaluation (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Cognitive rehabilitative strategy for children and adults with cognitive impairments
Recommendation 9.1.
For children with SCD and abnormal screening for developmental or cognitive status, the ASH guideline panel recommends the following:
A developmental, cognitive, and medical evaluation to diagnose any related disorders and to identify modifiable risk factors for developmental delays or cognitive impairments.
Following the cognitive domain-specific evidence-based guidelines for these conditions to provide appropriate interventions.
Recommendation 9.2.
For adults with SCD and abnormal screening for cognitive status, the ASH guideline panel recommends the following:
Cognitive and medical evaluation to diagnose any related disorders and to identify modifiable risk factors for cognitive impairments.
Following the cognitive domain-specific evidence-based guidelines for these conditions to provide appropriate interventions.
Screening for silent cerebral infarcts in children and adults with HbSS or HbSβ0 thalassemia
Recommendation 10.1.
Given the high prevalence of silent cerebral infarcts in children with HbSS or HbSβ0 thalassemia (1 in 3), and their association with cognitive impairment, poor school performance, and future cerebral infarcts, the ASH guideline panel recommends at least a 1-time MRI screening, without sedation, to detect silent cerebral infarcts in early-school-age children, when MRI can commonly be performed without sedation (strong recommendation based on moderate certainty in the evidence about effects ⊕⊕⊕◯).
Recommendation 10.2.
Given the high prevalence of silent cerebral infarcts in adults with HbSS or HbSβ0 thalassemia (1 in 2) and their association with cognitive impairment, poor school performance, and future cerebral infarcts, the ASH guideline panel suggests at least a 1-time MRI screening without sedation to detect silent cerebral infarcts in adults with HbSS or HbSβ0 thalassemia (conditional recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Values and preferences
Overall, the ASH Guideline Panel on Cerebrovascular Disease placed a higher value on maintaining cognitive function than on being alive with reduced cognitive function (significantly less than baseline functioning). Given the high prevalence of neurological morbidity (strokes, silent cerebral infarcts, and cognitive impairment) in children and adults with SCD, a critical component of the recommendations includes involving the individual with SCD and the individual's family in care decisions. All panel members strongly believed that full disclosures should occur with families regarding the cumulative high risk of neurological morbidity in SCD, the utility of screening for neurological disease (abnormal TCD, silent cerebral infarcts, cognitive impairment), and biologically plausible therapies that may attenuate future neurological injury without a phase 3 randomized controlled trial.
Explanations and other considerations
These recommendations take into consideration acceptability, feasibility, cost-effectiveness, and impact on health equity. When developing these recommendations, the ASH guideline panel acknowledged variability in knowledge about risks and benefits of treatments, as well as variability for patients, their family members, and provider perceptions of the balance between harms vs benefits.
Good practice statements
Good practice statement 1.
To adopt a health care system strategy for tracking TCD assessments and treatment of children with SCD and abnormal TCD measurements because these children are at extremely high risk for ischemic strokes. Tracking TCD surveillance and treatment in both low-middle– and high-income settings will significantly decrease strokes in children with SCD.
Good practice statement 2.
To consult with a neurologist and neuroradiologist (when available) for evaluation in all suspected acute neurological events and neuroimaging studies. For suspected ischemic strokes, timely and appropriate red blood cell transfusion (within 2 hours of presentation to medical care) should be provided.
Good practice statement 3.
To adopt a multidisciplinary (ie, hematology, neurology, neurosurgery, and neuroradiology) team management approach when a neurosurgical revascularization procedure is being considered for SCD-related moyamoya syndrome as adjunctive therapy to regular blood transfusion therapy for secondary stroke prevention.
Good practice statement 4.
To inform children and adults with HbSS and HbSβ0 thalassemia and their families about the affected individual’s silent cerebral infarct status based on at least 1 MRI of the brain without sedation, coupled with discussion about potential disease-modifying therapy to prevent infarct recurrence.
Good practice statement 5.
To have ongoing standardized cognitive or behavior surveillance and, when impairments are identified, refer the patient to a specialist who may better evaluate the magnitude of the cognitive impairments and provide rehabilitative approaches.
Introduction
Aims of these guidelines and specific objectives
The purpose of these guidelines is to provide evidence-based recommendations to facilitate prevention, diagnosis, and treatment of neurological morbidity, including strokes, silent cerebral infarcts, and cognitive morbidity, in children and adults with SCD. To achieve this goal, the panel reviewed and critically appraised the literature and provided evidence-based recommendations. Through improved provider and patient education using evidence-based recommendations, these guidelines aim to provide support for shared decision-making between providers, patients, and their families, which will result in decreased neurological morbidity and mortality of children and adults with SCD.
The target audience includes patients, hematologists, general practitioners, internists, other clinicians, and decision makers. Policy makers interested in these guidelines include those involved in developing local, national, or international plans with the goal of improving access to evidence-based care. Local or national panels may also use this document as a basis for implementation of strategies to prevent neurological morbidity in children and adults with SCD in their health care system. When health care systems and health care providers adopt these guidelines, there will be a decrease in neurological morbidity in children and adults with SCD.
Description of the health problem
Stroke, silent cerebral infarcts, and cognitive morbidity are the most common permanent sequelae of SCD in children and adults. Prior to 1990 in the United States, a large prospective cohort study demonstrated that by 40 years of age, ∼20% and ∼10% of adults with phenotype HbSS or HbSC had a cerebrovascular accident, respectively (Figure 1).1 For most children with HbSS living in low-middle–income settings, ∼11% will have a stroke by 18 years of age.1
The most common cause of permanent neurological injury in HbSS or HbSβ0 thalassemia is silent cerebral infarcts, occurring in ∼39% of children3 and >50% of adults4 (Figure 2). Silent cerebral infarcts require MRI to detect and a formal neurological examination to exclude the presence of an overt stroke. Both stroke and silent cerebral infarcts are associated with significant risk of infarct recurrence and clinically relevant cognitive impairments, which may indirectly alter employment status and quality of life.
In 2013, the American Heart Association/American Stroke Association (AHA/ASA) for the first time endorsed a definition of stroke that includes silent cerebral infarctions and silent cerebral hemorrhages typically identified by MRI of the brain.23 This change in definition reflects a shift in emphasis toward a radiological demonstration (tissue-based definition) of infarction or hemorrhage because permanent neurological injury may occur despite symptoms resolving in <24 hours. For patients with cerebral ischemia, the AHA/ASA stated that treatment should address the cause of the ischemic event and not be governed only by whether infarction has developed (in the case of TIA) or the size of the infarct.
The traditional definition of stroke endorsed by the World Health Organization (WHO) requires clinical symptoms for >24 hours and has been in use since the 1970s.24 Our panel affirmed the importance of silent cerebral infarcts given the known impact on cognition and an established biomarker for infarct recurrence in children and adults with HbSS or HbSβ0 thalassemia25-27 and in the general population.28 However, we recognize that the MRI-based definition is challenging in low-middle–income settings where MRI is not widely available. Hence, the WHO definition of stroke is clinically relevant29 and generalizable to individuals living in both low-middle– and high-income settings.
Methods
The methodology of this guideline is published in detail elsewhere.30 The guideline panel developed and graded the recommendations and assessed the certainty of the supporting evidence following the GRADE approach.16-22,31 The overall guideline development process, including funding of the work, panel formation, management of conflicts of interest, internal and external review, and organizational approval, was guided by ASH policies and procedures derived from the GIN–McMaster Guideline Development Checklist (http://cebgrade.mcmaster.ca/guidecheck.html) and was intended to meet recommendations for trustworthy guidelines by the Institute of Medicine and the GIN.12-15
The Mayo Evidence-Based Practice Research Program conducted or updated systematic reviews based on clinical questions developed and defined by the ASH guideline panel. This was a multidisciplinary group and included multiple stakeholders (4 hematologists who provide medical care for children and adults with SCD, 3 neurologists who provide medical care for children, 2 neuroradiologists who review the neuroimaging of children and adults with SCD, a pediatric psychologist with expertise in SCD, an individual with SCD who has had a stroke, a parent of a child with SCD who had a stroke after she was not offered TCD screening, and a physician with expertise in evidence-based medicine methodology). Following the GRADE approach, randomized trials and observational studies provide an initial level of certainty in evidence classified as either high or low, and this classification may be subsequently modified based on additional factors.32 Then, evidence-to-decision (EtD) factors are applied to make a recommendation.
Recommendations are either strong or conditional. Strong recommendations imply a high certainty of net benefit, such that the recommended action should be applied to most patients as a standard of care. Conditional recommendations imply that the balance of benefits and harms is less clear. Although the recommended action should be offered to the majority of patients, there will be important variation in context, and, in some cases, an alternative action is reasonable.32
Values and preferences and patient engagement when addressing all 19 recommendations
The values invoked in developing these 19 recommendations reflect the combined views of all panelists, including the 2 patient representatives. In all 19 recommendations, there was near unanimous agreement regarding the importance of prevention, detection, and treatment of CNS morbidity. The panelists rated the importance of outcomes on a scale of 1 to 9, where ratings of 7 to 9 reflect outcomes of critical importance to the decision at hand. As a guiding principle, all members of the panel placed a higher value on maintaining cognitive function than on being alive with minimal cognitive function (significantly less than baseline functioning).
Organization, panel composition, planning, and coordination
The work of this panel was coordinated with that of 4 other guideline panels (addressing other aspects of SCD) by ASH and the Mayo Evidence-Based Practice Research Center (funded by ASH under a paid agreement). Project oversight was provided by a coordination panel, which reported to the ASH Guideline Oversight Subcommittee. ASH vetted individuals and appointed them to the guideline panel. The Mayo Center vetted and retained researchers to conduct systematic reviews of evidence and coordinate the guideline development process including the use of the GRADE approach.30 The membership of the panels and the Mayo Center team is described in supplemental File 1.
In addition to systematically synthesizing evidence, the Mayo Center supported the guideline development process, including determining methods, preparing meeting materials, and facilitating panel discussions. The panel’s work was done using web-based tools (www.gradepro.org), as well as face-to-face and online meetings.
Guideline funding and management of conflicts of interest
Development of these guidelines was wholly funded by ASH, a nonprofit medical specialty society that represents hematologists. ASH staff supported panel appointments and coordinated meetings but had no role in choosing the guideline questions or determining the recommendations.
Members of the guideline panel received travel reimbursement for attendance at in-person meetings, and the patient representatives received honorariums of $100 per day for in-person meetings and $25 per conference call. The panelists received no other payments. Through the Mayo Clinic Evidence-Based Practice Research Program, some researchers who contributed to the systematic evidence reviews received salary or grant support. Other researchers participated to fulfill requirements of an academic degree or program.
Conflicts of interest of all participants were managed through disclosure, panel composition, and recusal, according to recommendations of the Institute of Medicine33 and the Guidelines International Network.14 Participants disclosed all financial and nonfinancial interests relevant to the guideline topic. ASH staff and the ASH Guideline Oversight Subcommittee reviewed the disclosures and composed the guideline panel to include a diversity of expertise and perspectives and avoid a majority of the panel having the same or similar conflicts. Greatest attention was given to direct financial conflicts with for-profit companies that could be directly affected by the guidelines. A majority of the panel, including the joint chairs, had no such conflicts. None of the Mayo-affiliated researchers who contributed to the systematic evidence reviews or who supported the guideline development process had any such conflicts.
Recusal was also used to manage conflicts of interest.14,34-36 During deliberations about recommendations, any panel member with a current, direct financial interest in a commercial entity that marketed any product that could be affected by a specific recommendation participated in discussions about the evidence and clinical context but was recused from making judgments or voting about individual domains (eg, magnitude of desirable consequences) and the direction and strength of the recommendation. The EtD framework for each recommendation describes which individuals were recused from making judgments about each recommendation.
In 2019, after the guideline panel had agreed on recommendations, it was discovered that 1 panelist had a direct financial conflict of interest with an affected company (meals in 2017) that had not been previously reported. The panelist had been recused for a similar disclosure during the guideline meeting held to form recommendations. Members of the Guideline Oversight Subcommittee reviewed this late disclosure and determined no additional action was required.
Supplemental File 2 provides the complete disclosure-of-interest forms of all panel members. Individuals disclosed: direct financial interests for 2 years prior to appointment in part A of the forms, indirect financial interests in part B, and interests that were not mainly financial in part C. Part D describes new interests disclosed by individuals after appointment. Part E summarizes ASH decisions about which interests were judged to be conflicts and how they were managed, including through recusal.
Supplemental File 3 provides the complete disclosure-of-interest forms of researchers who contributed to these guidelines.
Formulating specific clinical questions and determining outcomes of interest
The panel met in-person and via conference calls to generate possible questions to address. The panel then used an iterative process to prioritize the questions described in Table 1. Questions were formulated using the standard format of population, intervention, comparison, and outcomes (PICO).
Following the approach described in detail elsewhere,37 the panel also selected outcomes of interest for each question a priori. In brief, the panel first discussed all possible outcomes before rating their relative importance for decision-making following the GRADE approach.37 The panel acknowledged considerable variation in the clinical impact of patient outcomes; they considered outcomes critical for clinical decision-making across questions (Table 2).
Evidence review and development of recommendations
Researchers at the Mayo Clinic Evidence-Based Practice Center conducted new systematic reviews or updated existing systematic reviews to answer the main 10 PICO questions. When existing reviews were used, judgments of the original authors about risk of bias were either randomly checked for accuracy and accepted or conducted de novo if they were not available or not reproducible. In addition to conducting systematic reviews of intervention effects, the researchers sought evidence related to baseline risks, values, preferences, and costs and summarized these findings within the EtD frameworks.16,31 Subsequently, the certainty in the body of evidence (also known as quality of the evidence or confidence in the estimated effects) was assessed for each effect estimate of the outcomes of interest following the GRADE approach based on the following domains: risk of bias, precision, consistency and magnitude of the estimates of effects, directness of the evidence, risk of publication bias, presence of large effects, dose-response relationship, and an assessment of the effect of residual, opposing confounding factors. The certainty was categorized into 4 levels ranging from very low to high.17-19
For each guideline question, the Mayo Center prepared a GRADE EtD framework, using the GRADEpro Guideline Development Tool (www.gradepro.org).16,21,22,31 The EtD table summarized the results of systematic reviews of the literature that were updated or performed for this guideline. The EtD table addressed effects of interventions, resource utilization (cost-effectiveness), values and preferences (relative importance of outcomes), equity, acceptability, and feasibility. The guideline panel reviewed draft EtD tables before, during, or after the guideline panel meeting, made suggestions for corrections, and identified missing evidence. To ensure that recent studies were not missed, in addition to searches presented in supplemental File 4, panel members were asked to suggest any studies that might have been considered missed that fulfilled the inclusion criteria for the individual questions.
During a 2-day in-person meeting followed by online communication and conference calls, the panel developed clinical recommendations based on the evidence summarized in the EtD tables. For each recommendation, the panel took a population perspective and came to a consensus on the following: the certainty in the evidence, the balance of benefits and harms of the compared management options, and the assumptions about the values and preferences associated with the decision. The guideline panel also explicitly considered the extent of resource use associated with alternative management options. The panel agreed on the recommendations (including direction and strength), remarks, and qualifications by consensus or, in rare instances, by voting (an 80% majority was required for a strong recommendation), based on the balance of all desirable and undesirable consequences. The final guidelines, including recommendations, were reviewed and approved by all members of the panel. The approach is described in detail in a previous article describing the methods of development.30
Interpretation of strong and conditional recommendations
The recommendations are labeled as either “strong” or “conditional” according to the GRADE approach. The words “the guideline panel recommends” are used for strong recommendations, and “the guideline panel suggests” for conditional recommendations. Table 3 provides GRADE’s interpretation of strong and conditional recommendations by patients, clinicians, health care policy makers, and researchers.
Interpretation of good practice statements
As described by the GRADE Guidance Group, good practice statements endorse interventions or practices that the guideline panel agreed have unequivocal net benefit yet may not be widely recognized or used.22 Good practice statements in these guidelines are not based on a systematic review of available evidence. Nevertheless, they may be interpreted as strong recommendations.
Document review
Draft recommendations were reviewed by all members of the panel, revised, and then made available online on 24 September 2018, or for external review by stakeholders including allied organizations, other medical professionals, patients, and the public. Eighteen individuals or organizations submitted comments. The document was revised to address pertinent comments, but no changes were made to recommendations. The guidelines were reviewed by the ASH Guideline Oversight Subcommittee on 10 October 2019. On 21 October 2019, the ASH Committee on Quality confirmed that the defined guideline development process was followed, and, on 25 October 2019, the officers of the ASH Executive Committee approved submission of the guidelines for publication under the imprimatur of ASH. The guidelines were then subjected to peer review by Blood Advances.
How to use these guidelines
ASH guidelines are primarily intended to help clinicians make decisions about diagnostic and treatment alternatives. Other purposes are to inform policy, education, and advocacy and to state future research needs. They may also be used by patients. These guidelines are not intended to serve or be construed as a standard of care. Clinicians must make decisions based on the clinical presentation of each individual patient, ideally through a shared process that considers the patient’s values and preferences with respect to the anticipated outcomes of the chosen option. Decisions may be constrained by the realities of a specific clinical setting and local resources, including but not limited to institutional policies, time limitations, or availability of treatments. These guidelines may not include all appropriate methods of care for the clinical scenarios described. As science advances and new evidence becomes available, recommendations may become outdated. Following these guidelines cannot guarantee successful outcomes. ASH does not warrant or guarantee any products described in these guidelines.
Statements about the underlying values and preferences as well as qualifying remarks accompanying each recommendation are its integral parts and serve to facilitate more accurate interpretation. They should never be omitted when quoting or translating recommendations from these guidelines. Implementation of the guidelines will be facilitated by the related interactive forthcoming decision aids. The use of these guidelines is also facilitated by the links to the EtD frameworks and interactive summary of findings tables in each section.
Recommendations
Screening with TCD and primary stroke prevention
Should transfusion (vs no transfusion or hydroxyurea therapy) be used for children aged 2 to 16 years with HbSS or HbSβ0 thalassemia and abnormal TCD measurements?
Between 2 and 16 years of age, should children with HbS/Lepore disease, HbSE disease, HbS/O Arab disease, or HbS/D disease phenotypes or other compound heterozygous SCD phenotypes other than HbSC have TCD screening at the same frequency and interval as children with HbSS or HbSβ0 thalassemia?
Should annual screening with TCD be used for children between 2 and 16 years of age with HbSS or HbSβ0 thalassemia phenotypes from low-middle–income settings?
Recommendation 1.1
For children with HbSS or HbSβ0 thalassemia (ages 2-16 years), the ASH guideline panel recommends annual TCD screening (strong recommendation based on moderate certainty in the evidence about effects ⊕⊕⊕◯).
Recommendation 1.2
For children who have compound heterozygous SCD other than HbSC and have evidence of hemolysis in the same range as those with HbSS, the ASH guideline panel suggests TCD screening (conditional recommendation based on very low certainty in the evidence about effects ⊕◯◯◯).
Recommendation 2.1
For children with HbSS or HbSβ0 thalassemia (ages 2-16 years) who have abnormal TCD velocities and live in a high-income setting (where regular blood transfusion therapy, typically every 3-4 weeks, is feasible to maintain the maximum HbS level <30% and maintain the hemoglobin level >9.0 g/dL), the ASH guideline panel recommends regular blood transfusion for at least a year (vs no transfusion) with the goal of keeping maximum HbS levels below 30% and maintaining hemoglobin levels >9.0 g/dL to reduce the risk of stroke (strong recommendation based on moderate certainty in the evidence about effects ⊕⊕⊕◯).
Recommendation 2.2
For children who have compound heterozygous SCD other than HbSC, who have evidence of hemolysis in the same range as those with HbSS, an abnormal TCD velocity, and who live in a high-income setting (where regular blood transfusion therapy is feasible), the ASH guideline panel suggests regular blood transfusion for at least a year (vs no transfusion) with the goal of keeping maximum HbS levels below 30% to reduce the risk of stroke and maintaining the minimum hemoglobin >9.0 g/dL (conditional recommendation based on very low certainty in the evidence about effects ⊕◯◯◯).
Recommendation 2.3
For children with SCD (ages 2-16 years) and abnormal TCD results who have been receiving transfusion therapy for at least 1 year, and are interested in stopping transfusion, according to the clinical trial risk stratification with an MRI and MRA of the brain (see Technical remarks in supplemental File 5), the ASH guideline panel suggests that hydroxyurea treatment at the maximum tolerated dose can be considered to substitute for regular blood transfusions.
Remarks:
For children with abnormal TCD results and without MRA-defined vasculopathy or silent cerebral infarcts who have received at least 1 year of transfusions, based on the entry criteria of the TCD With Transfusions Changing to Hydroxyurea (TWiTCH) Trial, hydroxyurea therapy treatment, at the maximum tolerated dose, should be considered as a replacement for regular blood transfusion therapy.
For children with abnormal TCD results, MRA-defined vasculopathy, or silent cerebral infarcts, based on the exclusion criteria of the TWiTCH Trial, continue regular blood transfusions indefinitely.
Remarks for recommendations 1 and 2:
Recommendations 1 and 2 are applicable to countries or settings in which regular blood transfusion is feasible and acceptable.
The suggested threshold for treatment is 2 nonimaging TCD measurements >200 cm/s, a time-averaged mean of the maximum velocity (TAMMV) of ≥200 cm/s or a single measurement >220 cm/s in the distal internal carotid artery or proximal middle cerebral artery.
If the imaging TCD technique is used for assessment, then 2 measurements greater than a time-averaged mean maximum (TAMX) of ≥185 cm/s or a single measurement >205 cm/s is required in the distal internal carotid artery or proximal middle cerebral artery.
Predictive values of the TCD measurements in the other intracranial arteries have not been rigorously addressed and should not be used to stratify children into high- and low-risk groups for future strokes.
For recommendations 1.2 and 2.2, the threshold for hemolysis requiring regular TCD surveillance should be determined based on the individual patient characteristics. Consideration should be given to hemoglobin level, reticulocyte count, and degree of hemolysis in relationship to HbSS.
For recommendations 1.2 and 2.2, we could not define a laboratory threshold to determine who should undergo TCD. Also, no evidence was available demonstrating that children with HbSC should undergo TCD screening for primary stroke prevention.
Recommendation 3
For children (ages 2-16 years) with HbSS or HbSβ0 thalassemia or compound heterozygous SCD, who have abnormal TCD screening, and live in low-middle-income settings (where regular blood transfusion therapy and chelation therapy are not available or affordable), the ASH guideline panel suggests hydroxyurea therapy with at least 20 mg/kg per day at a fixed dose or the maximum tolerated dose (conditional recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Remark:
Recommendation 3 is applicable to low-middle–income settings and locations in which regular blood transfusion is not feasible. For children with abnormal TCD velocities in this setting, the optimal hydroxyurea dose and the appropriate infrastructure support required to safely administer hydroxyurea and to follow the patients for expected toxicities have not been determined.
Specific background.
Applying the results of the Optimizing Primary Stroke Prevention in Sickle Cell Anemia Trial (STOP) has contributed to one of the greatest advances in management of SCD. TCD screening coupled with regular blood transfusion therapy for those with an abnormal TAMMV TCD measurement is associated with a 92% reduction in stroke incidence compared with observation alone.38 The threshold for regular blood transfusion therapy is 2 nonimaging TCD measurements ≥200 cm/s or a single TAMMV measurement >220 cm/s in the proximal portion of the middle cerebral artery or the distal portion of the internal carotid artery (Figure 1 in supplemental File 5). Two TCD measurements are required for values ≥200 cm/s and <220 cm/s because of the ultrasonographer’s large coefficient of variation up to 12% of TCD measurement in the same child with HbSS measured only 3 hours apart.39 Furthermore, a large intrasubject standard deviation of the TCD measurement occurs; in 812 children with HbSS and HbSβ0 thalassemia and TCD measurements with at least 2 examinations in <6 months with no intervention, the standard deviation of the TAMMV was 14.9 cm/s.40
The peak systolic TCD velocity has not been applied as a predictor of initial stroke in a randomized controlled trial. These peak velocities are generated automatically from the TCD machine and cannot be used to stratify children for primary stroke prevention. Unfortunately, the TCD peak systolic velocity is sometimes confused with TAMMV, and as a result, patients can be inadvertently transfused.
STOP 2 demonstrated that, for STOP participants who received transfusions for 30 months or longer and whose TCD measurements became normal, continued regular blood transfusion was required to prevent strokes or reversion to abnormal TCD measurements.41 Thus, children with abnormal TCD measurements are presumed to have an indefinite risk of strokes. In summary, the results for STOP and STOP 2 demonstrated a clear benefit of regular blood transfusion compared with no transfusions (observation) (Figure 3). STOP 2 excluded children with severe stenotic lesions on cerebral MRA.

STOP trial results. Results of the STOP trial and STOP 2.38,41 TCD screening coupled with regular blood transfusion therapy for those with an abnormal TCD measurement (TAMMV > 200 cm/s) was associated with a 92% reduction in stroke incidence compared with observation alone.38 The threshold for treatment is 2 nonimaging TCD measurements >200 cm/s, TAMMV of ≥200 cm/s, or a single measurement >220 cm/s.38 Reprinted from Adams et al with permission.38
STOP trial results. Results of the STOP trial and STOP 2.38,41 TCD screening coupled with regular blood transfusion therapy for those with an abnormal TCD measurement (TAMMV > 200 cm/s) was associated with a 92% reduction in stroke incidence compared with observation alone.38 The threshold for treatment is 2 nonimaging TCD measurements >200 cm/s, TAMMV of ≥200 cm/s, or a single measurement >220 cm/s.38 Reprinted from Adams et al with permission.38
The optimal interval for reassessment of children with conditional TCD measurements (170-199 cm/s) has not been determined, but reassessment is commonly done within 6 months, and often sooner. The HbSS and HbSβ0 thalassemia phenotypes were both eligible for STOP and STOP 2, in part because of clinical challenges of distinguishing HbSS from HbSβ0 thalassemia using clinical laboratory values42 and the fact that both diagnoses have been included in primary stroke prevention stroke trials.38,41
If the imaging TCD technique rather than the nonimaging technique is used, then 2 measurements greater than the TAMX (≥185 cm/s) or a single TAMX measurement >205 cm/s is required.25 To ensure that proper velocity thresholds are used for clinical decision-making, clinicians should determine which type of TCD (imaging or nonimaging) is used at their center. Regardless of whether the imaging or nonimaging TCD is used, the threshold for treatment should not be based on peak systolic velocity (Figure 1 in supplemental File 5).
Children evaluated for abnormal TCD measurement should not have a recent blood transfusion because of the known association between TCD velocities and transfusions.43 Typically, the TCD measurement should be done at least 3 months after the last transfusion and when the child is at their baseline state of health.
Treatment with regular blood transfusion commonly requires iron chelation therapy to attenuate excessive stores of iron that accumulate with blood transfusion. If a child who had abnormal TCD screening measurement meeting criteria for transitioning to maximum tolerated dose of hydroxyurea, after 1 year of regular blood transfusion therapy, a discussion with the family should include whether hydroxyurea is preferable to regular blood transfusion therapy.44 Prior to consideration of transitioning from regular blood transfusion therapy to maximum tolerated dose of hydroxyurea, MRI of the brain should be completed to exclude silent cerebral ischemic lesions (see Table 1 PICO #10), and intracranial MRA should be completed to determine the presence and extent of cerebral vasculopathy, per the TWiTCH protocol.44
Given that the incidence rate of strokes was extremely low in the blood transfusion arm of the STOP Trial, <1 stroke per 100 patient-years,38 no formal assessment of stroke risk factors in the treatment arm of STOP can be used to determine the subgroup of children likely to have a stroke while receiving regular blood transfusion therapy.38 In the STOP 2 Trial for those randomly allocated to receive regular blood transfusion therapy, 21.6% (19 of 88) had persistent abnormal TCD measurements, with a mean follow-up of 2.4 years with no stroke occurrence.45
A small population of children with SCD (<3%) who are compound heterozygotes for HbS and do not have HbSC are at high risk of stroke. The utility of TCD screening in children with compound heterozygous SCD is not well defined. Given the relationship between TCD values and hemoglobin levels in children with HbSS,43 and the association between low hemoglobin levels and strokes,1 the evidence suggests that TCD screening and treatment will likely prevent strokes for children with compound heterozygous SCD and have evidence of hemolysis in a range similar to that of children with HbSS. The decision as to which children with compound heterozygous SCD should receive TCD screening and subsequent treatment should be made on an individual basis. In children with compound heterozygous SCD, we cannot identify a single marker of hemolysis or a threshold for markers of hemolysis that should be screened. For children with HbSC, the risk of abnormal TCD measurements and stroke is less than for those with HbSS.46
Hemoglobin levels in children with HbSβ+ thalassemia vary. The range is from hemoglobin levels of 7 g/dL to values within the normal range. The low hemoglobin levels are accompanied by magnitudes of hemolysis markers similar to those seen in children with HbSS. Consequently, we cannot state with high certainty which children with HbSβ+ thalassemia should receive TCD screening.
In a low-middle–income setting, without any primary stroke prevention strategy, ∼11% of the children with HbSS or HbSβ0 thalassemia will have a stroke before their 18th birthday.1 For ASH’s guidelines to have a significant impact on primary stroke prevention in children with HbSS or HbSβ0 thalassemia, a meaningful strategy must be aligned with public health policy that can be implemented in children with HbSS living in low-middle–income settings.
In an optimal implemented primary stroke program in high-income settings, <1% of the children who receive TCD screening coupled with regular blood transfusion therapy for abnormal TCD measurements will have strokes.2 For children with an abnormal TCD measurement, the risk of ischemic strokes is exceptionally high: 10.7 strokes per 100 patient-years.38
In untreated adults with atrial fibrillation, the stroke incidence rate is ∼4.4 events per 100 patient-years.47 The higher incidence of ischemic stroke risk for children with abnormal TCD measurements, when compared with adults with untreated atrial fibrillation, clearly demonstrates the potential public health impact of preventing strokes in children with HbSS or HbSβ0 thalassemia in both low-middle– and high-income settings.
The clinical experience of the panel, coupled with evidence from the literature, indicates that regular blood transfusion therapy is not a feasible option for the majority of children living in Africa or other low-middle–income settings. When presented with the benefits of blood transfusion for primary and secondary stroke prevention in Nigeria, caregivers uniformly elected for children not to receive regular blood transfusion.48 The reasons for not accepting regular blood transfusions include the annual cost of transfusions, the cost of iron chelation therapy, and the risk of a blood-borne infection or of life-threatening transfusion reactions.
Summary of the evidence.
The systematic review identified 3 randomized controlled trials at low risk of bias addressing the first question (transfusion of children with HbSS or HbSβ0 thalassemia and abnormal TCDs), no comparative studies addressing the second question (transfusion of other genotypes), and 1 nonrandomized study for question 3 (hydroxyurea in children with abnormal TCD measurements in low-middle–income settings).
The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/a34d09131753ee2f22fcaec5b2510f11, https://guidelines.gradepro.org/profile/b9ab114a172cfa3dab8c60ae5d7ba0b0, and https://guidelines.gradepro.org/profile/fe67f197d72505e0299c9557312b83b9.
Rationale and expected benefits.
The primary premise for our recommendations is that there is clear evidence from 3 National Heart, Lung, and Blood Institute (NHLBI)–funded controlled trials describing38,41,44 the benefit of identifying children between 2 and 16 years of age with HbSS or HbSβ0 thalassemia and abnormal TCD measurements (TAMMV >200 cm/s). The initial treatments for abnormal TCD measurements were regular blood transfusion, with therapy transitioned to maximally tolerated hydroxyurea therapy after 1 year of transfusion, for patients with MRA-defined vasculopathy. Secondary benefits of regular blood transfusion therapy include a decrease in the severity of the disease. No dissent was observed in the panel regarding the importance of identifying the risk of an initial stroke and providing caregivers with the option to select the best treatment of primary stroke prevention for the child.
The controlled clinical trials primarily addressed the question of whether children with abnormal TCD measurements (TAMMV, >200 cm/s) should receive regular blood transfusion, hydroxyurea therapy, or no therapy to reduce the high risk of stroke. One randomized controlled trial, STOP,38 included children with HbSS and HbSβ0 thalassemia screened for abnormal TCD measurements (TAMMV, >200 cm/s) and then randomly allocated to be treated with regular blood transfusion therapy or observation for primary stroke prevention.
For participants in STOP, one follow-up trial was completed, STOP 2,41 which included those randomly allocated in STOP who received regular blood transfusions for 30 months or longer and whose TCD measurements became <200 cm/s during that time. The former eligible STOP participants were randomly assigned to continue regular blood transfusion therapy or observation. The results demonstrated a clear benefit of ongoing regular blood transfusion therapy despite TCD measurements decreasing to <200 cm/s.41 Our recommendations are based on the strength of the evidence of these trials, which demonstrate the benefit of transfusion for primary stroke prevention.
TWiTCH is the only randomized controlled trial providing evidence for the safety of transitioning children with abnormal TCD velocities to the maximum tolerated dose of hydroxyurea. Children in this trial had no evidence of MRA-defined vasculopathy, had been receiving regular blood transfusion therapy for at least 1 year, and were escalated to the maximum tolerated dose of hydroxyurea.44 We do not have sufficient evidence to determine whether the group of children with abnormal TCD measurements and MRA-defined vasculopathy would remain stroke-free if transitioned from regular blood transfusion therapy to hydroxyurea because they were excluded from the TWiTCH trial.
In STOP, the group of children with the highest rate of stroke were those in the observation arm who had both abnormal TCD measurements and silent cerebral infarct at baseline. In this group, 51% (15 of 29) had a stroke, compared with 22% (9 of 40) in the observation group with abnormal TCD measurements and no silent cerebral infarct.49 Early evidence from small observational studies suggests that hydroxyurea alone may not attenuate cerebral infarct recurrence in adults with silent cerebral infarct.26,27
A consortium of French investigators demonstrated that children with HbSS or HbSβ0 thalassemia treated with regular blood transfusion for abnormal TCD velocities had lower TCD velocities 1 year after matched sibling donor hematopoietic stem cell transplantation (HSCT) compared with children treated with hydroxyurea therapy.50 These data suggest that HSCT is a reasonable option for children with SCD and abnormal TCD when performed in a clinical trial setting.50 However, this work should be considered preliminary because the long-term benefit vs the risk of using HSCT for primary stroke prevention has not been systematically studied, including the late effects of myeloablative and nonmyeloablative therapy in children and adults with SCD. Formal clinical trials are required to determine the optimal HSCT strategy for primary stroke prevention in HbSS or HbSβ0 thalassemia.
The primary premise for our PICO #3 recommendation (see Table 1) is based on the observation that ∼95% of children with HbSS live in low-middle–income settings,7 coupled with the urgency to prevent strokes in children, in the absence of a randomized controlled trial conducted in low-middle–income settings. Among children with HbSS and abnormal or conditional TCD measurement living in low-middle–income and high-income settings, data from observational studies consistently demonstrate that hydroxyurea lowers TCD measurements.51-60
The largest observational study that focused exclusively on primary stroke prevention in a low-middle–income country was conducted in Ibadan, Nigeria, and demonstrated the benefit of hydroxyurea for primary stroke prevention.60 Lagunju et al showed that children with conditional (n = 60) and abnormal (n = 44) TCD velocities taking hydroxyurea that started at 10 mg/kg, escalated to the maximum tolerated dose (20-35 mg/kg), and followed for a mean of 3.6 years had a mean drop in TAMMV from 198 cm/s to 169.3 cm/s. One stroke occurred in a child whose TAMMV remained abnormal despite adherence to hydroxyurea, and the calculated incidence rate for overt stroke was 0.27 per 100 person-years.60 There was also a child with TIA whose TAMMV remained abnormal after 15 months; this child had hydroxyurea discontinued because of mucositis, but had achieved a dose of 20 mg/kg per day. Leukopenia and neutropenia were not seen at this hydroxyurea dose.60 Another study, conducted in Kano, Nigeria, was a feasibility trial for primary stroke prevention for children with abnormal TCD measurements. In this study, 27 children with abnormal TCD measurements were given a fixed dose of hydroxyurea 20 mg/kg per day.39 Among the children with abnormal TCD measurements, hydroxyurea resulted in a mean TAMMV decrease of 18 cm/s and no strokes; with a total of 74 person-years, median follow up of approximately 2 years, and expected number of strokes of approimately 8 (10.7 per 100 patient-years).
To obtain additional evidence that hydroxyurea decreases TCD measurements, the panel reviewed 10 studies in children with HbSS or HbSβ0 thalassemia who had TCD measurements at baseline and several months after starting hydroxyurea therapy (Figure 4). The decrease in TCD measurements can occur as early as 3 months after starting hydroxyurea therapy with a sustained impact of hydroxyurea therapy on decreasing TCD measurements for at least 36 months. In a pooled analysis of 10 studies, the average drop in TCD measurement after starting hydroxyurea, 21 cm/s, was a clinically relevant decline (Figure 4).
![Pooled analysis of the 10 studies documenting TCD measurement before and after hydroxyurea therapy in children with HbSS or HbSβ0thalassemia. This meta-analysis demonstrates the average drop in TCD measurement after starting hydroxyurea therapy of 21 cm/s (95% confidence interval [CI], 14.8-29.0). The plot also suggests that the decrease in TCD measurements can be seen as early as 3 months after the start of hydroxyurea therapy with a sustained impact of hydroxyurea therapy on decreasing TCD measurements for at least 36 months. The analysis is updated from a previous one by DeBaun and Kirkham,58 plus additional references.51-57,59,60,165 ♦ represents the pooled estimate from a random-effect model, the edges of the diamonds represent the 95% CI; ▪ represents individual studies. MTD, maximum tolerated dose.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/8/10.1182_bloodadvances.2019001142/2/m_advancesadv2019001142cf4.png?Expires=1767900086&Signature=mOuTgC8U5sVRbw4SfnIpGcdR4Q7oEjhW4U0iD2I0WIzSvkvlbvn876tpjC6sKjLD1EF8ulwrevWNEwj21wgX2B8E20XA04jKXCavhU2ENwCGP6yQIVoLRFbf4dRTQ7WOdVhIB9-xm-sYEJZYhU~CVXuhTJsCFvNYdVpGzRiAp-NN3D7QWZ5dMtQvacIWYmHIfP5fSi8gXkY-qMPQ0GTzOixAzwNNCtJpnI80RAHLJwWT299XhRlDyLe8eG6jB2FXQIKMW4-2oKtPW13OB6LoLCsXCcupAVG-8tjFU9AWcLgdBUJCcbtsfCTeHHJAMlxYlqG3kx2y8kh3NhqkJ49UCQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Pooled analysis of the 10 studies documenting TCD measurement before and after hydroxyurea therapy in children with HbSS or HbSβ0thalassemia. This meta-analysis demonstrates the average drop in TCD measurement after starting hydroxyurea therapy of 21 cm/s (95% confidence interval [CI], 14.8-29.0). The plot also suggests that the decrease in TCD measurements can be seen as early as 3 months after the start of hydroxyurea therapy with a sustained impact of hydroxyurea therapy on decreasing TCD measurements for at least 36 months. The analysis is updated from a previous one by DeBaun and Kirkham,58 plus additional references.51-57,59,60,165 ♦ represents the pooled estimate from a random-effect model, the edges of the diamonds represent the 95% CI; ▪ represents individual studies. MTD, maximum tolerated dose.
Pooled analysis of the 10 studies documenting TCD measurement before and after hydroxyurea therapy in children with HbSS or HbSβ0thalassemia. This meta-analysis demonstrates the average drop in TCD measurement after starting hydroxyurea therapy of 21 cm/s (95% confidence interval [CI], 14.8-29.0). The plot also suggests that the decrease in TCD measurements can be seen as early as 3 months after the start of hydroxyurea therapy with a sustained impact of hydroxyurea therapy on decreasing TCD measurements for at least 36 months. The analysis is updated from a previous one by DeBaun and Kirkham,58 plus additional references.51-57,59,60,165 ♦ represents the pooled estimate from a random-effect model, the edges of the diamonds represent the 95% CI; ▪ represents individual studies. MTD, maximum tolerated dose.
Further data on the safety of hydroxyurea therapy in Africa was clearly demonstrated in a large prospective controlled trial recently reported from 4 sub-Saharan African centers (Luanda, Angola; Kinshasa, Democratic Republic of the Congo; Kilifi, Kenya; and Mbale, Uganda). The rate of clinical adverse events during the pretreatment phase (2 months) was compared with the treatment phase (mean, 29 months). During the treatment phase, there was a significant decrease in the incidence rates for death, malaria, and acute vaso-occlusive events; data on stroke were not presented separately.61
Taken together, these studies demonstrate that hydroxyurea therapy39,60,61 is safe for children in Africa, effective in decreasing TCD velocities, and reduces the incidence rate of mortality and morbidity. These studies provide a compelling rationale for the use of hydroxyurea for primary stroke prevention in children with abnormal TCD measurements in low-middle–income settings61 as additional trials with CNS complications as end points are conducted.39
The hydroxyurea dose used in Nigeria for primary stroke prevention ranges from a fixed moderate dose of 20 mg/kg,39 which typically does not require monthly full blood count monitoring, as performed in clinical trials, to maximum tolerated dose (typically ∼25-35 mg/kg per day) that requires monitoring every 2 to 3 months with complete blood counts.60,61 A randomized clinical trial for primary stroke prevention in Nigeria for children with HbSS and abnormal TCD is comparing moderate-dose hydroxyurea (20 mg/kg per day) to low-dose hydroxyurea (10 mg/kg per day) with results anticipated in 2021 (NCT02560935).
The optimal health care visit schedule for monitoring hydroxyurea therapy for primary stroke prevention in low-middle–income settings is not known. The primary purpose for this visit is for the health care provider to reinforce adherence to the therapy, adjust the dose of hydroxyurea due to the increasing weight of the growing child, and evaluate for toxicity, particularly myelosuppression. After stabilization on the maximum tolerated dose of hydroxyurea, monitoring blood counts every 8 weeks appears to be safe.61 Insufficient data were available to make a recommendation on the clinical utility of TCD screening in adolescents >16 years of age and adults with HbSS and HbSβ0 thalassemia.
Summary of harms and burden.
The potential harms associated with regular blood transfusion therapy have been quantified in controlled clinical trials for primary and secondary prevention strokes in children with HbSS or HbSβ0 thalassemia. These adverse events include, but are not limited to, the following, in decreasing order of prevalence: excessive iron stores62,63 that may eventually require chelation therapy,64 red blood cell alloimmunization,25,65 and adverse blood transfusion reactions.64
The family makes a significant time commitment when they agree to regular blood transfusion therapy. Typically, blood transfusions occur monthly and often require 2 visits (the first for crossmatching of the red blood cell units and the second for the actual blood transfusion). We did not find a study describing the full range of challenges of regular blood transfusion therapy for families, but the panel, including the 2 patient representatives, believed strongly that family preferences and the inconvenience and financial resources associated with regular blood transfusion therapy should be considered when making a decision to include this therapy.
In low-middle–income settings without public health care insurance systems to pay for hydroxyurea, the costs of hydroxyurea and complete blood cell counts assessments for myelosuppression may be prohibitive for most families. We did not identify a study reviewing the challenges of regular hydroxyurea therapy for families living in low-middle–income settings. The consensus among the panel was that some form of local or federal government subsidy for primary stroke prevention is required to have a sustainable program to treat the maximum number of children with abnormal TCD measurements living in low-middle–income settings. For instance, in Nigeria alone, 150 000 children are born with HbSS each year.66 Conceivably, 10% of each cohort of children born in the same year (15 000 children before 16 years of age) will have an abnormal TCD and will require primary stroke prevention. All panel members strongly believed that regardless of location in a high-income country or a low-middle–income country, the health care systems’ first obligation for following the panel guidelines was to prevent strokes in children with HbSS and HbSβ0 thalassemia.
EtD criteria and implementation consideration.
We combined HbSS and HbSβ0 thalassemia phenotypes because of the clinical challenges of distinguishing HbSS from HbSβ0 thalassemia based on clinical laboratory values42 and the fact that both diagnoses were eligible for primary stroke prevention trials.38,41
Patient representatives on the panel disclosed that regular blood transfusion therapy is less acceptable to some individuals with SCD and their caregivers. However, based on the extensive experience of the panel, blood transfusion therapy is acceptable to many caregivers and children with HbSS or HbSβ0 thalassemia phenotypes and abnormal TCD measurements. Transfusion is more feasible in high-income settings than in low-middle–income settings. Despite the lack of adequate cost-effectiveness studies, preventing strokes in children will always be less expensive than the long-term direct and indirect consequences of stroke and stroke-related death and disability, regardless of whether stroke prevention treatment is performed with transfusion and chelation therapy or hydroxyurea. The decision criteria are likely the same for children with other SCD compound heterozygotes at increased risk for stroke.
Hydroxyurea therapy requires at least the same health care system resources in a low-middle–income setting as those in high-income settings, including laboratory surveillance at the same interval as in a high-income setting (initially every 2 weeks and eventually every 2 or 3 months). Most likely, hydroxyurea therapy is more acceptable than transfusion for primary stroke prevention for patients and families. Health equity for stroke prevention in children living with SCD in low-middle– and high-income settings can potentially reduce the existing health disparities in childhood stroke prevalence between children with and without SCD.
The objective of regular blood transfusion is to maintain hemoglobin levels above 9 g/dL, but below 13 g/dL and pretransfusion HbS below 30%. Some patients will be difficult to transfuse effectively to keep the HbS <30% on a consistent basis.67,68 If the HbS cannot be kept consistently <30% with either simple transfusion, manual exchange transfusion, or automated exchange transfusion, higher HbS-level thresholds of ∼35% to 40% are acceptable provided that the patient is consistently transfused at 3- to 4-week intervals.
After 1 year of regular blood transfusion therapy, a gradual transition from transfusion to hydroxyurea can be considered. This involves a period of both hydroxyurea therapy and transfusion therapy, with eventual discontinuation of transfusion therapy. The transition may occur provided that the patient does not have intracranial MRA-defined vasculopathy as per TWiTCH.44 Prior to consideration of transitioning from regular blood transfusion therapy to maximum-tolerated-dose hydroxyurea, MRI of the brain should be undertaken to exclude silent cerebral ischemic lesions (see Table 1 PICO #10) and intracranial MRA to determine the presence and extent of cerebral vasculopathy. Children with cerebral vasculopathy were excluded from TWiTCH; therefore, transition from blood transfusion therapy to hydroxyurea is not recommended for these children. A discussion with the family should include whether hydroxyurea at the maximum tolerated dose is preferable to regular blood transfusion with chelation therapy to attenuate excessive stores of iron.44
Research needs.
The panel identified the following additional areas in need of research.
Best practices and implementation strategies for primary stroke prevention after using TCD as a screening tool should be determined. Over a 6-year study period among 4775 children with HbSS or HbSβ0 thalassemia from 6 US states, 22% to 44% of children received TCD screening.69
Alternative options for primary stroke prevention other than initial regular blood transfusion therapy for a year for some, then followed by maximum tolerated dose of hydroxyurea therapy, should be identified for children living in high-income settings.
Imaging strategies to identify the subgroup of children with an abnormal TCD measurement who are most likely to have a stroke should be improved. The current number needed to treat is 7 (ie, 7 children with abnormal TCD measurements must receive at least monthly blood transfusion therapy for at least a year to prevent 1 stroke). Strategies to personalize the risk of stroke for children with abnormal TCD measurements would be preferred to the current standard of red blood cell transfusion therapy for at least a year for children living in high-income settings for at least a year.
The optimal hydroxyurea dose (20 mg/kg per day vs 10 mg/kg per day vs the maximum tolerated dose of hydroxyurea) for primary stroke prevention in children with abnormal TCD measurement living in low-middle–income settings should be determined.
Use of a liquid formulation of hydroxyurea that is stable at room temperature when stored at home and can be provided to children <5 years of age unable to swallow a capsule is needed.
The best strategies to partner with local, state, and federal health care authorities in low-middle–income settings to provide hydroxyurea therapy for primary stroke prevention programs should be determined.
Training and quality assurance of TCD practitioners to increase the pool of qualified TCD practitioners, particularly in low-middle–income settings, are needed.
Should simple blood transfusion vs exchange transfusion be used for children and adults with SCD and suspected acute symptomatic stroke, including TIA?
Recommendation 4.1
For children or adults with SCD and acute neurological deficits, including TIA, the ASH guideline panel recommends prompt blood transfusion. The transfusion should be given immediately upon recognition of symptoms without delay beyond 2 hours of acute neurological symptom presentation. The type of transfusion (simple, modified exchange, or apheresis) is dependent on individual patient factors and local transfusion resources (strong recommendation based on high certainty in the evidence about effects ⊕⊕⊕⊕).
Recommendation 4.2
For children or adults with SCD and acute neurological deficits including TIA, the ASH guideline panel suggests exchange transfusion vs simple transfusion. When exchange transfusion is not available within 2 hours of presentation for medical care and hemoglobin is ≤8.5 g/dL, simple transfusion can be performed to avoid delays in treatment while a manual exchange transfusion or an automated apheresis is planned (conditional recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Remarks:
Optimal timing of therapy is to have prompt (within 2 hours of presentation to medical care) transfusion in children and adults with SCD presenting within 72 hours of symptom onset of a suspected acute stroke, new neurologic deficit, or TIA.
For children and adults with a new neurologic deficit or TIA presenting to medical care >72 hours after onset and without recent worsening, an assessment for anemia and percentage of sickle hemoglobin with consideration of transfusion on a case-by-case basis is suggested.
For individuals with hemoglobin levels >8.5 g/dL presenting with focal neurological deficits or TIA, exchange transfusion therapy to decrease the possibility of hyperviscosity syndrome is suggested.
Specific background.
Ischemic strokes in children and adults with SCD are one of the most important and common medical emergencies, particularly in regions where primary stroke prevention is not standard care. When a patient with SCD presents with an ischemic stroke or a TIA, timely response is required to minimize further ischemic injury. Optimal timing of intervention with blood transfusion therapy and brain-imaging modality has not been rigorously studied. However, in SCD, the principles of management of acute ischemic strokes and TIAs, coupled with observational studies, provide evidence for best practices
Summary of the evidence.
The systematic review identified 1 nonrandomized study that compared simple transfusion to exchange transfusion.
The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/bc68e8f1c23c576c468a5dca4557cb68.
Rationale and expected benefits.
In the absence of randomized clinical trials, no set of guidelines is likely to address the full spectrum of brain imaging and interventions for decreasing ischemic injury to the brain that represent evidence-based best practices; however, the principles of cerebral hemodynamics specific to SCD, coupled with multidisciplinary experiences of the panel, provide practical approaches.
Children and adults with SCD presenting with focal neurological deficits suggestive of stroke or TIA require rapid evaluation and close consultative interaction between hematologists, neurologists, and acute-care providers because the diagnosis of an acute ischemic stroke can be challenging (Figure 2 in supplemental File 5). If a hematologist or a health care practitioner skilled in managing exchange transfusion and acute stroke management in HbSS is not immediately available, appropriate care should be initiated with low-flow oxygen, IV fluids, complete blood count, and crossmatching; the patient should be sent to a facility with the required expertise to manage acute ischemic strokes and SCD. The differential diagnosis is broad (Figure 2 in supplemental File 5), and management of acute ischemic brain injury in children and adults with SCD continues to evolve.
The final diagnosis of ischemic stroke or TIA is based on a complete neurological history. An MRI of the brain may facilitate a diagnosis of acute cerebral ischemia, but cannot replace a history and examination The absence of abnormality seen on the diffusion-weighted image on MRI of the brain does not definitively exclude the diagnosis of an ischemic stroke.70 If a patient with SCD presents with an acute-onset focal neurological deficit and the health care provider believes that the patient has had an ischemic stroke or TIA, intervention should be the same to minimize the potential risk of ongoing ischemic brain injury.
Only one retrospective observational study in children with SCD provides the empiric data for the recommendation of an exchange transfusion (apheresis or manual) vs only simple transfusion for acute management of ischemic strokes. In this retrospective cohort study, children with HbSS or HbSβ0 thalassemia and acute ischemic stroke patients receiving simple transfusion for an acute ischemic stroke had a higher rate of recurrent strokes than children who received exchange transfusion (relative risk [RR], 5.0; 95% confidence interval [CI], 1.3-18.6).71
The preponderance of evidence supporting the recommendation for transfusion for acute ischemic stroke is from detailed cerebral hemodynamic studies in children and adults with SCD.72,73 In SCD, cerebral blood flow is increased compared with that in the general population,74,75 and flow is inversely related to arterial oxygen content (flow increases as oxygen content decreases).75 Oxygen delivery to the brain is the product of cardiac output and arterial oxygen content, which is primarily determined by the product of hemoglobin concentration and hemoglobin oxygen saturation. Both children and adults with SCD have altered cerebral hemodynamics resulting from the unique properties of sickled red blood cells, acute and chronic anemia,1,74,76 and cerebral vasculopathy.77 Concomitant complications of SCD such as acute chest syndrome can also reduce oxygen delivery, and cerebral metabolic demand is increased in conditions such as fever and seizures.76 Both overt ischemic strokes and silent cerebral infarcts in SCD typically occur in areas between cerebral large vessel vascular territories, also called the cerebral border zone, with the lowest cerebral blood flow.78
For children and adults presenting with a focal neurological deficit suggestive of an ischemic event, including a TIA, increasing the hemoglobin level with a red blood cell transfusion is the best option to achieve the goal of improving oxygen delivery to the brain. If the hemoglobin level is less than ∼8.5 g/dL, the panel recommends increasing the hemoglobin to ∼10.0 g/dL with a simple transfusion within 2 hours after presentation to medical care. After the hemoglobin has reached ∼10.0 g/dL with simple transfusion or if the baseline hemoglobin level is greater than ∼8.5 g/dL, the panel recommends an automated-exchange red blood cell transfusion (apheresis).79 This procedure will require a timely dialogue, initiated soon after presentation to the medical facility, between the transfusion service, the hematologist, and intensivist to ensure a multidisciplinary approach to management. If red blood cell exchange with apheresis is not a viable option, the patient should be stabilized and either transferred promptly to a facility that can perform apheresis or manual red blood cell exchange should be undertaken on site.80,81 The optimal strategy for manual red blood cell exchange for acute ischemic events has not been determined, but several options exist for reasonable starting points for providers that cannot offer apheresis in low-middle– and high-income settings.80,81
Unique attributes of SCD that are important for acute ischemic injury (suspected stroke or TIA) include the observation that as hemoglobin increases to a point, there is a corresponding increase in oxygen delivery.82,83 However, beyond this point any increase in hemoglobin concentration decreases the arterial oxygen delivery. For patients with HbSS or HbSβ0 thalassemia receiving simple transfusions, the point of diminishing benefit of arterial oxygen delivery is estimated to be between 10 and 11 g/dL82,83 (Figure 5).80

Relationship between hemoglobin level and oxygen delivery in individuals with SCD. The maximal hemoglobin (Hbmax) to deliver oxygen transport in sickle cell patients is 10 to 11 g/dL because SCD alters red cell viscosity and decreases oxygen transport. However, when the HbS level is low (∼20%), this impairment of oxygen transport is improved, and a higher hemoglobin level (such as 13 g/dL) may be beneficial. Red cell apheresis can rapidly lower the HbS to levels that maximize oxygen delivery, in contrast to the risks of simple transfusion resulting in increased viscosity and decreased oxygen delivery.82,83 Professional illustration by Patrick Lane, ScEYEnce Studios. Adapted from Swerdlow with permission.80
Relationship between hemoglobin level and oxygen delivery in individuals with SCD. The maximal hemoglobin (Hbmax) to deliver oxygen transport in sickle cell patients is 10 to 11 g/dL because SCD alters red cell viscosity and decreases oxygen transport. However, when the HbS level is low (∼20%), this impairment of oxygen transport is improved, and a higher hemoglobin level (such as 13 g/dL) may be beneficial. Red cell apheresis can rapidly lower the HbS to levels that maximize oxygen delivery, in contrast to the risks of simple transfusion resulting in increased viscosity and decreased oxygen delivery.82,83 Professional illustration by Patrick Lane, ScEYEnce Studios. Adapted from Swerdlow with permission.80
Evidence suggests that oxygen delivery to the brain in SCD is dependent not only on the total hemoglobin concentration, but also on the percentage of HbS.84 Thus, the goal of the red blood cell exchange with apheresis is to lower the percentage of HbS while preventing a rise in hemoglobin level above a threshold that may cause viscosity-related complications. Thresholds for postapheresis HbS percentage and hemoglobin level are typically set at 15% to 20% and 10 g/dL, respectively. The HbS level of 15% to 20% allows a threshold of <30% HbS to persist for ∼4 weeks. When the HbS level is <20%, the total hemoglobin level can generally be >10 g/dL, up to ∼12 to 13 g/dL, without concerns for viscosity-related complications, and the optimal recommended range of hemoglobin level postapheresis is 10 to 12 g/dL. When the hemoglobin level is <5.0 g/dL, a simple transfusion to increase the total hemoglobin to ∼10.0 g/dL may be required. Generally, red blood cell exchange with apheresis is not recommended for patients with SCD and hemoglobin levels <5.0 g/dL, in part because of concerns of lowering the hemoglobin level during priming of the apheresis machine. We identified an algorithm for acute management of suspected ischemic stroke or TIA in children and adults (Figures 6 and 7, respectively).
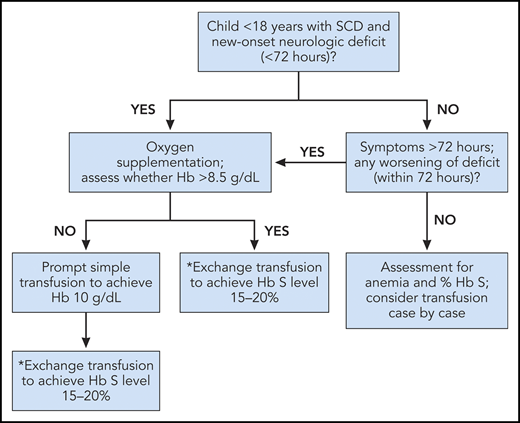
Algorithm of management of acute suspected ischemic stroke in children with SCD. The goal is prompt transfusion beginning within 2 hours of presentation to medical care to achieve hemoglobin of 10 g/dL and an HbS level of 15% to 20%. *See text for PICO question 4 or established inclusion and exclusion criteria. Professional illustration by Patrick Lane, ScEYEnce Studios.
Algorithm of management of acute suspected ischemic stroke in children with SCD. The goal is prompt transfusion beginning within 2 hours of presentation to medical care to achieve hemoglobin of 10 g/dL and an HbS level of 15% to 20%. *See text for PICO question 4 or established inclusion and exclusion criteria. Professional illustration by Patrick Lane, ScEYEnce Studios.

Algorithm of management of acute suspected ischemic stroke in adults with SCD. The goal is prompt transfusion beginning within 2 hours of presentation to medical care to achieve hemoglobin of 10 g/dL and an HbS level of 15% to 20%. *See PICO questions 4 and 7 or established inclusion and exclusion criteria. Professional illustration by Patrick Lane, ScEYEnce Studios.
Algorithm of management of acute suspected ischemic stroke in adults with SCD. The goal is prompt transfusion beginning within 2 hours of presentation to medical care to achieve hemoglobin of 10 g/dL and an HbS level of 15% to 20%. *See PICO questions 4 and 7 or established inclusion and exclusion criteria. Professional illustration by Patrick Lane, ScEYEnce Studios.
Based on the concept that “time is brain” and the urgency of rapid stroke care in the general adult population, we suggest similar urgency for treatment in SCD as soon as possible after presentation to medical care.85 Although we suggest a threshold time interval for consideration of exchange transfusion of 72 hours, this is based only on expert opinion. Few data are available to inform the question of the time interval for potential clinical benefit from red blood cell transfusion therapy after stroke onset in children and adults with SCD.
Supportive care in the acute poststroke period should reflect AHA guidelines, which suggest avoiding hyperglycemia, hypoglycemia, hyperpyrexia (treating temperatures of >38°C with antipyretics), and hypotension.86 As in adult stroke, early evidence in childhood stroke suggests that hypertension in the acute period after stroke is associated with worse outcomes.87 There is not enough evidence in children with SCD to make formal recommendations regarding treatment of blood pressure, but many centers allow blood pressure above prestroke baseline and treat blood pressures that are consistently above the 95th percentile for age and height.88 Given the observation that blood pressures in individuals with SCD are generally lower than in the general population,89 knowing the patients baseline blood pressure (prestroke) may facilitate blood pressure management at the time of stroke.
Summary of harm and burden.
The main complications of prompt treatment with simple blood or an exchange transfusion are those associated with blood transfusion and with the requirement for central line placement for red blood cell exchange with apheresis. Complications of regular blood transfusion include blood transfusion reactions,64 blood-borne infection, alloimmunization,25,65 and excessive iron stores,62,63,90 which typically requires iron chelation therapy.64 Complications of central line placement include inadvertent vascular injury, local infection at the site of the central line placement, and systemic infection and catheter-related venous thrombosis. Overall, timely treatment with blood transfusion therapy for acute ischemic injury of the brain was deemed to outweigh the risk of treatment.
EtD criteria and implementation considerations.
Prevention of recurrent stroke or extension of stroke will decrease the magnitude of stroke-related disability and mortality in children and adults with SCD, improving health equity. In terms of feasibility and acceptability, exchange transfusion often requires admission to an intensive care unit (ICU) and central line placement; however, the procedure can also be done in a non-ICU setting and with peripheral venous access if peripheral veins are adequate. Management of central line thrombosis or infection is challenging. Exchange transfusion by automated apheresis is not always immediately available. If apheresis is not available within 2 hours, strong consideration should be given to manual exchange or transfer to a center where automated apheresis can be performed.
Research needs.
The panel identified the following additional areas in need of research.
Evidence to define the optimal interval between onset of ischemic stroke or TIA and transfusion is needed. The time point at which there is no longer a benefit or at which risk outweighs benefit is unknown.
Development of additional therapeutic strategies or alternatives to blood transfusion is needed for better prevention of progressive brain injury after an initial acute ischemic stroke.
A more precise understanding of the mechanisms of cerebral hemodynamics in children and adults with SCD is needed to develop targeted therapies and to improve risk stratification for initial and subsequent cerebral infarct and cerebral hemorrhage.
Should red blood cell transfusion targeted to keep HbS levels below 30% (vs no treatment) or hydroxyurea therapy be used for children with SCD with a history of stroke?
Recommendation 5
For children with HbSS or HbSβ0 thalassemia and a history of prior ischemic stroke, the ASH guideline panel recommends blood transfusion goals for secondary stroke prevention of increasing the hemoglobin greater than 9 g/dL at all times and maintaining the HbS level at <30% of total hemoglobin until the time of the next transfusion (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Remarks:
The ASH guideline panel acknowledges that for children who cannot be transfused or refuse transfusion, hydroxyurea therapy is an inferior alternative to regular blood transfusion for secondary stroke prevention, but superior to no therapy at all for secondary stroke prevention.
Adolescents who had a stroke as a child should continue transfusion into adulthood for secondary stroke prevention.
Adults who suffer their first stroke as an adult should receive the recommended evaluation for stroke-modifiable risk factors according to AHA guidelines. Secondary stroke prevention should include regular blood transfusion and other AHA-recommended measures.
Specific background.
Strokes in children and adults with HbSS or HbSβ0 thalassemia are one of the most common and devastating complications of the disease that produce lifelong sequelae, including but not limited to cognitive morbidity, increased risk of future cerebral infarcts, and earlier death.
Summary of the evidence.
The systematic review identified 12 studies, one of which was randomized and 4 of which were nonrandomized comparative studies. The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/c9d01e0189434d904716b2360f56c103.
Rationale and expected benefits.
Children and adults with HbSS or HbSβ0 thalassemia and strokes (overt) have ongoing risk for infarct recurrence even when receiving regular blood transfusion therapy. In 2 large multicenter retrospective cohort studies, children with HbSS or HbSβ0 thalassemia receiving regular blood transfusion therapy had ongoing risk of future symptomatic ischemic strokes. In the first large multicenter retrospective cohort study, 60 children with strokes (ischemic) receiving regular blood transfusion therapy were followed for a total of 191 patient-years. The incidence of stroke recurrence was 4.2 per 100 patient-years. There was a statistically significant reduction in stroke incidence compared with historical controls who had strokes and did not receive regular blood transfusion therapy.91 In a second large retrospective multicenter study, 137 children with strokes were followed for a total of 1382 patient-years (mean, 10.1 years; minimum, 5 years; maximum, 24 years) with an incidence rate of 2.2 events per 100 patient-years.76 HbS levels at the time of stroke recurrence were available for only 19% of patients, and most of the HbS values were <30%.
In a prospective multicenter single-arm trial of 40 children with strokes followed for a median of 5.4 years (total, 222 patient-years), the incidence of strokes was 3.1 events per 100 patient-years.92 All participants received regular blood transfusion with a mean pretransfusion HbS level of 29%, indicating that the goal of keeping maximum HbS concentration <30% was met. Furthermore, in this cohort, there was progressive vasculopathy on MRA imaging, with recurrent overt or silent cerebral infarct (RR, 12.7; 95% CI, 2.65-60.5; P = .01). Taken together, these 3 studies provide evidence that regular blood transfusion therapy is partially effective for secondary prevention of strokes in children and adults with HbSS or HbSβ0 thalassemia. However, recurrent stroke risk while patients are receiving regular blood transfusion therapy remains significant.
Hydroxyurea therapy is inferior to regular blood transfusion for secondary stroke prevention in children with HbSS or HbSβ0 thalassemia. No data exist for the other SCD phenotypes. In a single-center prospective feasibility study, 35 children on regular transfusion with a previous history of stroke were transitioned to hydroxyurea at the maximal tolerated dose and had serial phlebotomy to reduce transfusion iron overload. The stroke recurrence rate was 5.7 events per 100 patient-years. Phlebotomy significantly lowered iron burden and normalized hepatic iron.93 However, the analysis was not adjusted for the period of time with the highest rate of recurrent stroke, that is, the first 2 years.11,76,94 Based on these results, a phase 3 noninferiority randomized controlled trial, Stroke With Transfusions Changing to Hydroxyurea (SWiTCH), was conducted.95 In this trial, hydroxyurea at the maximal tolerated dose combined with phlebotomy was found to be inferior to continuing transfusion together with the ongoing requirement of iron chelation therapy because of excessive iron stores. The rate of secondary stroke was 5.6 events per 100 patient-years in the hydroxyurea arm compared with 0 events per 100 patient-years for children who continued with transfusion (the standard-care arm).95 The NHLBI leadership closed SWiTCH after interim analysis revealed equivalent liver iron content, indicating futility for the composite primary end point of allowing an increase in stroke, but requiring superiority for removing iron.
Where transfusion is not available, hydroxyurea therapy is better for secondary stroke prevention than no treatment at all. In a prospective nonrandomized study in Nigeria,96 13 children received hydroxyurea therapy, whereas 18 caregivers declined hydroxyurea therapy for their children. Maximum dose of hydroxyurea ranged from 20 to 25 mg/kg per day. The secondary stroke incidence of 7 events per 100 person-years in the hydroxyurea group was significantly lower than the 28 per 100 person-years in the nontreatment group (P = .001; odds ratio [OR], 3.80; 95% CI, 1.55-9.31).96
Overall, studies indicate that regular blood transfusion and hydroxyurea therapy are palliative for secondary prevention of strokes in children and adults with HbSS or HbSβ0 thalassemia, with blood transfusion being superior to hydroxyurea therapy and both therapies being superior to no therapy.
For children and adults with strokes and severe cerebral vasculopathy (moyamoya syndrome), alternative treatments such as revascularization surgery may be considered as adjunct therapy to regular blood transfusion therapy for secondary stroke prevention. HSCT is considered a curative50,97,98 and definitive option for secondary stroke prevention for children with matched related donors97 or haploidentical bone marrow transplantation with posttransplantation cyclophosphamide.98,99 Recent studies have revealed that new cerebral infarcts are detected after central adjudication of neuroimaging and neurology examination are performed as part of the HSCT protocol.100,101 Individuals and their families should be informed of the potential long-term benefits and long-term risks of secondary stroke prevention therapies.
Current and future therapeutic options for secondary stroke prevention should be considered in the context of a multicenter clinical trial setting with peer review, a data safety monitoring board to assess safety and futility, a study design that includes long-term follow-up of at least 5 years, and central adjudication of all neurological events. Without such approaches, there will continue to be less-than-optimal data to inform families about best strategies for secondary stroke prevention.
Summary of harms and burden.
The potential harms associated with regular blood transfusion therapy have been quantified in controlled clinical trials for primary and secondary prevention of strokes in children with HbSS or HbSβ0 thalassemia.
These adverse events include, but are not limited to, the following, in decreasing order of prevalence: excessive iron stores62,63,90 that may eventually require chelation therapy,64 red blood cell alloimmunization,25,65 and adverse blood transfusion reactions.64
The family makes a significant time commitment when they agree to regular blood transfusion therapy. Typically, blood transfusion occurs monthly and often requires 2 visits (the first for crossmatching of the red blood cell units and the second for the actual blood transfusion). We did not find a study describing the full range of challenges of regular blood transfusion therapy for families, but the panel, including the 2 patient representatives, believed strongly that family preferences and the inconvenience and financial resources associated with regular blood transfusion therapy should be considered.
EtD criteria and implementation considerations.
Despite the absence of a randomized controlled clinical trial comparing regular blood transfusion to maintain HbS level <30% and minimum hemoglobin >9.0 g/dL, a strong recommendation was made despite low-quality evidence based on the large difference in outcomes (stroke rate per 100 patient-years) that favored transfusion to maintain the HbS level at <30% when compared with no treatment of secondary stroke prevention. Additionally, the committee relied heavily on the studies describing the cerebral hemodynamics specific to SCD, the unique rheological studies of SCD, coupled with multidisciplinary experiences of the panel. The panel placed a high value on the uncertain, but potentially quality-of-life preserving, benefit of the intervention. Moreover, the burden and harms of transfusion were not felt to be prohibitive given the panelists’ and patients’ values focusing on reducing the risk of stroke over possible burdens and harms. Regular blood transfusion programs are resource-intensive. Patients’ and parents’ acceptance are variable. Caring for children in situations in which caregivers have strong beliefs against blood transfusion therapy may require that a court order be obtained in the best interest of the minor to provide the maximum benefit of decreasing future stroke recurrences.
Research needs.
The panel identified the following additional areas in need of research.
Optimal therapeutic strategies for secondary stroke prevention in children and adults (blood transfusion therapy vs blood transfusion therapy plus revascularization surgery vs HSCT) with long-term follow-up in children and adults with SCD are needed.
Optimal therapeutic strategies or secondary stroke prevention in low-middle–income settings where blood transfusion therapy is not available are needed.
Optimal transfusion targets and methods for secondary stroke prevention are needed.
Risk stratification to identify the group of children and adults with strokes likely to have infarct recurrence should be carried out.
Optimal treatment and stroke recurrence rate for children and adults other than those with HbSS or HbSβ0 thalassemia should be determined.
Should cerebral revascularization surgery (including EDAS, EDAMS, pial synangiosis, or direct anastomosis) plus regular blood transfusion therapy vs regular blood transfusion therapy alone be used for patients with SCD and moyamoya syndrome?
Recommendation 6
For adults and children with SCD, moyamoya syndrome, and a history of stroke or TIA, the ASH guideline panel suggests evaluation for candidacy for revascularization surgery in addition to continuing regular blood transfusions (conditional recommendation based on very low certainty in the evidence about effects ⊕◯◯◯).
Remarks:
Without evidence, but with compelling extensive experience, the panel endorses a multidisciplinary team evaluation including a hematologist, neurologist, neuroradiologist, and neurovascular surgeon for evaluation of the pros and cons of surgical evaluation and optimization of patients’ health before, during, and after surgery. The evaluation neither supports nor negates performing 1 of the 5 different revascularization procedures, but rather provides the background information for shared decision-making to consider surgery based on the available evidence for benefits and risks. However, if neurosurgery is considered, then a multidisciplinary evaluation is strongly preferred.
For individuals who undergo revascularization surgery, standardized care protocols and long-term outcome tracking (for a minimum of 5 years) through a local, national, or international prospective registry are encouraged.
Which of the 5 revascularization approaches, plus ongoing blood transfusion therapy, is more effective in secondary stroke prevention than regular blood transfusions alone is unclear. The quality of the studies does not allow for a pooled analysis nor an evidence-based recommendation-specific procedure with clear benefit, hence the reason for prospective standardized protocols and long-term outcome tracking.
Specific background.
Progression of cerebral infarcts and cerebral vasculopathy is common despite regular blood transfusion therapy for secondary stroke prevention. Some patients progress to a moyamoya vasculopathy with high risk of TIA, ischemic and hemorrhagic stroke,102 and cognitive decline.103 Outcome studies of children and adults with SCD who have moyamoya and have had a stroke have been generally limited to small or single-center series, with little follow-up beyond 5 years.104-111 No rigorous prospective controlled trial has been done to compare the benefits and risks of revascularization surgery plus regular blood transfusion therapy to regular blood transfusion therapy alone for secondary stroke prevention in children and adults with SCD and moyamoya syndrome.
Several studies have compared the incidence of strokes before and after revascularization surgery; however, such studies are intrinsically limited. The highest incidence rate for cerebral infarct recurrence occurs within 2 years of initial stroke, with or without preventive treatment.11,76,94 Table 1 in supplemental File 5 has the most recent summary of the revascularization procedures performed in SCD and the list of adverse outcomes that occur primarily in the first month after revascularization procedures. No pooled analysis of the studies could be completed because of the heterogeneity of the 5 different neurosurgery procedures (pial synangiosis, encephalo-duro-arterio-myo-synangiosis [EDAMS], encephalo-duro-arterio-synangiosis [EDAS], encephalo-myo-arterio-synangiosis, multiple burr holes), and the lack of uniform neurological assessment (surveillance of MRI of the brain and neurology assessment for infarct recurrence) during the follow-up period.
Future studies designed to assess the added utility of surgical revascularization in SCD-related moyamoya syndrome for secondary stroke prevention should include surveillance MRIs of the brain to identify silent cerebral infarcts, formal neurological assessment by a neurologist to identify subtle neurologic deficits associated with overt strokes because these subtle changes may alter the recommendation for future therapy, and adjustment for the high-risk period of stroke recurrence. To date, none of the published studies of cerebral revascularization in SCD have included these strategies to improve the scientific rigor of these studies, thus limiting the inferences of the potential benefit.
Summary of the evidence.
The systematic review identified 13 nonrandomized studies (Table 1). Five of the included studies were comparative (surgical intervention plus transfusion vs regular blood transfusion without surgery). The other 8 studies were noncomparative. Most studies did not report the SCD phenotypes of the participants, a major consideration for stroke recurrence.
The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/5bf40c8e961a95877aefa0af3d23dd09.
Rationale and expected benefits.
Most studies addressing the benefit and risk with cerebral revascularization surgery (pial synangiosis, EDAMS, EDAS, encephalo-myo-arterio-synangiosis, multiple burr holes) have been single-center observational studies (Table 1 in supplemental File 5). Due to the small sample sizes, the variation in revascularization procedures, and the incomplete reporting of the outcomes, studies could not be grouped to provide a composite assessment of the benefit of cerebral revascularization surgery. The type of revascularization approach that is most effective is unclear and depends on the individual patient, clinical context, and availability of surgical expertise. Further secondary stroke prevention studies in children and adults with SCD are needed to explore the risk-to-benefit ratio for surgery in addition to other therapies such as HSCT.
Summary of harms and burden.
The potential harms associated with cerebral revascularization procedures are considerable and include intraoperative and postoperative complications associated with SCD and periprocedural ischemic and hemorrhagic stroke. Surgical risk may be higher in children and adults with SCD than in the general population, so studies of cerebral revascularization for moyamoya syndrome in other populations may not be generalizable to patients with SCD. Given the small, single-center studies, coupled with the limited follow-up time (most <5 years), particularly compared with the lifespan of adults with SCD, the long-term benefits and risks of these revascularization procedures in children and adults with strokes and moyamoya syndrome have not been adequately assessed. We included studies only of individuals with strokes and moyamoya syndrome. We did not have sufficient evidence to evaluate surgery in children and adults with SCD and moyamoya syndrome alone or moyamoya syndrome and silent cerebral infarcts.
Hydroxyurea is a myelosuppressive agent and may impair wound healing. Hydroxyurea, when given before or after revascularization surgery, may prevent sprouting and growth of new cerebral blood vessels, as well as impairing wound healing after revascularization surgery.112,113
EtD criteria and implementation considerations.
Data on values and preferences, cost-effectiveness, acceptability, and feasibility of revascularization procedures are lacking. Variability in surgical expertise can limit the broad implementation of this recommendation. We also expect a wide range of how much an individual may value the immediate risk of perioperative and postoperative complications vs possible reduction of risk of a long-term outcome such as stroke, cognitive decline, or both.
Research needs.
The panel identified the following additional areas in need of research.
Rigorous studies that include longitudinal outcomes after revascularization surgery for moyamoya syndrome in SCD are needed.
Multicenter prospective studies or registries for individuals with SCD and moyamoya syndrome should be conducted and implemented as a first step to collect outcome data.
Should IV thrombolysis with tPA vs no treatment with tPA be used for adults with SCD presenting with acute ischemic stroke and no hemorrhage on CT scan within 4.5 hours of onset of symptoms?
Recommendation 7
For adults with SCD presenting with symptoms of acute ischemic stroke and being considered for IV tPA (age ≥18 years, no hemorrhage on CT scan, within 4.5 hours of onset of signs, symptoms, and without contraindications for thrombolysis), the ASH guideline panel suggests management using a shared decision-making approach that follows these principles:
For all patients, the administration of tPA should not delay prompt simple or exchange blood transfusion therapy.
Patients may be evaluated for IV tPA based on its established inclusion and exclusion criteria detailed in stroke-management algorithms.
The following factors suggest likely benefit from IV tPA: older age, atrial fibrillation, diabetes, hypertension, and hyperlipidemia. Management of younger patients without these risk factors should emphasize early transfusion.
There are no validated risk stratification or reliable age cutoff criteria to guide the choice of initial therapy.
IV tPA is not recommended for children with SCD (<18 years of age).
Remarks:
For Recommendation 7, the ASH guideline panel recognizes that prompt identification of an adult with SCD presenting to the emergency department with focal neurological deficit and balancing the timely treatment with IV tPA and timely treatment with blood transfusion therapy is challenging (Recommendations 4.1 and 4.2).
Evidence does not exist as to which treatment option should be provided first (tPA or blood transfusion). Conceptually, prioritization of treatment should be informed by underlying stroke etiology (SCD vs non-SCD), but this may not be clear in the hyperacute setting.
Given the increased overall survival of adults with SCD into middle and old age with the cumulative effect of traditional cardiovascular risk factors leading to stroke, offering emergent treatment with tPA to older adults with SCD presenting with acute ischemic strokes within 4.5 hours of symptom onset is advised. However, no absolute age cutoff could be defined.
In some cases, the treatment with tPA may occur before the patient has been recognized as having SCD. In such instances, blood transfusion should be considered as soon as possible after SCD is identified.
A systematic review on endovascular thrombectomy was not performed because we are unaware of any interventional studies specific to SCD; although multiple large clinical trials support thrombectomy for stroke in selected patients with acute large vessel occlusion outside SCD.
Evidence supports endovascular thrombectomy for stroke with acute large vessel occlusion in the general population; however, we do not have evidence of the risks and benefits in adults with SCD.
The utility of endovascular thrombectomy in adults with SCD should be carefully evaluated due to the prevalence of cerebral vasculopathy and moyamoya syndrome and the absence of data describing the benefits and risks.
Specific background.
To date, only one study has examined the use of tPA in adults with SCD.114 Overall, the SCD population is younger at the time of a first stroke and includes a higher proportion of people with hemorrhagic stroke compared with all adults with strokes.
Summary of the evidence.
The systematic review identified 3 nonrandomized studies, 1 of which was comparative. The other 2 were case reports. The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/c960529ed37314a2f4804dbd6a48ee31.
Rationale and expected benefits.
For adults with SCD presenting within 4.5 hours of the time when they were last seen to be normal after ischemic stroke onset who meet established eligibility criteria, IV tPA improves functional outcomes at 3 to 6 months poststroke.115 One study of tPA for hyperacute stroke compared outcomes of adults with and without an SCD diagnosis using administrative data from a large US health provider.114 There was no difference in efficacy or safety outcomes between the 2 groups, but the study was limited by lack of confirmation of SCD phenotype and probable inclusion of individuals with sickle cell trait. Although recognizing that the expected benefit of IV tPA for adults with SCD may differ from that in general medical practice because stroke etiologies differ, the ASH guideline panel suggests that adults with SCD and acute ischemic stroke be considered for IV tPA following established guidelines because of the strong evidence for improved outcomes in the general population. However, evidence for the benefit of tPA in adults with SCD is scant, and the potential harm associated with tPA is significant.
Summary of harms and burden.
Major risks of tPA include symptomatic intracerebral hemorrhage in 3% to 6% and life-threatening systemic hemorrhage.116 Use of thrombolytics for acute stroke may also delay prompt blood transfusion. tPA administration in a young adult with SCD presenting with an ischemic stroke may delay the administration of regular blood transfusion therapy, a therapy with a clear benefit in this population. The timing of tPA and acute blood transfusion therapy should be individually based.
EtD criteria and implementation considerations.
The acceptability and feasibility of the panel’s recommendation not to delay transfusion are limited by prehospital emergency medical systems that direct patients with suspected stroke to certified stroke centers, where stroke teams are trained to reduce door-to-needle times for treatment with IV tPA. Rapid implementation of stroke protocols for IV tPA treatment may result in the missed opportunity for prompt transfusion, particularly if adults are not identified with SCD. Conversely, stroke protocols typically suggest placement of 2 large-bore IV lines on arrival to medical care, which could facilitate more rapid simple transfusion and possibly exchange transfusion. Joint protocols including both IV tPA and transfusion should be developed by stroke teams and hematologists for rapid identification and management of patients with SCD presenting with acute ischemic stroke. A proxy with power of attorney is often beneficial for shared decision-making in the emergency setting when an acute stroke occurs.
Research needs.
The panel identified the following additional areas in need of research.
Rigorous studies of the safety of tPA use in individuals confirmed to have SCD and acute ischemic stroke are needed.
Systematic data collection in adults with SCD receiving IV tPA that includes stroke risk factors, presumed stroke etiology after workup, and outcomes should be carried out.
Implementation science studies designed to identify the optimal clinical practice for administering both tPA and acute blood transfusion therapy to adults with SCD presenting to the emergency department with acute ischemic strokes are needed.
Should clinicians perform or refer for screening for developmental delay and cognitive impairment vs no screening in children and adults with SCD?
Recommendation 8.1
Given the high prevalence of developmental delay and cognitive impairment, coupled with the guidelines set by the American Academy of Pediatrics, the ASH guideline panel recommends that clinicians supervising care of children with SCD conduct surveillance using simplified signaling questions for the following:
Concerns about developmental delays in preschool-age children
Concerns about neurodevelopmental disorders in school-age children with SCD, such as academic or behavioral problems or symptoms of inattention, hyperactivity, or impulsivity.
Recommendation 8.2
For children with SCD who have abnormal surveillance results suggesting increased risk for developmental delay or cognitive impairments, the ASH guideline panel recommends screening or referral for formal screening by a psychologist or a pediatrician able to perform screening with the available validated tools (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Recommendation 8.3
Given the high prevalence of cognitive impairment in adults with SCD, coupled with the guidelines set by the American Academy of Neurology, the ASH guideline panel recommends that clinicians supervising care of adults with SCD conduct surveillance for cognitive impairment using simplified signaling questions (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Recommendation 8.4
For adults with SCD who have abnormal surveillance results suggesting cognitive impairment, the ASH guideline panel recommends formal referral to a psychologist or a primary care physician able to perform more in-depth cognitive evaluation (strong recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Remark:
For Recommendation 8, the ASH guideline panel provides specific examples of signaling questions that can be used for screening in Table 2 in supplemental File 5.
Specific background.
Cognitive impairment is perhaps the most insidious CNS morbidity of SCD. In children with SCD, there is a clear gradient in full-scale intelligence quotient (FSIQ) depending on the degree of neurological injury (normal MRI, silent cerebral infarct, or stroke). In the most comprehensive meta-analysis to date, children with HbSS with normal MRIs of the brain, silent cerebral infarct, and strokes had mean FSIQs of 91, 84, and 73, respectively.6 Few comprehensive cognitive studies in adults with SCD have been done that include assessment of FSIQ, employment status, and assessment of cerebral infarct status. Despite the high prevalence of cognitive impairment, the diagnosis is difficult to discern and challenging to ascertain because of the requirement of formal cognitive testing.
In several health care settings, the potential for reimbursement is increasing; for example, the American Medical Association Current Procedural Panel has developed a new billing code (96127) for behavioral health screening that can be used to screen for cognitive impairment.117 Physicians, nonpsychologist staff, and practitioners have also seen a dramatic increase in cognitive screenings conducted, indicating that surveillance and screening are possible in settings without ready access to specialists in psychological assessment.118 Both the American Psychological Association and the National Academy of Neuropsychology have acknowledged that in many health care settings there is a need for medical doctors or nonpsychologist staff or practitioners to provide wider access to cognitive or behavioral health screenings, and these organizations have provided clarification on the distinction between screening and a more comprehensive psychological assessment requiring a specialist.119,120
The specific tests for cognitive screening in children and adults with SCD, and the optimal strategies to screen and implement rehabilitation for cognitive impairments, have not been established. Consequently, recommendations are extrapolated from non-SCD populations. Evidence-based practices for screening for cognitive impairments and rehabilitative interventions for cognitive function, specifically executive dysfunction, are well-established and endorsed practices in professional societies, such as the American Academy of Pediatrics, the American Academy of Neurology, and the American Congress of Rehabilitation Medicine. The guideline panel relied heavily on well-established evidence-based practices from these 3 prominent professional societies to identify strategies for screening and treatment of cognitive morbidities.121-123
Summary of the evidence.
The systematic review did not identify any studies that compared screening to no screening. The review identified 48 studies that evaluated cognitive screening without a comparison group and reported outcomes of IQ score, the prevalence of cognitive impairment, developmental delay, or school performance. The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/72b18bf3a46115c0989f3068aa701629.
Rationale and expected benefits.
The use of formal screening tools improves the ability to detect developmental delays or cognitive impairment compared with informal observational methods.124,125 Furthermore, the combination of surveillance and screening appears to work better for identifying children in need of developmental services than either method in isolation.126 Many screening tools can be used in primary care or community settings, require less administration time than full assessments, and can be administered by staff with less training, so that access is increased and burden to patients is decreased. See Tables 2 and 3 in supplemental File 5 for established initial screening tools and ongoing surveillance questions, respectively.
In the general population and in children and adults with SCD, identification of children and adults with developmental or cognitive delays increases likelihood of access to remediation services. In the United States and in most European countries, laws mandate interventions for children with significant developmental delay and conditions that impact learning in a public setting. If the child is <3 years of age, a significant developmental delay results in referral to early intervention therapy and the child and family undergo assessment for an individualized family service plan.127 Services are typically provided in the home or “natural environment” and include occupational therapy, physical therapy, developmental therapy, and speech therapy. These services are provided to enhance the development of the child. If the child or young adults is between 3 and 21 years of age, and a significant impairment is identified, the local public school district is tasked with working with the family to develop an individualized education program. The program provides services in the public school educational setting including supplemental aides if indicated, learning accommodations, and modifications.127 Both the individualized family service plan and individualized education plans include measurable goals, assessments, and meetings to review the children’s progress. If a child is not making progress, the plans are typically modified to provide better support.
A recent systematic review of cognition across the lifespan of people with SCD confirmed that the magnitude of deficits increases as the injury to the brain increases. On average, performance declined from groups without infarct to those with silent cerebral infarct to those with overt stroke.6 In addition, medium to large differences in cognition between children with SCD and their siblings or controls existed. Fewer adult studies were included, but small to large differences between the adults with SCD and controls were documented, with the greatest difference in processing speed.6
Vascular cognitive impairment is of particular concern in adults with SCD and requires ongoing efforts to identify cognitive symptoms and medical risk factors.128 Detection of cognitive impairments is important for identifying treatable causes, helping patients and families to understand the cause of functional deficits, and discussing the prognosis to plan for future needs.123 Outpatient services such as speech therapy, occupational therapy, or rehabilitation psychology may be indicated and available depending on the results of a full evaluation.129
In general, children and adults with SCD are a vulnerable population. The overwhelming majority of patients in the Unites States with this condition are African American130 and living at or near poverty level131-133 ; associated social factors such as parental education attainment and measures of socioeconomic status impact cognitive performance.134-137 In addition, in children with HbSS or HbSβ0 thalassemia and disease-associated morbidity such as low oxygen hemoglobin saturation levels, silent cerebral infarcts and overt strokes are all associated with lower FSIQ scores.134,138
Summary of harms and burden.
There are few downsides to surveillance and screening for cognitive impairment, developmental impairments, or both, as part of routine health care. Available cognitive-screening methods improve detection yet lack sufficient sensitivity to be used as a substitute for clinical judgment.120 For cases in which the patient’s history, risk factors, and functional complaints indicate a high risk for cognitive impairment, screening may be skipped in favor of directly referring for a comprehensive assessment. False-positive results from screening can also result in distress and inconvenience for patients and their families.139,140 Such distress likely already exists for patients and families who already have concerns about developmental delays or cognitive impairment.
EtD criteria and implementation considerations.
The quality of evidence demonstrating benefits of cognitive and developmental screening specifically in individuals with SCD is low. However, such screening is noninvasive and can lead to timely referral to address those with developmental delays and cognitive impairment. The harm of not addressing such deficits in children and adults is severe on a personal and societal level. Therefore, a low certainty in benefit but high certainty of harm (particularly from other populations) justified a strong recommendation.141 When considering patient values and preferences, the panel members, including the 2 patient representatives, reported values with little variation in favor of screening.
Critical resources are needed to screen children and adults for developmental delay and cognitive impairment. Such resources are likely more available in medical centers with comprehensive care for individuals with SCD than in primary care settings. Resources and tools for screenings have been designed for primary care settings and are widely available; however, practitioners need to prioritize screening followed by periodic surveillance as part of routine health care. Patients may have to travel to obtain appropriate cognitive and developmental screening, particularly in managed-care settings.
Research needs.
The panel identified the following additional areas in need of research.
Better documentation of the prevalence and progression of cognitive impairments in adults with SCD is needed.
Evaluation of screening and surveillance approaches for cognitive and developmental concerns assessed within the SCD population should be developed, rather than relying on data from broader populations.
Research evaluating implementation practices that produce the best access to screening, surveillance, and assessment for developmental delays and cognitive impairments, or both, is needed.
Future research is required to determine which development and cognitive-screening tools have the highest clinical utility in low-middle– and high-income settings.
Should cognitive rehabilitation therapy vs no rehabilitation be used for children and adults with SCD and cognitive deficit?
Recommendation 9.1
For children with SCD and abnormal screening for developmental or cognitive status, the ASH guideline panel recommends the following:
A developmental, cognitive, medical evaluation to diagnose any related disorders and to identify modifiable risk factors for developmental delays or cognitive impairments;
Following the cognitive domain-specific evidence-based guidelines for these conditions to provide appropriate interventions.
Recommendation 9.2
For adults with SCD and abnormal screening for cognitive status, the ASH guideline panel recommends the following:
A developmental, cognitive, and medical evaluation to diagnose any related disorders and to identify modifiable risk factors for cognitive impairments;
Following the cognitive domain-specific evidence-based guidelines for these conditions to provide appropriate interventions.
Specific background.
As previously mentioned, cognitive impairment is perhaps the most insidious CNS morbidity of SCD. Evidence-based rehabilitative interventions for cognitive function, specifically executive dysfunction, are well-established and endorsed practices in professional societies, such as the American Academy of Pediatrics, the American Academy of Neurology, and the American Congress of Rehabilitation Medicine.122,123,142 The guideline panel relied heavily on these well-established evidence-based practices treatments of cognitive morbidities.
Summary of the evidence.
The systematic review did not identify any comparative studies in SCD that compared cognitive rehabilitation to no rehabilitation. The review identified 1 randomized controlled trial that compared academic tutoring with memory rehabilitation vs academic tutoring, 1 observational study that compared academic tutoring and specific learning and memory strategies with academic tutoring, and 1 observational study that compared computer-based cognitive training completers with noncompleters. The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/c35a65914af486fba822a798f2059f64.
Rationale and expected benefits.
Cognitive impairment in SCD characteristically affect specific mental processes such as memory, attention, executive function, processing speed, and visual-spatial function. These deficits can impair one’s learning, educational performance, work performance, medical adherence, and activities of daily living. However, these deficits will not be adequately identified without screening. Deficits in executive function are commonly found in children and adults with SCD.6 These deficits correlate with the frequent injury to the frontal lobe, a frequent area of the brain affected by silent cerebral infarcts.78,143
A systematic review of rehabilitation for impairment of executive functions found a solid evidence base for interventions that incorporate metacognitive strategy instruction that offers strategies to solve problems, increase awareness, and cue for signals that may improve problem solving and internal training. Most of the available studies were conducted among adults with traumatic brain injury or stroke.144 However, pilot studies that applied similar techniques to children with SCD generated positive results.145 Cognitive lapses, which occur most often in individuals with cognitive impairments, and aspect of executive dysfunction (eg, defects in mental flexibility and working memory) are the most common cause of poor adherence to medical therapies in the general population.146-148
Functional assessments of executive function can both identify deficits and inform interventions by determining what types of cues patients need while completing multistep activities (Figure 3 in supplemental File 5).149,150 Once these deficits are diagnosed, interventions can be implemented (Figure 4 in supplemental File 5).142
The intervention that makes the largest difference is the Cognitive Orientation to Daily Occupational Performance (CO-OP),151 a top-down approach that reduces impairments and improves health. “Developed for use with children who have occupational performance deficits, CO-OP is an individualized, client-centered approach focused on strategy-based skill acquisition” through a process of strategy use and guided discovery.122 CO-OP focuses treatments directly on improving performance in everyday life activity rather than treating the underlying impairments and hoping for secondary improvement in meaningful activities. Three randomized controlled trials have demonstrated that CO-OP is more effective than impairment-focused therapy delivered by occupational therapists.152-154
Failure to identify and address cognitive impairments contributes to difficulties with medical adherence. Formal assessments of cognition providing an objective evaluation of the patient’s cognitive capacities are often requested after cerebral injury is diagnosed by cerebral MRI scan. The assessment enables health care staff and the family to better understand the patient’s need for information, support, and rehabilitation. The identification of neurocognitive impairments is an important first step in obtaining rehabilitation therapy121 and is recommended by national organizations representing several relevant disciplines.129 In addition, hematologists and other medical providers can make use of behavioral and family supports (eg, CO-OP approaches) to better support adherence.129,148,155
Summary of harms and burden.
No harm is anticipated after providing cognitive rehabilitation. The panel did not identify any burden with cognitive rehabilitation. Rather, a commitment to time with occupational therapists is required for patients to receive appropriate services. Working with the occupational therapist for individuals with cognitive impairment requires a shift in the approach to treatment of both providers and individuals with the disease to increase awareness of cognitive impairments and to offer cognitive rehabilitation therapies. Individuals with SCD should be treated in a manner similar to that used for the thousands of individuals without SCD, but with brain injury due to stroke or trauma.
EtD criteria and implementation considerations.
The quality of evidence on the benefits of cognitive rehabilitation specifically in individuals with SCD was low. Hence, the panel formulated recommendations advising clinicians to follow disease-specific evidence-based guidelines for conditions identified through screening. The certainty of the net benefit of this recommendation was considered high. In addition, despite the lack of published literature, input from patient representatives on the panel supported the acceptability of cognitive rehabilitation therapy. Resources are likely more available in academic centers with expertise in SCD.
The American Congress of Rehabilitation Medicine endorses behavioral strategies to support individuals with executive function impairments. Executive functions are a set of processes that focus on managing oneself and one’s resources in order to achieve a goal, involving mental control and self-regulation.121 After recognizing executive dysfunction, the person can be trained to use task-specific approaches, problem-solving strategies, external cues, and internalized strategies.156
Systematic reviews of randomized controlled trials support the use of goal-management training, problem-solving techniques, and time pressure management.156,157 Challenges with following through on plans of daily life that include medical appointments, medications, education, and work are frequently underappreciated as cognitive impairments. Unfortunately, those around the affected individuals may label such impairments as problem behaviors or lack of motivation. Similarly, the affected individual may be unaware of these limitations as well. Despite endorsement from multiple different professional societies, implementing cognitive-screening strategies in high-risk children and adults with SCD will be challenging to implement without basic research strategies to identify barriers and facilitators within SCD care centers.
Research needs.
The panel identified the following additional areas in need of research.
Testing of specific cognitive rehabilitation strategies for people with SCD is needed.
The optimal setting for cognitive rehabilitation therapy should be identified.
The individuals most likely to benefit from cognitive rehabilitation therapy should be identified.
Should screening with MRI for silent cerebral infarcts vs no screening be used for children and adults with HbSS or HbSβ0 thalassemia?
Recommendation 10.1
Given the high prevalence of silent cerebral infarcts in children with HbSS or HbSβ0 thalassemia (1 in 3) and their association with cognitive impairment, poor school performance, and future cerebral infarcts, the ASH guideline panel recommends at least a 1-time MRI screening, without sedation, to detect silent cerebral infarcts in early-school-age children, when MRI can commonly be performed without sedation (strong recommendation based on moderate certainty in the evidence about effects ⊕⊕⊕◯).
Recommendation 10.2
Given the high prevalence of silent cerebral infarcts in adults with HbSS or HbSβ0 thalassemia and their association with cognitive impairment, poor school performance, and future cerebral infarcts, the ASH guideline panel suggests at least a 1-time MRI screening without sedation to detect silent cerebral infarcts in these adults (conditional recommendation based on low certainty in the evidence about effects ⊕⊕◯◯).
Remarks:
The definition of a silent cerebral infarct–like lesion is an MRI signal abnormality at least 3 mm in 1 dimension and visible in 2 planes on fluid-attenuated inversion recovery (FLAIR) or T2-weighted images (or similar image with 3-dimensional [3D] imaging) with no correlative neurological findings.
After an infarct-like lesion is identified, the panel recommends the following plan of action:
Neurological evaluation to assure that infarcts are classified as a silent cerebral infarct rather than overt stroke.
After a silent cerebral infarct is detected, there should be a discussion regarding:
Secondary prevention options including regular blood transfusions and HSCT.
Cognitive screening assessment, as per recommendation 8.
MRI surveillance every 12 to 24 months to assess for cerebral infarct progression. If new infarcts are present, then a discussion with the patient and family regarding the pros and cons of a step-up in therapy intensity to prevent cerebral infarct recurrence.
Specific background.
Silent cerebral infarcts are common in children and adults with HbSS or HbSβ0 thalassemia, with a prevalence of ∼39%3 and ∼50%, respectively (Figure 2)4 An individual with SCD is diagnosed as having a silent cerebral infarct if the following 3 features are present: (1) no history of focal neurologic deficits; (2) on an MRI of the brain, a T2-weighted image with FLAIR signal abnormality that is at least 3 mm in 1 dimension and that is visible in 2 planes (or similar image with 3D imaging); and (3) a normal neurological examination, preferably conducted by a neurologist, or an abnormality found on examination that could not be explained by the location of the brain lesion or lesions.25
This definition of silent cerebral infarct in children has been validated. A definition of silent cerebral infarcts that requires a 5-mm size with corresponding T1-weighted hypointensity on MRI, instead of 3-mm only, will lead to a large misclassification bias with fewer children being identified with silent cerebral infarcts.158 A minimum size for silent cerebral infarct of 3 mm has been used in adult SCD studies and is predictive of infarct recurrence (Figure 8).26
The diagnosis of a silent cerebral infarct can be challenging if the radiologist is unfamiliar with the definition of silent cerebral infarct in SCD. The definition of silent cerebral infarct cannot be extrapolated to include the common definition of lacunar strokes in the general population,143 which includes a T1 hypointensity in addition to a 5-mm FLAIR hyperintensity.158
When available, the imaging should be done on a 3.0 T magnet instead of a 1.5 T to improve the detection of silent cerebral infarcts (Figure 8). As new FLAIR sequences are acquired via whole-brain 3D imaging with no gaps between slices, the requirement for imaging in 2 planes to confirm a silent infarct may not be necessary.
Imaging examples of silent cerebral infarcts and mimics, specifically, Virchow-Robin spaces and periventricular leukomalacia, which may be useful for a clinician are displayed in Figure 8.
The Silent Cerebral Infarct Transfusion (SIT) Trial demonstrated that blood transfusion therapy is superior to observation for secondary stroke prevention (overt or silent cerebral infarct with a 56% RR reduction in cerebral infarct recurrence).25 However, the number needed to treat to prevent infarct recurrence with regular blood transfusions is 13. There are no controlled trial data demonstrating the noninferiority of hydroxyurea therapy to regular blood transfusion therapy for children or adults with silent cerebral infarcts.
SCI and mimics in SCD. (A) Axial (left) and coronal fluid-attenuated inversion recovery (FLAIR) images illustrate a qualifying silent cerebral infarct in the left parietal lobe (white arrows). An infarct-like lesion was defined as an MRI signal abnormality that was at least 3 mm in 1 dimension and that was visible in 2 planes on FLAIR images or similar image with 3D FLAIR sequence (not shown) and documented neurological examination performed by a neurologist demonstrating that the participant has a normal neurologic examination or an abnormality on examination that could not be explained by the location of the brain lesion(s). (B) Axial FLAIR (left), T2-weighted (middle), and T1-weighted (right) images of the same lesion demonstrate that the FLAIR sequence is better for the identification of SCIs (green circles). (C) Axial FLAIR images show that higher magnet strength (3.0 Tesla) improves image quality and identification of subtle lesions only seen at 3.0 Tesla (arrowhead). More obvious lesions are visible at 1.5 Tesla and 3.0 Tesla (arrows). (D) Linear and punctate T2 hyperintensities that suppress on FLAIR are consistent with prominent perivascular spaces (Virchow-Robin spaces). (E) Terminal zones of myelination on T2-weighted images. Axial FLAIR and T2-weighted images show ill-defined symmetrical T2-weighted hyperintensity in the deep parietal white matter. The T2-weighted image on the right illustrates that there are well-defined linear perivascular spaces extending throughout the area of subtle hyperintensity (green arrows). (F) Axial FLAIR images demonstrate a case of white matter injury in a premature infant (periventricular leukomalacia) that can mimic a SCI because of the increased signal on FLAIR (white arrows).
SCI and mimics in SCD. (A) Axial (left) and coronal fluid-attenuated inversion recovery (FLAIR) images illustrate a qualifying silent cerebral infarct in the left parietal lobe (white arrows). An infarct-like lesion was defined as an MRI signal abnormality that was at least 3 mm in 1 dimension and that was visible in 2 planes on FLAIR images or similar image with 3D FLAIR sequence (not shown) and documented neurological examination performed by a neurologist demonstrating that the participant has a normal neurologic examination or an abnormality on examination that could not be explained by the location of the brain lesion(s). (B) Axial FLAIR (left), T2-weighted (middle), and T1-weighted (right) images of the same lesion demonstrate that the FLAIR sequence is better for the identification of SCIs (green circles). (C) Axial FLAIR images show that higher magnet strength (3.0 Tesla) improves image quality and identification of subtle lesions only seen at 3.0 Tesla (arrowhead). More obvious lesions are visible at 1.5 Tesla and 3.0 Tesla (arrows). (D) Linear and punctate T2 hyperintensities that suppress on FLAIR are consistent with prominent perivascular spaces (Virchow-Robin spaces). (E) Terminal zones of myelination on T2-weighted images. Axial FLAIR and T2-weighted images show ill-defined symmetrical T2-weighted hyperintensity in the deep parietal white matter. The T2-weighted image on the right illustrates that there are well-defined linear perivascular spaces extending throughout the area of subtle hyperintensity (green arrows). (F) Axial FLAIR images demonstrate a case of white matter injury in a premature infant (periventricular leukomalacia) that can mimic a SCI because of the increased signal on FLAIR (white arrows).
Summary of the evidence.
The systematic review did not identify eligible studies that compared screening by means of MRI scan of the brain to no screening. The review identified 15 studies that reported the yield of screening, a study that showed the cumulative prevalence of silent cerebral infarct in children, and 1 randomized controlled trial that showed the benefit of regular blood transfusion therapy vs observation in decreasing cerebral infarct recurrence in children with HbSS or HbSβ0 thalassemia who underwent MRI of the brain without sedation and were noted to have silent cerebral infarcts.
The evidence summary and EtD framework can be found online at: https://guidelines.gradepro.org/profile/e53297aadb1f5346b6de0d64eebccacf.
Rationale and expected benefits.
There are 5 independent reasons to justify screening for silent cerebral infarct in children and adults with HbSS or HbSβ0 thalassemia:
Silent cerebral infarcts are prevalent; ∼39% of children3 and 50% of young adults will have a silent cerebral infarct.4
Silent cerebral infarcts are progressive in both children25,159 and adults26,27 (Figure 9A-B, respectively). The presence of silent cerebral infarcts predicts future neurological injury to the brain with an incidence rate that exceeds the accepted threshold for prevention of neurological injury in adults with atrial fibrillation not receiving anticoagulation.
Silent cerebral infarcts are associated with at least a 5-point FSIQ drop in children,134 and biologically plausible evidence to believe a similar degree of neurological morbidity in adults.
Once silent cerebral infarcts are identified, children and adults are eligible for evaluation for individual education plans and Americans with Disability Act services, respectively.
Most silent cerebral infarcts occur in the border zone regions of the brain, including in the frontal lobe,72 which disproportionately affect executive function. The American Congress of Rehabilitation Medicine has formally endorsed evidence-based strategies to support individuals with executive dysfunction.
Children with silent cerebral infarcts can be treated with regular blood transfusion to substantially reduce the incidence of a new stroke, silent infarct recurrence, or both.25
Preexisting SCIs. (A) Preexisting SCIs associated with subsequent neurological events in children with HbSS or HbSβ0 thalassemia. Time to first neurological event-stroke, seizure, TIA for children with normal or conditional TCD measurements (time averaged mean maximum velocity of <200 cm/s, nonimaging, or <185 cm/s imaging technique) and with (n = 68) and without SCIs on MRI (n = 353). (B) Associated with recurrent SCIs in adults with sickle cell anemia. A total of 54 adults with HbSS or HbSβ0 thalassemia had a minimum time of 6 months between at least 2 MRIs of the brain; in this group of adults, 43% (n = 23) had SCI at baseline and 57% (n = 31) had no SCI at baseline; individuals with overt stroke were excluded based on history and examination by a neurologist. Reprinted from Jordan et al159 (A) and Jordan et al26 (B) with permission.
Preexisting SCIs. (A) Preexisting SCIs associated with subsequent neurological events in children with HbSS or HbSβ0 thalassemia. Time to first neurological event-stroke, seizure, TIA for children with normal or conditional TCD measurements (time averaged mean maximum velocity of <200 cm/s, nonimaging, or <185 cm/s imaging technique) and with (n = 68) and without SCIs on MRI (n = 353). (B) Associated with recurrent SCIs in adults with sickle cell anemia. A total of 54 adults with HbSS or HbSβ0 thalassemia had a minimum time of 6 months between at least 2 MRIs of the brain; in this group of adults, 43% (n = 23) had SCI at baseline and 57% (n = 31) had no SCI at baseline; individuals with overt stroke were excluded based on history and examination by a neurologist. Reprinted from Jordan et al159 (A) and Jordan et al26 (B) with permission.
Summary of harms and burden.
The summary of harms and burden is the same as for PICO questions #1 and #2 (see Table 1). The potential harms associated with regular blood transfusion therapy have been quantified in controlled clinical trials for primary and secondary prevention strokes in children with HbSS or HbSβ0 thalassemia.
These adverse events include, but are not limited to, the following, in decreasing order of prevalence: excessive iron stores62,63,90 that may eventually require chelation therapy,75 red blood cell alloimmunization,11,25,65 and adverse blood transfusion reactions.64,75 The burden of indefinite regular blood transfusion therapy is significant for the family. Typically, the regular blood transfusion occurs monthly and often requires 2 visits (the first for crossmatching of the red blood cell units and the second for the actual blood transfusion). We did not find a study rating the intangible challenges of regular blood transfusion therapy for families, but the panel, including the 2 patient representatives, believed strongly that family preferences and the inconvenience and financial resources associated with regular blood transfusion therapy should be considered. Despite the burden, the panel believed that both children and adults should be aware of whether they have a silent cerebral infarct, an injury to the brain that places the individual at risk for future cerebral infarcts and may impact educational attainment in children and adults, as well as employment and family support in adults.
EtD criteria and implementation considerations.
Access to MRI may be limited in some geographic areas or low-middle-income settings. Cost-effectiveness data are unavailable, but can be extrapolated from other environmental exposures associated with neurologic sequelae, low IQ, and social outcomes.
A hematologist’s diagnosis of only silent cerebral infarct in a child with HbSS or HbSβ0 thalassemia will misclassify ∼7% of the children as having a silent cerebral infarct, when in fact they had a stroke.25 This misclassification of a stroke as a silent cerebral infarct may result in a different clinical course in treated and untreated individuals. In the presence of a silent cerebral infarct, annual surveillance with MRI may allow for increased therapeutic interventions; thus, a second screening MRI can be considered if new or cognitive impairment occurs or a change in academic performance is noted. A child who cannot undergo an MRI without sedation may be supported by child life services to attempt MRI without sedation.
Research needs.
The panel identified the following additional areas in need of research.
A therapeutic strategy for primary prevention of silent cerebral infarcts is needed.
Imaging strategies to identify subgroups of children and adults likely to have infarct recurrence are needed.
Alternative treatment strategies, other than regular blood transfusion, for secondary prevention of infarct recurrence in children and adults with silent cerebral infarcts should be developed.
The clinical benefit of HSCT or gene therapy vs regular blood transfusion therapy for secondary prevention of cerebral infarcts in children and adults with preexisting silent cerebral infarct should be determined.
The optimal treatment and infarct recurrence rate for children and adults with SCD phenotypes other than HbSS or HbSβ0 thalassemia, and with silent cerebral infarcts should be determined.
The clinical utility of screening for silent cerebral infarcts in low-middle–income settings with MRI scans is unknown. Furthermore, the neuroradiology expertise is far less available. If feasible, screening for silent cerebral infarcts in children and adults with HbSS in a low-middle–income country should be done for the same reason that the screening occurs in high-income settings.
What are others saying and what is new in these ASH guidelines?
SCD is a rare disease, with few guidelines developed for prevention, screening, and treatment of CNS manifestations in children and adults with SCD. For only 2 of 10 PICO questions did we have the benefit of phase 3 randomized controlled trials on which to base our recommendations: screening and treatment of primary stroke prevention (with TCD) and screening and treatment of silent cerebral infarct to prevent cerebral infarct recurrence. The responses to the remaining PICO questions were based on review of all available observational studies, including cerebral hemodynamic studies in SCD.
With the exception of primary and secondary stroke prevention, guidelines for management of common CNS problems in SCD were not discussed in the 2014 NHLBI Expert Panel Report on the Evidence-Based Management of Sickle Cell Disease. The SIT Trial25 was published after the 2014 panel review and was not referenced in the 2014 NHLBI Expert Panel Report.162 The recent AHA/ASA statement on pediatric stroke management86 provides guidance for initial management of suspected or confirmed acute ischemic strokes in children with SCD, as well as primary and secondary stroke prevention with regular blood transfusion. Their recommendations are similar to the ASH CNS panel recommendation.
For children and adults with SCD presenting with a focal neurological deficit, the panel recommends increasing the hemoglobin level with a red blood cell transfusion to achieve the goal of improving oxygen delivery to the brain. Given the challenge of distinguishing between MRI diffusion-weighted negative ischemic strokes and TIAs,163 the clinical decision to manage an individual with SCD and a suspected ischemic infarct should not be based solely on the results of the MRI, but rather should be a bedside decision where the risks and benefits of transfusion must be considered. In most cases, the benefit of transfusing a child or adult with SCD and with acute focal neurologic deficits will outweigh the risks.
The panel did not include the role of HSCT for primary and secondary stroke prevention, an emerging treatment strategy in high-income settings. The panel’s absence of any recommendation for HSCT for primary stroke prevention or secondary prevention of infarct recurrence does not reflect an absence of data or priority for the panel, but rather a decision to defer this subject matter to the ASH HSCT Guideline Panel.
The panel members determined that there was sufficient evidence to support guidelines for initial screening and subsequent surveillance for developmental delays and cognitive impairment for the general population, where the prevalence of impairments is significantly lower than in children and adults with SCD, to be applied to individuals with SCD. The panel’s strong recommendation for screening for developmental delay and cognitive impairment in children with SCD was based on 3 factors. First, the American Academy of Pediatrics recommends that all children be screened for both developmental delay and cognitive impairment122 ; second, children with SCD have a high prevalence of these impairments; and third, there is high impact for securing educational resources for children with significant impairments via individualized education plans. Similarly, the ASH guideline panel’s conditional recommendation for screening for cognitive impairment in adults with SCD was based on at least 2 dominant factors. The first was the recommendation by the American Academy of Neurology for screening for mild cognitive impairment in adults123 ; the second was the high prevalence of these impairments in adults with SCD.6 Research in implementation science is required to define the optimal reach (proportion of individuals that receive screening and ongoing surveillance) for detecting developmental delay and cognitive impairment in SCD. The ASH Guideline Panel endorsed the evidence-based recommendations by the American Congress of Rehabilitation Medicine for cognitive rehabilitation.121
Revision or adaptation of the guidelines
Plans for updating these guidelines
After publication of these guidelines, ASH will maintain them through surveillance for new evidence, ongoing review by experts, and regular revisions.
Updating or adapting recommendations locally
Adaptation of these guidelines will be necessary in many circumstances. These adaptations should be based on the associated EtD frameworks.164
Acknowledgments
The authors acknowledge their 2 patient representatives, Tabitha Barber and Maria Rivera, who provided invaluable insight into the impact of strokes in the lives of children and adults with SCD. They also provided key perspectives about families with regard to the importance of having autonomy, being well informed about their neurological morbidity, and knowing the full range of treatment options, even when an option is perceived to be inferior to standard care. The authors also acknowledge Starr Webb, Kendall Alexander, and Robert Kunkle from ASH for their outstanding leadership in bringing the panel together and unyielding support for this project. M.R.D. also acknowledges Leshana Saint-Jean for her outstanding support of organizing the references and proofing the document.
Authorship
Contribution: M.R.D. wrote the first draft of the manuscript; as an iterative process, L.C.J. and M.H.M. revised the manuscript based on the suggestions of the other authors; guideline panel members (A.A.K., J.S., E.V., C.K.F., R.C.M., P.T., M.A.K., L.D., and F.J.K.) critically reviewed the final version of the manuscript and provided suggestions for improvement; L.D. led the team of investigators from the Mayo Clinic Evidence-based Practice Center who conducted the evidence synthesis; all authors approved the content; M.R.D. was chair of the panel, with CNS content knowledge expertise; M.H.M. jointly chaired the panel, with content knowledge of guidelines and systematic review expertise; and M.R.D. and M.H.M. led multiple panel meetings.
Conflict-of-interest disclosure: All authors were members of the guideline panel, the systematic review team, or both. As such, they completed a disclosure-of-interest form, which was reviewed by ASH and is available as supplemental Files 2 and 3.
Correspondence: M. R. DeBaun, Vanderbilt-Meharry Center of Excellence in Sickle Cell Disease, Vanderbilt University Medical School, 2525 West End Ave, Suite 750, Nashville, TN 37203-1738; e-mail: m.debaun@vumc.org.



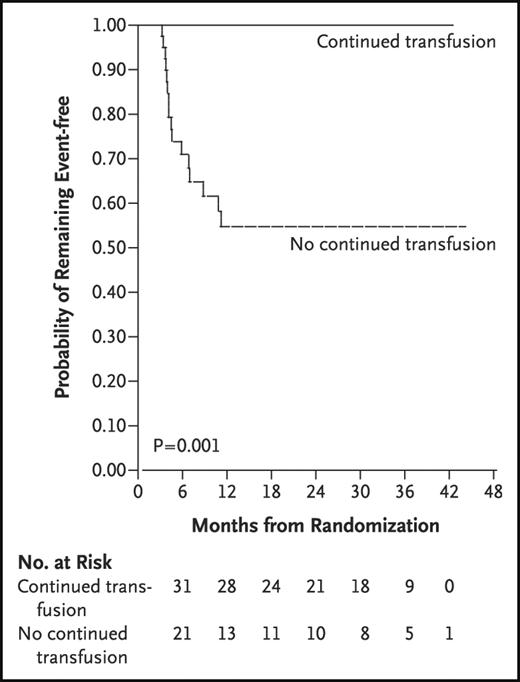
![Pooled analysis of the 10 studies documenting TCD measurement before and after hydroxyurea therapy in children with HbSS or HbSβ0thalassemia. This meta-analysis demonstrates the average drop in TCD measurement after starting hydroxyurea therapy of 21 cm/s (95% confidence interval [CI], 14.8-29.0). The plot also suggests that the decrease in TCD measurements can be seen as early as 3 months after the start of hydroxyurea therapy with a sustained impact of hydroxyurea therapy on decreasing TCD measurements for at least 36 months. The analysis is updated from a previous one by DeBaun and Kirkham,58 plus additional references.51-57,59,60,165 ♦ represents the pooled estimate from a random-effect model, the edges of the diamonds represent the 95% CI; ▪ represents individual studies. MTD, maximum tolerated dose.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/8/10.1182_bloodadvances.2019001142/2/m_advancesadv2019001142cf4.png?Expires=1768075288&Signature=ELLy3qqw0EJRNogfuM6HYWdjx2hF~4PVuOFkbiPO5r-fv8m6UZOhXSA9rFlw0PhpxAuoV9Ky4z-eK9fv~JJ6uk4abWfbXeKdMRfjmvdBXiEQCk0JwAbVbyvV~PTxck9QoCY0Sphk0rBjrAP7z7A8r~dqUx~UKabEmp3OIE6HFcVG6D5-HT0ZITJ46JbShJ~e9kvsFgLiloRqx4FJTPFLjE0WFFAz6qdcdETlsWG~3KCz9I6FiuV5PlPjxRUjjWRnfif1hTk3-1N9y2f5LdXrN0-J6Ed-d5uUsbJ7llCknpxtXBtAoMwY0te7ZE3Ghzb1vRlx84EDWLtEMFu49yOiyg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)