

Key Points
First evidence of ruxolitinib efficacy for subcutaneous panniculitis-like T-cell lymphoma with hemophagocytic lymphohistiocytosis.
Supporting rationale for ruxolitinib use, not more aggressive treatment, in this context, questioning this condition’s neoplastic nature.
Introduction
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a distinct subset of cutaneous lymphomas in which neoplastic cytotoxic α/β CD8+ T cells infiltrate subcutaneous adipose tissue.1-3 SPTCL may affect both children and adults.4 Systemic symptoms such as fever, asthenia, and weight loss, associated with cytopenias and liver dysfunction, are common. In addition, hemophagocytic lymphohistiocytosis (HLH) is observed in 20% of cases,3 which is associated with poor prognosis and may be the leading cause of death.3 There is no standardized therapy for SPTCL alone or in association with HLH. Chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisolone is frequently used, with an overall remission rate of 50%.1 Immunosuppressive regimens, particularly cyclosporine A (CsA), may be also effective.5 In some severe cases, stem cell transplantation has been attempted.1
Recently, germline mutations causing loss of function of T-cell immunoglobulin mucin 3 (TIM-3) were identified in 60% to 85% of SPTCL patients.6,7 In these patients, TIM-3 deficiency was shown to promote T-lymphocyte and -macrophage activation and the production of proinflammatory cytokines, challenging the malignant nature of skin T-lymphocyte infiltration.6
Ruxolitinib is a selective JAK1/JAK2 inhibitor licensed for treatment of myelofibrosis and polycythemia vera in adults.8,9 Studies in animal models of HLH support the efficacy of this drug to prevent and treat HLH in these models.10,11 Anecdotal experiences of successful use of ruxolitinib to control refractory primary HLH or secondary HLH are also reported in humans.12-19 JAK1/JAK2 inhibitors are also increasingly used in inflammatory diseases.
We herein report the use and efficacy of ruxolitinib in a patient with recurrence of SPTCL and HLH and in whom TIM-3 deficiency was recently identified.6
Case description and methods
The patient (reported as P4 in Gayden et al6 and carrying a homozygous p.Tyr82Cys variant in HAVCR2/TIM-3) is a teenage boy of French Polynesian origin. At 11 years of age, he experienced persistent fever and pain in the right flank. With 5 of 8 positive criteria (persistent fever, pancytopenia, hyperferritinemia [3000 μg/L], hypofibrinogenemia [<0.6 g/L], and hemophagocytosis on bone marrow aspirate), he was diagnosed with HLH.
The patient was treated with corticosteroids, CsA (4-6 mg/kg per day), 4 doses of etoposide (VP16 150 mg/m2 per dose), and 1 intrathecal methotrexate injection, which led to complete remission. Corticosteroids were tapered and stopped within 6 weeks. CsA was discontinued after 8 months with complete clinical and biological remission. At 13 years of age, he presented with a relapse of HLH and painful redness of the right flank. A positron emission tomography–computed tomography (PET-CT) scan showed diffuse enhancement of subcutaneous tissues revealing SPTCL, histologically confirmed with monoclonal T-cell receptor β-chain rearrangement of CD8 T cells. CsA was reinitiated (4-6 mg/kg per day), allowing partial remission with intermittent high fever requiring several courses of corticosteroids. Abatacept was added for a period of 6 months without any benefit.
In June 2017, at 16 years of age, the patient’s medical situation became unsatisfactory under CsA treatment (4 mg/kg per day). He had recurrent episodes of fever, persistence of HLH features, lymphopenia, diffuse pain, and persistent mild and diffuse panniculitis with subcutaneous enhancement on PET-CT scan (Figures 1 and 2A). Because the dose of cyclosporine could not be increased due to poor renal tolerance, corticosteroids (0.5 mg/kg per day) were added to alleviate symptoms. One month later, TIM-3 deficiency was identified in this patient, and shown to result in increased in vitro production of tumor necrosis factor-α and interleukin (IL-1β) by the deficient macrophages.6 Therefore, the IL-1 inhibitor anakinra (100 mg in daily subcutaneous injection) was initiated, whereas corticosteroids were stopped. This treatment led to overall clinical improvement and prevented fever recurrence6 but did not lead to normalization of biologic parameters (Figure 1). Serum levels of interferon-γ (IFN-γ)–induced CXCL10, IL-18, and soluble CD25 (sCD25), a cluster of inflammatory markers characteristic of primary HLH,20 remained elevated (Figure 2B). PET-CT scan performed 6 months after anakinra initiation showed increased diffuse subcutaneous enhancement (Figures 1 and 2A). Of note, CsA was responsible for mild renal impairment but doses could not be tapered because of fever relapse at each attempt.
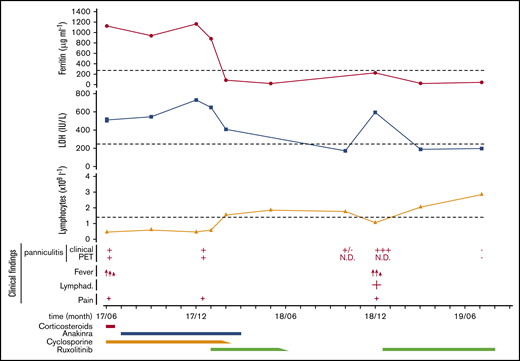
Plots showing the time course of SPTCL and HLH episodes in the patient during the past 24 months and the different therapeutic strategies implemented. Clinical findings depict the presence of high fever (red arrows), lymphadenopathies (Lymphad.; +), and pain (+), as well as the presence (+) or absence (−) of panniculitis, detectable either clinically (Clinical) or by PET-CT scan (PET). Dotted lines indicate the lower (lymphocyte count) or the higher (ferritin and LDH serum levels) normal limits. Anakinra was administrated at 100 mg per day, cyclosporine 4 to 6 mg/kg per day, prednisone at 0.5 mg/kg every day, and ruxolitinib first at 15 mg twice daily, then 20 mg twice daily at relapse. N.D., not determined.
Plots showing the time course of SPTCL and HLH episodes in the patient during the past 24 months and the different therapeutic strategies implemented. Clinical findings depict the presence of high fever (red arrows), lymphadenopathies (Lymphad.; +), and pain (+), as well as the presence (+) or absence (−) of panniculitis, detectable either clinically (Clinical) or by PET-CT scan (PET). Dotted lines indicate the lower (lymphocyte count) or the higher (ferritin and LDH serum levels) normal limits. Anakinra was administrated at 100 mg per day, cyclosporine 4 to 6 mg/kg per day, prednisone at 0.5 mg/kg every day, and ruxolitinib first at 15 mg twice daily, then 20 mg twice daily at relapse. N.D., not determined.
![Clinical and biological response of the TIM-3–deficient patient to various immunosuppressive treatments. (A) PET-CT scan analysis showing hyperactive lesions in patient’s subcutaneous tissues (arrows) before (December 2017; left) and their absence 7 months after (July 2019; right) initiation of ruxolitinib therapy. (B) Plots showing the patient’s high serum levels of inflammatory markers (sCD25, CXCL10 chemokine, and IL-18 cytokine) over time under various treatments (under ruxolitinib therapy [Ruxolitinib +] or not [Ruxolitinib −]), as compared with control (Ctrl). Data are means plus or minus standard deviation (SD) from 6 different controls. sCD25, CXCL10, and IL-18 were quantified by enzyme-linked immunosorbent assay. (C) Patient’s increased serum levels of IFN-γ and IL-6 during flare (Ruxolitinib −; December 2018) and their normalization under ruxolitinib therapy (Ruxolitinib +; July 2019) as compared with control. Data are means plus or minus SD from 6 different controls. (D) Increased proportion of highly activated (CD38+HLA-DR+) CD8 T cells from patient during flare (Ruxolitinib −; December 2018), as compared with control (Ctrl CD8 T cells), which normalized under ruxolitinib therapy (Ruxolitinib +; July 2019). (E) High increased proportion of effector memory CD8 T lymphocytes (CD45RA−CCR7−) at the expense of naive CD8 T lymphocytes (CD45RA+CCR7+) in the patient during flare (blue bars; December 2018) that normalized under ruxolitinib therapy (green bars; July 2019). The shaded area indicates our in-house normative values. EMRA, effector memory reexpressing CD45RA.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/7/10.1182_bloodadvances.2020001497/1/m_advancesadv2020001497f2.png?Expires=1767704317&Signature=m0BK5Hl3hErbZwuzIPnbHEnDDuV0M2bSRXhI8YSs2Jl1bHWGR~5Oie34pQcQjO84lVKM7AcmxXuShgvoMJPf5pH3Ry98aD~gczWRttc9GkYwCQgaITPkrzOzWz4em7sv6XCgx9G~8HjZY9EdXDZokjEFa6jmscZtf6bQoj~49hZKwWr7AnX0LMQw0POx5LOQtU4tnUW3gKhT3QvIR~Vg78~pIvWEvmoXQ3PSW-Wh0VHYd6IEiVFHcM-C4secIHU6prdFDT74AsfZMt3AV6ArPbBuk3eZpmjMEtm0LVWb6~jrCfIdYPLVTew1sp6H9Zqkklomo6tkwDmgFUlTcuTwOg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Clinical and biological response of the TIM-3–deficient patient to various immunosuppressive treatments. (A) PET-CT scan analysis showing hyperactive lesions in patient’s subcutaneous tissues (arrows) before (December 2017; left) and their absence 7 months after (July 2019; right) initiation of ruxolitinib therapy. (B) Plots showing the patient’s high serum levels of inflammatory markers (sCD25, CXCL10 chemokine, and IL-18 cytokine) over time under various treatments (under ruxolitinib therapy [Ruxolitinib +] or not [Ruxolitinib −]), as compared with control (Ctrl). Data are means plus or minus standard deviation (SD) from 6 different controls. sCD25, CXCL10, and IL-18 were quantified by enzyme-linked immunosorbent assay. (C) Patient’s increased serum levels of IFN-γ and IL-6 during flare (Ruxolitinib −; December 2018) and their normalization under ruxolitinib therapy (Ruxolitinib +; July 2019) as compared with control. Data are means plus or minus SD from 6 different controls. (D) Increased proportion of highly activated (CD38+HLA-DR+) CD8 T cells from patient during flare (Ruxolitinib −; December 2018), as compared with control (Ctrl CD8 T cells), which normalized under ruxolitinib therapy (Ruxolitinib +; July 2019). (E) High increased proportion of effector memory CD8 T lymphocytes (CD45RA−CCR7−) at the expense of naive CD8 T lymphocytes (CD45RA+CCR7+) in the patient during flare (blue bars; December 2018) that normalized under ruxolitinib therapy (green bars; July 2019). The shaded area indicates our in-house normative values. EMRA, effector memory reexpressing CD45RA.
Clinical and biological response of the TIM-3–deficient patient to various immunosuppressive treatments. (A) PET-CT scan analysis showing hyperactive lesions in patient’s subcutaneous tissues (arrows) before (December 2017; left) and their absence 7 months after (July 2019; right) initiation of ruxolitinib therapy. (B) Plots showing the patient’s high serum levels of inflammatory markers (sCD25, CXCL10 chemokine, and IL-18 cytokine) over time under various treatments (under ruxolitinib therapy [Ruxolitinib +] or not [Ruxolitinib −]), as compared with control (Ctrl). Data are means plus or minus standard deviation (SD) from 6 different controls. sCD25, CXCL10, and IL-18 were quantified by enzyme-linked immunosorbent assay. (C) Patient’s increased serum levels of IFN-γ and IL-6 during flare (Ruxolitinib −; December 2018) and their normalization under ruxolitinib therapy (Ruxolitinib +; July 2019) as compared with control. Data are means plus or minus SD from 6 different controls. (D) Increased proportion of highly activated (CD38+HLA-DR+) CD8 T cells from patient during flare (Ruxolitinib −; December 2018), as compared with control (Ctrl CD8 T cells), which normalized under ruxolitinib therapy (Ruxolitinib +; July 2019). (E) High increased proportion of effector memory CD8 T lymphocytes (CD45RA−CCR7−) at the expense of naive CD8 T lymphocytes (CD45RA+CCR7+) in the patient during flare (blue bars; December 2018) that normalized under ruxolitinib therapy (green bars; July 2019). The shaded area indicates our in-house normative values. EMRA, effector memory reexpressing CD45RA.
The research reported in this manuscript has been approved by the local investigational review board and the French Advisory Committee on Medical Research (IRB registration #00001072, no. CPP: 2015-03-03/DC 2014-2272).
Results and discussion
Because of the positive IFN-γ signature (discussed in "Case description and methods"), ruxolitinib was initiated (15 mg twice daily/80 kg) allowing rapid improvement of the clinical and biological manifestations of HLH (Figures 1 and 2B). It was possible to taper CsA over 4 weeks and to stop anakinra 2 weeks later without relapse. After 4 months of treatment (including 2 months of ruxolitinib monotherapy), the patient was clinically well and biologically in remission (Figures 1 and 2B). This teenage patient then decided on his own to taper and stop his medication (Figure 1). After 8 months, he came back with fever, marked asthenia, and a large localized panniculitis of his left leg (giant patches of 11 × 13 cm in length) associated with a homolateral enlarged inguinal lymph node (4 × 3 cm). Ruxolitinib discontinuation thus correlated with disease relapse. Clinical manifestations were associated with elevated serum levels of lactate dehydrogenase (LDH), IFN-γ, IL-6, IL-18, CXCL10, and sCD25, whereas lymphocyte counts again decreased (Figure 1 and 2B-C). A large proportion of circulating CD8 T lymphocytes (34.5%) was highly activated in vivo, as assessed by the coexpression of CD38 and HLA-DR molecules at their surface (Figure 2D). A detailed analysis of the circulating T-cell populations over time revealed a CD8 lymphopenia with the presence of a large proportion of effector memory CD8 T cells (CD45RA−CCR7−) in the patient during flare at the expense of the naive CD8 population (CD45RA+CCR7+) (Figure 2D). The significant fraction of effector CD8 T cells during flare might reflect a disproportionate T-lymphocyte response to putative infectious trigger occurring in the context of impaired TIM-3 checkpoint inhibition.
Reinitiation of ruxolitinib therapy alone (20 mg twice daily) again resulted in rapid clearance of the fever (within 72 hours) and progressive resolution of the panniculitis within 2 weeks and of HLH features within 3 weeks (Figure 1). The levels of ferritin and inflammatory markers in the serum fully normalized, and lymphocyte counts and distribution reached normal values when analyzed 3 months later (Figures 1 and 2B-D). Ten months later under the same treatment, the patient remained in complete remission of SPTCL (assessed by PET-CT scan) and HLH (Figures 1 and 2A-B). The main adverse events associated with ruxolitinib therapy are hematologic (potential development of cytopenia, anemia, and thrombocytopenia8 ) and infectious (shingles and herpes reactivation being the most frequent). In addition, transaminitis was also reported.8 No such toxicity events were observed in this patient after 14 months of ruxolitinib treatment. Close monitoring of clinical and biological markers of disease activity will allow ruxolitinib to be tapered to the minimal dose required to maintain long-term remission.
Until recently, the etiology of SPTCL was mostly unknown. The recent description of a genetic defect impairing the function of TIM-3 in a large proportion of SPTCL patients provided some clues to understanding the underlying pathological mechanism of this condition.6 TIM-3 acts as a negative immune checkpoint that regulates the effector function of T lymphocytes and myeloid cells.21 TIM-3 downregulates the production of proinflammatory cytokines by these cells, which significantly increased levels in SPTCL patients.6 Therefore, targeting inflammatory cytokine pathways emerged as a rational therapeutic approach. The clear efficacy of ruxolitinib therapy, both on patient’s HLH and panniculitis manifestations, supports this assumption, although a more extensive clinical trial would be needed to fully validate it. In addition, the efficacy of ruxolitinib therapy in this patient who displayed a severe form of the disease questions the “neoplastic” nature generally attributed to this condition, and invites reconsideration of this entity as an inflammatory condition rather than as a lymphoma.
Data-sharing requests may be e-mailed to the corresponding authors, B.N. (benedicte.neven@aphp.fr) and G.d.S.B.(genevieve.de-saint-basile@inserm.fr).
Acknowledgments
This work was supported by INSERM, l’Agence Nationale de la Recherche (NewHLH/ANR-18-CE15-0017), La Ligue contre le Cancer (RS19-75-79), and the Histiocytosis Association of America.
Authorship
Contribution: B.N., F.E.S., and G.d.S.B. conceived and designed the study; F.G., M.F., and A.C. acquired and analyzed data; B.N., R.L., and D.M. evaluated and treated patients; and G.d.S.B., F.E.S., A.F., and B.N. drafted the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bénédicte Neven, Unité d’Immuno-Hematologie et Rhumatologie, Département de Pédiatrie, AP-HP, Hôpital Necker-Enfants Malades, F-75015 Paris, France; e-mail: benedicte.neven@aphp.fr; and Geneviève de Saint Basile, Imagine Institute, Université de Paris, INSERM U1163, F-75015 Paris, France; e-mail: genevieve.de-saint-basile@inserm.fr.

