Key Points
Acquired aplastic anemia is a T-cell–mediated autoimmune bone marrow aplasia, without a known etiologic trigger.
Clonal expansion of CD8+ effector T lymphocytes can occur following vaccination and accompany graft dysfunction or aplastic anemia relapse.

Introduction
Acquired aplastic anemia (AA) is an immune-mediated bone marrow aplasia caused by cytotoxic T lymphocyte attack on early hematopoietic cells.1 In most patients, no trigger or specific mechanism of autoimmunity is identified.2 Without understanding the causes of AA autoimmunity, rationally designed prevention and treatment are not possible. Barriers to identifying triggers of AA include the rarity of AA and the inability to analyze patient samples before AA onset at the time of exposure to the putative trigger.
Several cases of AA onset or relapse following immunizations have been reported.3-8 Based on these clinical observations, the British Society for Standards in Haematology recommended against vaccinating AA patients treated with immunosuppression because of the concern for triggering AA relapse.9 However, beyond clinical descriptions of temporal association, evidence to support this recommendation is lacking. Here, we describe AA relapse post–bone marrow transplant (BMT) after vaccination and present a detailed immunologic analysis demonstrating a massive clonal expansion of CD8+ T effector memory–like (TEM) lymphocytes postvaccination.
Case description
A 31-year-old male with severe AA was treated with fludarabine (120 mg/m2), cyclophosphamide (1200 mg/m2), and rabbit anti-thymocyte globulin conditioning,10 followed by an infusion of 2.1 × 108 total nucleated (1.1 × 106 CD34+) bone marrow cells per kilogram from his HLA-identical sister. Graft-versus-host disease prophylaxis was methotrexate and cyclosporine. Neutrophil and platelet engraftment occurred on days +26 and +33, respectively. At 6 months posttransplant, the patient had nearly normal blood counts, with a white cell count of 3.8 × 109 cells per liter with 64% neutrophils, hemoglobin of 11.8 g/dL, an absolute reticulocyte count of 71 × 109 cells per liter, and platelet count of 217 × 109 cells per liter. Chimerism analysis revealed >99% CD33/CD66b+ myeloid donor chimerism, but only 7% CD3+ donor T cells.
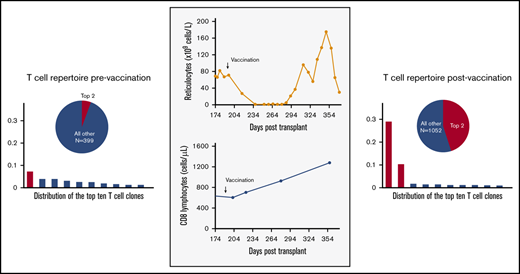
Six months after transplant, while continuing cyclosporine, the patient received concurrent pneumococcal conjugate and inactivated influenza vaccines. Within 1 week of vaccination, the patient’s absolute reticulocyte count dropped to 27 × 109 cells per liter, and it fell to undetectable levels by 4 weeks after vaccination, with new red cell transfusion dependence (Figure 1). The patient developed progressive thrombocytopenia, with a nadir of 81 × 109 platelets per liter. The bone marrow was hypocellular with reduced erythroid elements. Work-up for potential etiologies, including viral infections, was unrevealing (supplemental Materials and methods; supplemental Table 1), without evidence of graft-versus-host disease. CD8+ lymphocyte counts doubled from 605 × 106 cells per liter to 1280 × 106 cells per liter (CD4/CD8 ratio of 0.1-0.2), with a clonal T-cell receptor (TCR)–γ rearrangement. The patient was diagnosed with presumed immune-mediated graft dysfunction and treated with an increasing cyclosporine dose, with improvement in blood counts. Because of persistent poor mixed chimerism, a donor lymphocyte infusion was administered, with eventual conversion to full donor chimerism.
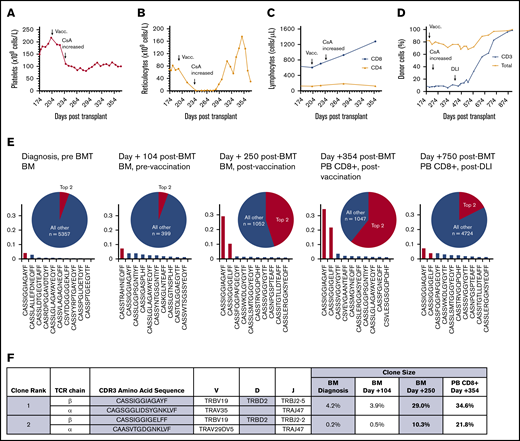
Clonal expansion of CD8+ lymphocytes temporally associated with immunization and graft dysfunction in a patient with AA. (A) Time course of platelet count showing decreasing platelet counts postimmunization and stabilization of thrombocytopenia after an increase in cyclosporine (CsA) dose. (B) Time course of absolute reticulocyte count, demonstrating a precipitous decline in the immediate postimmunization period, with an improvement in reticulocytopenia following an increase in cyclosporine dose. (C) Time course of CD4+ and CD8+ lymphocyte counts showing a selective expansion of CD8+ T lymphocytes postvaccination. (D) Time course of the percentage of donor chimerism in total PB and CD3+ T lymphocytes, showing poor mixed CD3 chimerism, with improved chimerism after treatment with donor lymphocyte infusion (DLI). (E) Frequency histograms of the 10 top productive TCRβ rearrangements, as determined by bulk NGS sequencing of the TCR Vβ gene in the patient’s bone marrow (BM) at diagnosis before stem cell transplantation (pre BMT), at day +104 posttransplant prior to vaccination (Vacc.), at day +250 posttransplant after vaccination, and in sorted PB CD8+ T lymphocytes at day +354 posttransplant after vaccination. The patient was subsequently treated with donor lymphocyte infusion (DLI), with improvement in the percentage of donor chimerism, as shown in panel D, and diminution of the top expanded clones, as shown in PB CD8+ lymphocytes on day +750 posttransplant. The pie charts illustrate the proportion of the top 2 expanded clones (red) relative to all of the remaining clones (blue). The total number of remaining productive TCR Vβ gene rearrangements identified by bulk NGS are listed (n). (F) The 2 dominant CD8+ T lymphocyte clonotypes, with their corresponding frequencies pre- and postvaccination. Frequencies in BM at diagnosis, at day +104 posttransplant prevaccination, at day +250 posttransplant after vaccination, and in sorted PB CD8+ T lymphocytes at day +354 posttransplant were obtained by bulk TCRβ sequencing. TCRα pairing was determined through 10× genomics single-cell immune profiling of sorted PB CD8+ T lymphocytes on day +354 posttransplant.
Clonal expansion of CD8+ lymphocytes temporally associated with immunization and graft dysfunction in a patient with AA. (A) Time course of platelet count showing decreasing platelet counts postimmunization and stabilization of thrombocytopenia after an increase in cyclosporine (CsA) dose. (B) Time course of absolute reticulocyte count, demonstrating a precipitous decline in the immediate postimmunization period, with an improvement in reticulocytopenia following an increase in cyclosporine dose. (C) Time course of CD4+ and CD8+ lymphocyte counts showing a selective expansion of CD8+ T lymphocytes postvaccination. (D) Time course of the percentage of donor chimerism in total PB and CD3+ T lymphocytes, showing poor mixed CD3 chimerism, with improved chimerism after treatment with donor lymphocyte infusion (DLI). (E) Frequency histograms of the 10 top productive TCRβ rearrangements, as determined by bulk NGS sequencing of the TCR Vβ gene in the patient’s bone marrow (BM) at diagnosis before stem cell transplantation (pre BMT), at day +104 posttransplant prior to vaccination (Vacc.), at day +250 posttransplant after vaccination, and in sorted PB CD8+ T lymphocytes at day +354 posttransplant after vaccination. The patient was subsequently treated with donor lymphocyte infusion (DLI), with improvement in the percentage of donor chimerism, as shown in panel D, and diminution of the top expanded clones, as shown in PB CD8+ lymphocytes on day +750 posttransplant. The pie charts illustrate the proportion of the top 2 expanded clones (red) relative to all of the remaining clones (blue). The total number of remaining productive TCR Vβ gene rearrangements identified by bulk NGS are listed (n). (F) The 2 dominant CD8+ T lymphocyte clonotypes, with their corresponding frequencies pre- and postvaccination. Frequencies in BM at diagnosis, at day +104 posttransplant prevaccination, at day +250 posttransplant after vaccination, and in sorted PB CD8+ T lymphocytes at day +354 posttransplant were obtained by bulk TCRβ sequencing. TCRα pairing was determined through 10× genomics single-cell immune profiling of sorted PB CD8+ T lymphocytes on day +354 posttransplant.
Methods
The diagnosis of AA was determined by standard criteria.11-13 Peripheral blood (PB) mononuclear cells, stained for CD3, CD4, CD8, CD27, CD45RA, CCR7, CD95, CD127, FoxP3, and TCRVB19 (VB17 using older nomenclature), were analyzed on a BD LSR II flow cytometer. The TCR-β chain repertoire was analyzed by next-generation sequencing (NGS) of the TCR Vβ gene rearrangements on bulk bone marrow or sorted PB mononuclear cells. Paired α and β chain TCR sequences were determined by single-cell immune profiling and analyzed for convergent antigen specificity using the GLIPH (grouping of lymphocyte interactions by paratope hotspots) algorithm.14 See supplemental Materials and methods for additional details.
Results and discussion
To explore the mechanism of immune-mediated graft dysfunction, we performed serial analysis of the TCR-β repertoire in the patient’s bone marrow DNA collected at AA diagnosis, after engraftment prevaccination, and postvaccination at graft dysfunction (Figure 1E). NGS of the TCR Vβ gene rearrangements identified a dominant recipient-derived clone (TCRVB19-D2-J2-5) that accounted for 4.2% of T-cell rearrangements at diagnosis; it persisted after BMT and expanded following vaccination to account for 29.0% of all T-cell rearrangements and 34.6% of CD8+ T-cell rearrangements. A second recipient-derived T-cell clone with a similar, but nonidentical, rearrangement (TCRVB19-D2-J2-2) also expanded markedly postvaccination, increasing from <1% prevaccination to 10.3% of Vβ gene rearrangements and accounting for 21.8% of CD8+ T-cell rearrangements after vaccination. Single-cell immune profiling confirmed the same dominant TCR-β clonotypes and identified the paired TCR-α chains (Figure 1F; supplemental Table 2). Using the GLIPH algorithm,which uses multiple metrics of convergence, including conserved motifs, gene usage, and similarity of complementarity-determining region 3 (CDR3) sequences to cluster TCRs into groups with a high probability of sharing antigen specificity,14 the expanded TCR clones in our patient clustered into a single antigen convergence group (supplemental Materials and methods; supplemental Dataset 1).
Taking advantage of the shared TCRVB19 between the 2 dominant clones, staining with anti-TCRVB19 specific antibody demonstrated that TCRVB19+ lymphocytes made up 52.3% of the patient’s CD8+ population and exhibited a CD27− TEM phenotype (CCR7− CD27− CD95+), consistent with antigen-stimulated differentiated cells (Figure 2A-B). CD4+ FoxP3+ regulatory T-cell numbers were low (supplemental Figure 1).
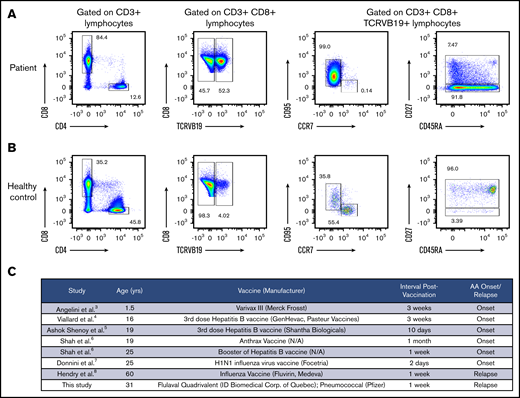
Clonally expanded T lymphocytes have T effector memory and T effector memory RA phenotype. (A) Immunophenotyping analysis of the patient’s PB lymphocytes on day +354 posttransplant revealed an expansion of the TCRBV19+ T-cell population with the CD27− T effector memory phenotype (CD3+ CD8+ CD45RA− CD27− CCR7− CD95+) and the T effector memory RA phenotype (CD3+ CD8+ CD45RA+ CD27− CCR7− CD95+). The 2 T-cell clones of interest accounted for 92.5% of all TCRBV19+ PB CD8 lymphocytes, as determined by bulk TCR Vβ sequencing. (B) Immunophenotyping of PB lymphocytes from a healthy donor. (C) A systematic review of published cases of AA onset or relapse temporally related to vaccinations.
Clonally expanded T lymphocytes have T effector memory and T effector memory RA phenotype. (A) Immunophenotyping analysis of the patient’s PB lymphocytes on day +354 posttransplant revealed an expansion of the TCRBV19+ T-cell population with the CD27− T effector memory phenotype (CD3+ CD8+ CD45RA− CD27− CCR7− CD95+) and the T effector memory RA phenotype (CD3+ CD8+ CD45RA+ CD27− CCR7− CD95+). The 2 T-cell clones of interest accounted for 92.5% of all TCRBV19+ PB CD8 lymphocytes, as determined by bulk TCR Vβ sequencing. (B) Immunophenotyping of PB lymphocytes from a healthy donor. (C) A systematic review of published cases of AA onset or relapse temporally related to vaccinations.
Our results demonstrate a striking clonal expansion of recipient-derived CD8+ TEM lymphocytes temporally associated with immunization and graft dysfunction in a patient with AA. The dominant CD8+ T-cell clone after the vaccination corresponded to the most abundant T-cell clone in the patient’s bone marrow at initial AA diagnosis. The temporal relationship between immunization and graft dysfunction, accompanied by the postvaccine clonal expansion of the recipient CD8+ TEM lymphocytes, suggests that immunization played a role in triggering graft dysfunction. Other factors that contributed to this patient’s graft dysfunction are the mixed T-cell chimerism, which can occur with a reduced-intensity BMT using anti-thymocyte globulin– and alemtuzumab-containing conditioning,10,15,16 subtherapeutic cyclosporine level, and low CD34+ cell dose. Reduced regulatory T cells likely contributed to postvaccine TEM cell expansion.17,18
Several cases of AA onset or relapse closely following vaccinations or viral infections have been reported3-8,19 (Figure 2C). However, a causal relationship has not been established, and no study has explored immunological changes induced by immunizations in relapsed AA patients. Our data provide the first insight into the mechanism underlying vaccine-associated immune-mediated marrow failure by demonstrating CD8+ TEM cell expansion after vaccination.
One potential explanation for postvaccine marrow failure is the cross-reactivity between the antigens contained in the vaccine and the putative AA autoantigen(s) present on hematopoietic cells. A similar mechanism has been proposed for hepatitis-associated AA.20 Of the 2 coadministered vaccines, the selective expansion of CD8+ lymphocytes, whose normal function is immune surveillance against intracellular pathogens, including viruses and tumors, suggests that the influenza vaccine could be the culprit. Two previous cases of AA following influenza vaccination7,8 and 1 case following influenza infection19 have been described. Usage of Vβ19 was reported in CD8+ T-cell immune responses to influenza matrix protein in HLA-A2 individuals exposed to influenza A,21,22 and the CDR3 sequences of the patient’s expanded clones are similar to known influenza-reactive CDR3 sequences23 (supplemental Figure 2). The selective expansion and antigen convergence of the 2 dominant clones suggest antigen-specific clonal expansion, likely in response to influenza vaccination, and argue against a nonspecific immune response. Future studies are needed to evaluate the reactivity of the expanded T-cell clones against specific vaccine-associated epitopes and to determine their cytotoxicity against the patient’s hematopoietic cells.
Clinical interpretation of our results requires caution. Our results support the recommendation against routine vaccinations in AA patients treated with immunosuppression9 and suggest that AA patients with low CD3+ chimerism after BMT may similarly be at risk for immune-mediated marrow failure after vaccination. We recommend careful consideration of the risks and benefits of immunizations in AA patients and advise delaying routine vaccinations in BMT recipients with high residual host CD3+ content until immune reconstitution of donor T cells. In patients in whom cytotoxic T-cell responses need to be minimized, a recombinant flu vaccine would be preferable to whole inactivated virus vaccine, which contains a wider range of antigenic targets and potential T-cell epitopes.24 Finally, in AA, unlike in malignant disorders, post-BMT cyclosporine should be continued at therapeutic levels for 6 to 9 months, followed by a cautious taper while monitoring for late graft failure.9
Acknowledgments
The authors thank the patient for participation in the study. They also thank Vivianna Van Deerlin for assistance with clinical chimerism analysis.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant K08 HL132101 (D.V.B.) and US Department of Defense grant BM170031 (T.S.O.).
Authorship
Contribution: C.R. and D.V.B. performed experiments, analyzed the data, performed the literature review, and wrote the manuscript; W.M. and E.T.L.P. performed and interpreted TCR NGS; N.L.S., M.L.B., C.X., and P.Y. performed T-cell immunophenotyping and clonal T-cell analysis; A.C.H., B.M.C., and E.T.L.P. contributed immunology expertise; R.H. assisted with GLIPH bioinformatic analysis; P.N., J.-M.F., D.L., and T.S.O. enrolled the patient, processed and provided critical patient samples, and oversaw the human subject aspects of the research; T.S.O. provided bone marrow failure and BMT expertise; D.V.B. conceived and oversaw the study; and all authors edited and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daria V. Babushok, Division of Hematology-Oncology, Department of Medicine, University of Pennsylvania, Room 808 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: daria.babushok@pennmedicine.upenn.edu.