Key Points
Germline RUNX1 mutations lead to significant phenotypic heterogeneity in families.
Germline RUNX1-mutated acute myeloid leukemia has a different somatic mutation profile from the sporadic RUNX1-mutated type.

Abstract
First reported in 1999, germline runt-related transcription factor 1 (RUNX1) mutations are a well-established cause of familial platelet disorder with predisposition to myeloid malignancy (FPD-MM). We present the clinical phenotypes and genetic mutations detected in 10 novel RUNX1-mutated FPD-MM families. Genomic analyses on these families detected 2 partial gene deletions, 3 novel mutations, and 5 recurrent mutations as the germline RUNX1 alterations leading to FPD-MM. Combining genomic data from the families reported herein with aggregated published data sets resulted in 130 germline RUNX1 families, which allowed us to investigate whether specific germline mutation characteristics (type, location) could explain the large phenotypic heterogeneity between patients with familial platelet disorder and different HMs. Comparing the somatic mutational signatures between the available familial (n = 35) and published sporadic (n = 137) RUNX1-mutated AML patients showed enrichment for somatic mutations affecting the second RUNX1 allele and GATA2. Conversely, we observed a decreased number of somatic mutations affecting NRAS, SRSF2, and DNMT3A and the collective genes associated with CHIP and epigenetic regulation. This is the largest aggregation and analysis of germline RUNX1 mutations performed to date, providing a unique opportunity to examine the factors underlying phenotypic differences and disease progression from FPD to MM.
Introduction
In 2016, germline predisposition to hematological malignancy (HM) debuted in the World Health Organization classification of myeloid neoplasms and acute leukemia.1 It has been 20 years this year since causative germline mutations affecting the runt-related transcription factor 1 (RUNX1) were identified in familial platelet disorder (FPD) with predisposition to myeloid malignancy (FPD-MM; OMIM 601399).2 Since that time, many separate studies have reported on germline RUNX1 mutations in families with FPD-MM, highlighting its significant clinical presence (reviewed in Brown et al3 and Sood et al4 ). Although thrombocytopenia and platelet dysfunction are present in almost all RUNX1 mutation carriers, we and others have observed that the age at onset of HM and the type of malignancy varies among family members, and in some cases, RUNX1 mutation carriers have no apparent HM development over their lifespan. The reasons for and extent of this heterogeneity of penetrance are currently unknown and require further exploration. The somatic genetic changes that lead to leukemia in individuals most likely contribute to this heterogeneity. One recent study of FPD-MM reported that somatic mutation of the second RUNX1 allele is the most commonly acquired somatic mutation in a cohort of germline RUNX1-mutated individuals.5 Interestingly, several other smaller studies did not observe the same frequency of somatic RUNX1 mutation,6-8 possibly because of cohort size. Patients with sporadic acute myeloid leukemia (AML) with somatic RUNX1 mutations have an adverse prognosis, and biallelic RUNX1 mutations are associated with an even poorer outcome, indicating a dosage effect.9-11 It is clear, therefore, that additional studies investigating somatic mutations and conditions of single and biallelic RUNX1-mutated tumors in FPD-MM are needed, to better understand and predict the development of this disease.
In this study we examined the genetic, phenotypic, and clinical data of 10 new families with germline RUNX1 mutations. In line with the increasing importance of correctly identifying truly pathogenic variants in a sea of genomic variants, we applied the newly defined American Society of Hematology-Clinical Genome Resource (ASH-ClinGen) Myeloid Expert Committee modifications of the American College of Medical Genetics classification rules for RUNX112 to these 10 families. In addition, we reviewed the existing literature on RUNX1 to gain insight into the molecular mechanisms underlying the FPD-MM disease heterogeneity and progression. First, we merged the data on these 10 families with data on all published germline RUNX1 families to date, to generate a comprehensive overview of all genetic mutations and associated disease phenotypes. Second, to better understand the disease progression to AML, we compared the somatic variants in AML patients with germline RUNX1 mutations to those reported in sporadic AML patients with somatic RUNX1 mutations.
Methods
More detailed information on the methods used for all analyses is available in the supplemental Methods.
Genetic analysis methods
The families with germline RUNX1 variants (Figure 1; Table 1) were collected over 15 years. As genetic testing has changed frequently since the introduction of next-generation sequencing (NGS), different genetic tests have been applied for different samples and families. For the families described in this article, RUNX1 germline mutations were identified using single-nucleotide polymorphism (SNP) array and multiplex ligation-dependent probe amplification (MLPA; families 1 and 2), Sanger sequencing (families 3, 4, and 5) NGS gene panel (29 genes; families 7 and 8), and exome sequencing (families 7 and 10). Somatic mutation detection was performed using MLPA (family 3), NGS gene panel (families 1, 4, 5, 7, and 8), and whole-exome sequencing (WES; families 2, 6, and 10).
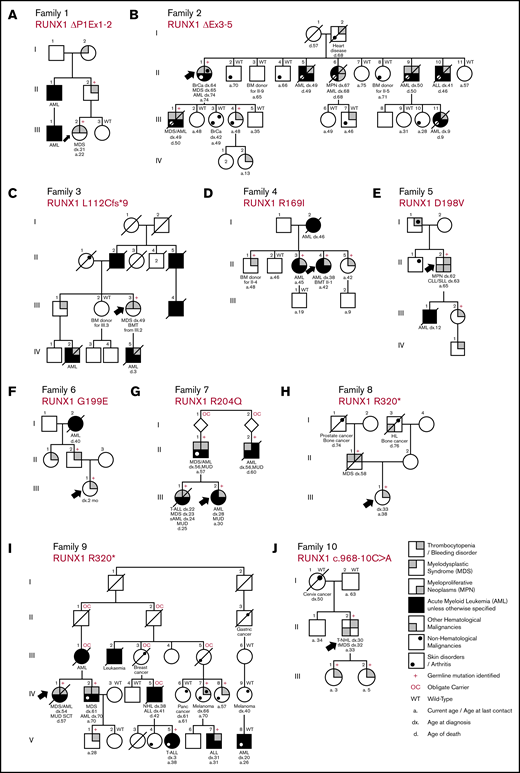
Pedigrees showing the genotypes and phenotypes detected in the new families with germline RUNX1 mutations and inherited HMs. Family 1: RUNX1 ΔP1Ex1-2 (A); family 2: RUNX1 ΔEx3-5 (B); family 3: RUNX1 L112Cfs*9 (C); family 4: RUNX1 R169I (D); family 5: RUNX1 D198V (E); family 6: RUNX1 G199E (F); family 7: RUNX1 R204Q (G); family 8: RUNX1 R320* (H); family 9: RUNX1 R320* (I); family 10: RUNX1 c.968-10C>A (J). Δ indicates a partial gene deletion; *, stop-gain mutation; amino acid changes (p.) are mentioned for missense variants, and the splice-site variant is annotated to the cDNA position. a, age at last date of contact; BM, bone marrow; BMT, bone marrow transplant; BrCa, breast cancer; CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; d, age at death; dx, age at diagnosis; fs, frameshift mutation; MUD, matched unrelated donor; Panc cancer, pancreatic cancer; sAML, secondary AML; SCT, stem cell transplant; T-ALL, T-cell acute lymphoblastic leukemia; tMDS, therapy-related MDS; T-NHL, T-cell non-Hodgkin lymphoma.
Pedigrees showing the genotypes and phenotypes detected in the new families with germline RUNX1 mutations and inherited HMs. Family 1: RUNX1 ΔP1Ex1-2 (A); family 2: RUNX1 ΔEx3-5 (B); family 3: RUNX1 L112Cfs*9 (C); family 4: RUNX1 R169I (D); family 5: RUNX1 D198V (E); family 6: RUNX1 G199E (F); family 7: RUNX1 R204Q (G); family 8: RUNX1 R320* (H); family 9: RUNX1 R320* (I); family 10: RUNX1 c.968-10C>A (J). Δ indicates a partial gene deletion; *, stop-gain mutation; amino acid changes (p.) are mentioned for missense variants, and the splice-site variant is annotated to the cDNA position. a, age at last date of contact; BM, bone marrow; BMT, bone marrow transplant; BrCa, breast cancer; CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; d, age at death; dx, age at diagnosis; fs, frameshift mutation; MUD, matched unrelated donor; Panc cancer, pancreatic cancer; sAML, secondary AML; SCT, stem cell transplant; T-ALL, T-cell acute lymphoblastic leukemia; tMDS, therapy-related MDS; T-NHL, T-cell non-Hodgkin lymphoma.
WES
WES was performed using SeqCap EZ MedExome (Roche) on the Illumina Next-Seq sequencing system. Alignment and variant calling were performed as previously described.13 Detection of somatic copy number variants (CNVs), including a copy neutral loss of heterozygosity (CN-LOH) in family 7 patient III-2, was performed using the Sequenza program.14
NGS myeloid gene panel
Amplicons for all coding regions of 29 myeloid genes ASXL1, BAP1, BRAF, CBL, CEBPA (poor coverage), DNMT3A, EGFR, EZH2, GATA2, IDH1, IDH2, JAK1, JAK2, KIT, KRAS, MET, MPL, MYD88, NOTCH1, NPM1, NRAS, PTPN11, RUNX1, SF3B1, SRP72, SRSF2, TET2, U2AF1, and XPO1. Samples were prepared via the Ion AmpliSeq (ThermoFisher Scientific) custom Fluidigm Access Array (Fluidigm). Ion Proton sequencing and analysis13 and curation of mutations in myeloid genes13,15 were performed as previously described.
Results
Novel RUNX1 alterations and phenotypes in FPD-MM
In the 10 families with germline RUNX1 mutations described herein, we surveyed 67 family members, identifying 2 partial gene deletions, 3 novel mutations, and 5 recurrent mutations as the germline RUNX1 alterations leading to FPD-MM. Their clinical, phenotypic and genetic information are summarized in Figure 1 and Table 1 and described below. Additional detailed information on clinical findings and samples screened are found in the supplemental Tables and supplemental Clinical Information.
RUNX1 deletion mutations
Deletions make up a significant yet very likely underrepresented proportion of germline RUNX1 cases because of the limitations of sequencing technologies in identifying deletion events. We describe 2 families with FPD-MM (Figure 1A-B) that were negative for RUNX1 mutation by sequencing, but alternative technologies (MLPA and SNP microarray), showed them to harbor germline partial deletions of RUNX1, thus explaining the family histories. In family 1 (Figure 1A), with thrombocytopenia and MM, a deletion removing the P1 promoter and exons 1 and 2 of the RUNX1c isoform was identified (breakpoints mapped by PCR and Sanger; supplemental Figure 1). Similar, deletions that remove this genomic region of RUNX1 are reported in 3 other FPD-MM families (supplemental Tables 2 and 3), suggesting that removal of this portion of RUNX1c is sufficient to manifest the FPD-MM phenotype. Family 2 (Figure 1B) is a large family with a history of thrombocytopenia and MMs but also includes cases of acute lymphoblastic leukemia (ALL; II-10) and breast cancer (II-1). MLPA and SNP microarray of III-1 identified a novel 47 766-base-pair deletion in RUNX1, removing exons 3, 4, and 5 of RUNX1c, predicting altered splicing resulting in an in-frame p.E20_G170 deletion in the RUNT domain (supplemental Figure 2) and also removing the P2 promoter and exons 1, 2, and 3 of the RUNX1a and b transcripts.
RUNX1 missense mutations
In 4 FPD-MM families, we found missense mutations in the RUNX1 RUNT homology domain (families 4, 5, 6, and 7; Figure 1D-G), 3 of which affect RUNX1 hotspot residues as defined by the new ASH-ClinGen RUNX1 classification rules (families 4, 5, and 7; supplemental Table 3). In family 4 (Figure 1D), Sanger sequencing of RUNX1 in the proband identified a novel germline heterozygous missense (c.506G>T; p.R169I) mutation in RUNX1. Consistent with its hotspot designation, recurrently somatically mutated in both sporadic myelodysplastic syndrome (MDS)/AML and breast carcinoma (supplemental Figure 4) and is essential for RUNX1 DNA binding activity.16 In families 5 and 6, RUNX1 c.593A>T; p.D198V and c.596G>A; p.G119E mutations were identified, respectively (Figure 1E-F). These mutations have both been identified previously in the context of inherited thrombocytopenia without reported leukemia predisposition.7,17 In this study, however, we saw them associated with a family history of AML and also, in family 5, with a myeloproliferative neoplasm (MPN) that transformed into to a lymphoproliferative disorder (II-2). Family 7 (Figure 1G) is a complex family with thrombocytopenia, multiple cases of MDS and AML, and a case of T-ALL. WES of the proband identified a RUNX1 single-nucleotide substitution (c.611G>T), leading to a p.R204Q variant in the RUNT domain. A germline RUNX1 p.R204Q mutation has been described that is associated with both AML and T-ALL in carriers.18 Somatic mutations at this position (including somatic p.R204Q) have also frequently been reported in AML, supporting its hotspot designation.11,19
RUNX1 frameshift and premature termination mutations
We identified 4 families with mutations that cause premature termination of the RUNX1 protein (families 3, 8, 9, and 10; Figure 1C,H-J). In family 3 (Figure 1C), we identified a novel (c.415delC; p.L112Cfs*10) mutation which removes most of the RUNT domain and the entire transactivation domain (supplemental Figure 3B). In families 8 and 9 (Figure 1H-I), an identical germline RUNX1 c.958C>T; p.R320* stop-gain variant was identified. The extensive history taken in family 9 includes a wide range of malignancy phenotypes (myeloid and lymphoid) and ages at onset, as well as a prevalent psoriatic skin disorder (Figure 1I). The p.R320* mutation has been described in another FPD-MM family20 and is also a recurrent site of somatic mutation in sporadic AML.11 Comparing the genomes of individuals from families 8 and 9 (family 8, II-1 and II-1, to family 9, IV-1), did not find shared haplotypes on chromosome 21, indicating that they are likely to be unrelated families (ie, separate genetic events; supplemental Figure 8). Family 10 was recruited because of a proband with a personal history of thrombocytopenia and T-cell non-Hodgkin lymphoma (NHL) and young daughters who also had thrombocytopenia. He had no ancestral family history of thrombocytopenia or HMs. WES identified an intronic RUNX1 c.968-10C>A variant that was predicted to generate a cryptic splice acceptor generating a transcript encoding p.A324Lfs*7, which confirmed through RNA studies (supplemental Figure 9). Analysis of WES of the parents of the proband confirmed parentage and, in the father, identified the RUNX1 variant present at 2% allele burden in blood (supplemental Figure 9), indicating that he is likely to be a mosaic carrier, due to a postzygotic de novo mutation, a rarely observed occurrence in cases of germline RUNX1 mutation.
Additional genetic variants in germline RUNX1 mutation carriers
In total, 16 hematological samples (4 preleukemic; 11 leukemic, and 1 NHL) from the 10 families with germline RUNX1 variants were available for additional genetic profiling. Myeloid panels and WES (see “Methods” for details), were performed, with results presented in Table 2 and supplemental Figure 10. In the malignant samples, RUNX1 was the most frequently somatically mutated gene. We identified 4 samples with somatic alteration of RUNX1 (36%), including 1 patient with both a somatic mutation and duplication of the germline RUNX1-mutated chromosome (family 3, III-3). Two individuals, 1 with MDS and 1 with an MPN, had acquired the common JAK2 p.V617F mutation and, in the latter case, this was associated with homozygosity for the germline JAK2 46/1 haplotype (GGCC; family 5, II-2). Across all the families, singleton somatic tumor-associated mutations were also identified in PHF6, SH2B3, TET2, MEIS1, BCOR1, BCORL1, KRAS, and EZH2. Somatic mutation of U2AF1 was identified in both a patient with MDS (family 1, II-2) as well as a preleukemic carrier (family 5, III-2). No other somatic mutations were identified in preleukemic samples tested. In addition to the germline JAK2 46/1 haplotype in 1 patient, we identified germline variants of interest in ASXL1, GATA2, IDH1, and CEBPA (Table 2).
Aggregation of germline RUNX1 mutations and associated phenotypes
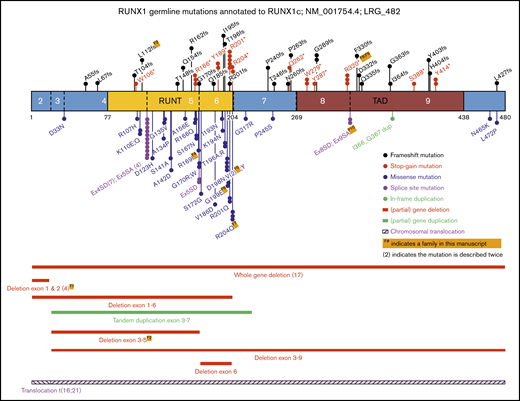
To contextualize our cohort findings, we updated our review of the literature,3 to identify as many known germline RUNX1 mutations as possible, including studies focusing on inherited thrombocytopenia, both with and without reported leukemia predisposition. As shown in Figure 2 and supplemental Table 1, also including the families in this study, to date, 104 different germline RUNX1 alterations, across 130 families, have been reported to be associated with thrombocytopenia and predisposition to HMs.2,5-8,17,18,21-70 Collectively, we identified approximately equivalent numbers of missense, stop-gain, frameshift, and deletion alterations across the RUNX1 gene. Missense mutations were significantly enriched in the RUNT domain (26 of 31; 84%), compared with truncating mutations (stop-gain and frameshift) that were annotated throughout the protein (15 of 36; 42% in RUNT domain; P < .0005; Fisher’s exact test). Canonical splice-site variants affecting the −1 and +1 positions between exons 4 and 5 were the most frequently observed germline RUNX1 mutations (11 independent families, 4 different nucleotide changes; Figure 2; supplemental Table 1). The most frequently mutated amino acid was p.R201, observed 8 times, with 5 cases resulting in a stop-gain p.R201* (supplemental Table 1).
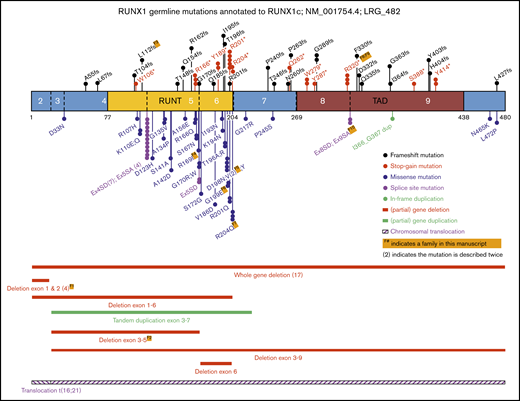
Schematic representation of RUNX1 showing the known germline genetic alterations associated with FPD-MM spectrum phenotypes. Shown are all mutations reported in this study, combined with published RUNX1 single-nucleotide variants (SNVs), small insertions and deletions (Indels), and CNVs annotated to RUNX1C; NM_001754.4; LRG_482. Full-mutation annotations can be found in supplemental Table 1. The numbers of the coding exons and known functional domains are shown within the protein, and the number of amino acids are shown below the diagram. The frequency of recurrent mutations is indicated by (N). For all mutations, the protein changes (p.) are shown, unless specified as splice-site variants (SA or SD). SA, splice acceptor; SD, splice donor.
Schematic representation of RUNX1 showing the known germline genetic alterations associated with FPD-MM spectrum phenotypes. Shown are all mutations reported in this study, combined with published RUNX1 single-nucleotide variants (SNVs), small insertions and deletions (Indels), and CNVs annotated to RUNX1C; NM_001754.4; LRG_482. Full-mutation annotations can be found in supplemental Table 1. The numbers of the coding exons and known functional domains are shown within the protein, and the number of amino acids are shown below the diagram. The frequency of recurrent mutations is indicated by (N). For all mutations, the protein changes (p.) are shown, unless specified as splice-site variants (SA or SD). SA, splice acceptor; SD, splice donor.
Aggregation of somatic mutations in germline RUNX1 carriers, with and without malignancy
Expanding on our analysis of the landscape of somatic mutations in germline RUNX1 mutated malignancies,3 we combined gene panel and exome data from families reported in this study with aggregation of reported somatic mutations from previously published studies. Collectively somatic variant profiling was available for 72 individuals with germline RUNX1 mutations (supplemental Figure 11), including 23 preleukemic individuals, 1 with MPN, 9 with isolated MDS, 7 with MDS/AML, 28 with AML, 3 with T-ALL, and 1 with tMDS. Of all identified somatic variants, RUNX1 mutations were the most frequently observed in MDS and AML patients (40%). The most common somatic variant was a duplication of the allele with the germline RUNX1 mutation (8 patients), resulting in a (partial) trisomy (5 patients) or uniparental disomy (3 patients) of chromosome 21. As early-onset clonal hematopoiesis of indeterminate potential (CHIP) has been previously described in germline RUNX1-mutated individuals without malignancy,6 we examined variants in genes with a known role in CHIP.71,72 Mutations were found in 5 (22%) preleukemic individuals and 18 (40%) patients with MMs, and, in the preleukemic individuals, they consisted of mutations in DNMT3A and TET2 (supplemental Figure 11). Somatic mutations in the other 2 most frequently mutated genes across the cohort (BCOR and PHF6) were identified in 6 (17.6%) of the myeloid tumors and were absent from the preleukemic samples. Strikingly, missing from the data set were mutations in ASXL1, which occurred in only 1 patient with AML. As ASXL1 mutations are highly associated with somatic mutation of RUNX1 in sporadic AML,11 this finding led us to examine our data for other FPD-MM–specific somatic mutation features.
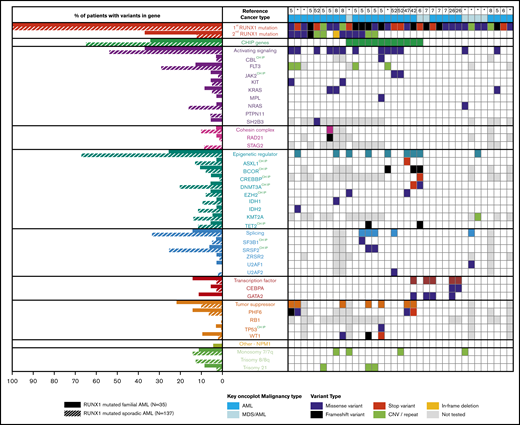
Comparing somatic variants of familial to sporadic RUNX1-mutated AML
For germline cases that with a diagnosis of AML, combining published somatic mutation profiles (n = 27) with the genomic data from our AML patients (n = 8) resulted in a subcohort of 35 AML patients with germline RUNX1 mutations (FPD-AML). From a published cohort of sporadic AML patients, individuals carrying at least 1 somatic RUNX1 variant were identified (n = 137).9 Somatic variation patterns of the FPD-AML cohort (n = 35) showed an increase (Figure 3) of somatic variation affecting the second RUNX1 allele and GATA2, compared with the sporadic AML cohort (n = 137). In addition, we identified a decrease in mutations in FPD-AML individually for the DNMT3A, NRAS, and SRSF2 and 2 gene groups, including the genes associated with CHIP and known epigenetic regulators, noting that 6 of the 9 epigenetic regulators also fall into the CHIP gene group. As expected from our previous observations, there were fewer ASXL1 mutations in the FPD-AML cohort (2.9% compared with 13.1% in the sporadic cohort). With regard to recurrent cytogenetic abnormalities, both sporadic and germline RUNX1-mutated AMLs demonstrated a similar frequency of monosomy 7, whereas sporadic RUNX1-mutated AML had an increased frequency of trisomy 8 (Figure 3).
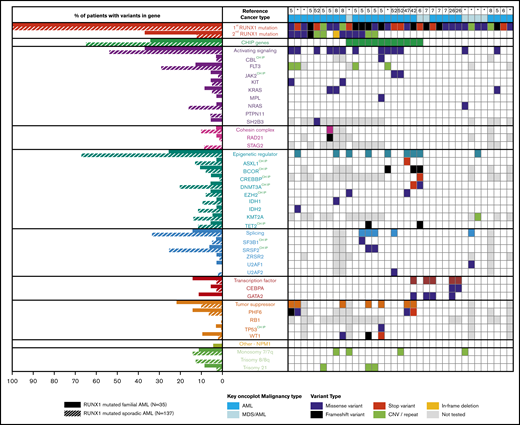
Somatic mutation differences in inherited (germline) and sporadic (somatic) RUNX1 mutated AML. Aggregation of somatic mutations in germline RUNX1 carriers who developed AML as identified through literature review (right). Comparison of the frequency of somatic mutation in myeloid genes in individuals with inherited or sporadic AML (left). *Somatic mutations identified in samples from this study.
Somatic mutation differences in inherited (germline) and sporadic (somatic) RUNX1 mutated AML. Aggregation of somatic mutations in germline RUNX1 carriers who developed AML as identified through literature review (right). Comparison of the frequency of somatic mutation in myeloid genes in individuals with inherited or sporadic AML (left). *Somatic mutations identified in samples from this study.
Discussion
RUNX1 mutation types
In the current study, we describe 10 new families with germline RUNX1 mutations, including 2 new partial gene deletions, 4 novel mutations, and 3 recurring mutations leading to FPD-MM. After combining these 10 new families with an updated review of the literature,3 130 families with germline RUNX1 genetic alterations have now been reported. An aggregated diagram of the known alterations is presented in Figure 2, and the details and citations are available in supplemental Table 1.
Collectively these data show a consistent pattern of stop-gain and frameshift mutations throughout the RUNX1 protein, with missense mutations enriched in the RUNT domain and in particular impacting residues that directly affect DNA binding and nuclear localization. In addition to the 103 single-nucleotide variants and small indels, 21% (n = 27) of all reported germline RUNX1 mutations are structural variants, including 25 (partial) gene deletions, 1 tandem duplication, and 1 translocation (Figure 2). Twenty-seven is likely to be an underestimation, because additional analyses to identify these events (such as SNP arrays, RNA analysis, and CNV calling on exome/gene panel data) are often not performed on mutation-negative patients. These findings continue to be consistent with a general model of RUNX1 as a tumor-suppressor gene in this setting. This is layered with a complexity that findings from tumor sequencing suggest that RUNX1 operates as both a haploinsufficient tumor suppressor (one mutation only required) and a classic Knudsonian tumor suppressor (2 mutations required)73 and further implies that mutations in other pathways may act to phenocopy the effect of a second RUNX1 mutation in some cases or provide alternative pathways to leukemogenesis. Recently, the properties of FPD-MM–associated germline RUNX1 mutations were assessed by a joint ASH-ClinGen Myeloid Malignancy Variant Curation Expert Panel to provide gene-specific modifications of the American College of Medical Genetics and Genomics/Association for Molecular Pathology classification guidelines.12,74 Using these up-to-date rules, all of the variants described in our 10 families are classified as either pathogenic or likely pathogenic (supplemental Table 3).
Association of RUNX1 mutations with phenotypic heterogeneity
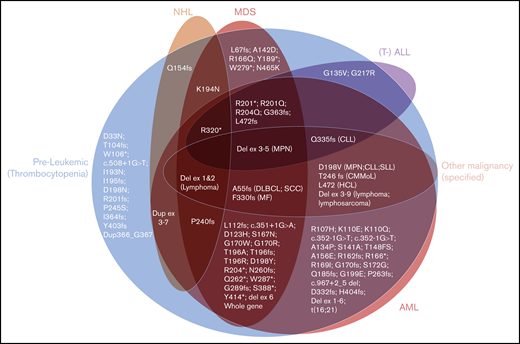
As expected, long-standing thrombocytopenia was the most common phenotype described in carriers of RUNX1 mutations in our 10 families, with platelet counts ranging from 50 × 109/L to 143 × 109/L (Table 1). The majority of HMs were of myeloid subtype (12 of 15 in confirmed carriers), with 4 individuals observed with lymphoid malignancies (Table 1). This indicates that lymphoid malignancies are a significant subtype of diseases associated with germline RUNX1 mutations, which is consistent with our aggregation of the literature showing that 25% of families have at least 1 individual with a lymphoid malignancy (supplemental Table 1; Figure 4). Interestingly, in our families, 2 of the lymphoid malignancies were observed in individuals who had multiple HMs that switched phenotype (family 5, III-2, and family 7, III-1). The aggregated data set was used to assess the correlation between type or the location (protein domains) of the germline RUNX1 mutations and development of malignancy (Figure 4). This analysis did not reveal a significant correlation between the type or location of mutations and malignancy. There was also a lack of correlation between the subtype of HM and type of RUNX1 mutation (Figure 4). This is consistent with the pattern of HM malignancy types in our families, and the literature (supplemental Table 1), where HM subtype heterogeneity is a feature that falls within families, as well as between families with different germline mutations. Twelve (15%) of the 82 published mutations have been reported only in isolated thrombocytopenia without HM. These 12 mutations show similar types (missense, frameshift, and stop-gain) and similar distribution across the RUNX1 gene compared with the mutations described in HM. This is further supported by comparing these 12 thrombocytopenic mutations individually to mutations known to predispose to HM; the majority (n = 7) of the thrombocytopenia-only mutations affect identical amino acids (n = 2) or amino acids directly adjacent (n = 5) to the ones mutated in HM. We conclude from this analysis that the identification of mutations through different contexts (inherited thrombocytopenia studies vs malignancy as the initial presentation) may reflect ascertainment bias, and identification of all such mutations should trigger consideration of assessment and counseling of potential HM risk.
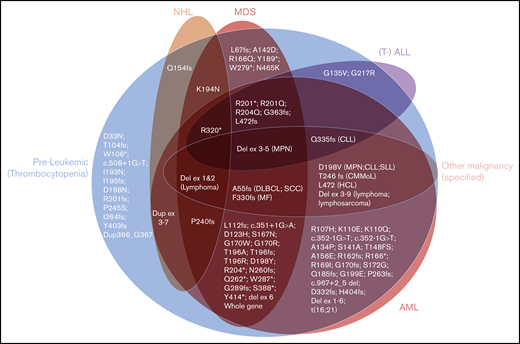
The spectrum of hematological phenotypes reported with different germline RUNX1 mutations. The protein (p.) changes are shown, for frameshift (fs), stop-gain (*), and missense variants. Splice-site variants are shown with the cDNA change (c.), and (partial) gene deletions are abbreviated. CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; CMMoL, chronic myelomonocytic leukemia; Del, deletion; DLBCL, diffuse large B-cell lymphoma; Dup, duplication; Ex, Exon; HCL, hairy cell leukemia; MF, myelofibrosis; SCC, squamous cell carcinoma.
The spectrum of hematological phenotypes reported with different germline RUNX1 mutations. The protein (p.) changes are shown, for frameshift (fs), stop-gain (*), and missense variants. Splice-site variants are shown with the cDNA change (c.), and (partial) gene deletions are abbreviated. CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; CMMoL, chronic myelomonocytic leukemia; Del, deletion; DLBCL, diffuse large B-cell lymphoma; Dup, duplication; Ex, Exon; HCL, hairy cell leukemia; MF, myelofibrosis; SCC, squamous cell carcinoma.
Recurrent comorbidities in RUNX1 families in this study
In addition to thrombocytopenia and HM, other clinical characteristics (eg, solid tumors, eczema, and arthritis) were observed in multiple families. Various solid tumors (breast, prostate, bone, gastric, pancreas, and skin) have been diagnosed in 3 of the families described (Figure 1; Table 1; families 2, 8, and 9). Although somatic RUNX1 mutations have been reported in breast cancer,75 there is no conclusive evidence that these solid tumor malignancies form part of the predisposition spectrum for germline RUNX1 mutations. Nonmalignant phenotypes of eczema/psoriasis and/or arthritis were present in families 1, 6, and 8 (Figure 1; Table 1), with varying degrees of cosegregation with the germline RUNX1 mutation. Eczema has been reported in association with a germline RUNX1 variant,63 and interestingly, genome-wide association studies for both phenotypes, including psoriasis, psoriatic arthritis, rheumatoid arthritis, and juvenile arthritis have all implicated RUNX1 through variants in binding sites of target genes or intronic SNPs that may be associated with regulation of RUNX1 expression directly.76-80 Because of the commonality and variation of these phenotypes in the population, benchmarking them as part of the RUNX1 spectrum will require further detailed studies of their clinical and laboratory characteristics in carriers.
Phenotypic heterogeneity: a role for additional germline genetic modifiers?
The phenotypic heterogeneity in families with RUNX1 germline mutations implies that additional mutations may be important modifiers to both penetrance and phenotype of HM development. A first indication of this was the report of the germline JAK2 haplotype81 in combination with a pathogenic germline RUNX1 mutation associated with JAK2-V617F–positive malignancies in family members.52 Here, we also report an individual with both a germline RUNX1 mutation and the JAK2 haplotype, with JAK2-V617F–positive MPN (family 5). In another family (family 4), we observed that coinheritance of a predicted damaging germline ASXL1 variant correlated with the development of malignancy in this family (Table 2; supplemental Table 2), whereas their sibling (II-5) without malignancy is carrier of a germline GATA2 variant, P161A, that reduces GATA2 activity in vitro (supplemental Figure 4C). As data accumulate, a systematic study of coinherited variants in genes of interest may help explain phenotypic variability and assist in risk assessment at an individual level.
Unique genetic features of FPD-MM relative to sporadic and somatic disease
The majority of the RUNX1 amino acid hotspots (top 5 most mutated) overlap for germline and somatic RUNX1 mutations (p.R166, p.D198, p.R201, and p.R204). However, the frequently observed germline canonical (+1 and −1) splice-site variants between exons 4 and 5 (n = 11; 11%) are absent from the 151 variants in the sporadic somatic RUNX1 mutation data set (P < .0001; Fisher’s exact test).9 Conversely, 1 of the most recurrent (7 times) somatic missense RUNX1 variants in sporadic AML (p.R162K) has not been observed in the 103 germline single-nucleotide mutations (Figure 2; P < .05).
Our somatic aggregation analysis of FPD-AML showed the importance of somatic mutation of the second RUNX1 allele with progression to myeloid malignancy, present in 18 of 45 cases (40%). This number is likely to be higher in reality because the CN-LOH (17% of somatic RUNX1 mutations; supplemental Figure 10) and noncanonical splice variants are often missed from genomic analyses. The germline mutation type does not influence the acquisition rate of the somatic RUNX1 mutation; 67% (12 of 18) of individuals with somatic RUNX1 mutations had a truncating germline RUNX1, which is similar to the overall myeloid malignancy cohort (62%). This aggregated data set also indicates an enrichment of somatic variants in FLT3 and WT1, which are only observed co-occurring with somatic RUNX1 mutations. We did not observe somatic RUNX1 mutations in any preleukemic cases, either in our data or from the aggregated literature data, suggesting that acquisition of somatic RUNX1 mutation, although frequent in tumors, is a later event in the disease process. In preleukemic cases, somatic mutations were observed in DNMT3A, TET2, and U2AF1. Mutations in these genes also appeared in tumors, indicating the potential of evolution of somatic gene–mutated preleukemic to leukemic states, as previously suggested.5,6,47 However, they were not the predominant tumor-associated mutations across the cohort, which highlights an ongoing knowledge gap of acquisition of molecular changes in the preleukemic state for FPD-MM. In the tumor state, more data were available for analysis, and our comparison of FPD-AML with sporadic AML highlighted differences in somatic mutation frequencies. In sporadic disease, AML derived from DNMT3A-mutated lymphomyeloid clonal state are significantly enriched with RUNX1 mutations.82 In our analysis, we found that compared with sporadic RUNX1-mutated AML, FPD-MM was less associated with mutations in the clonal hematopoiesis genes DNMT3A and SRSF2, indicating that, although, in the sporadic setting, RUNX1 may act as a late trigger event for both inherited and acquired preleukemic states, in forcing RUNX1 to be the first mutation, germline RUNX1-mutated malignancies acquire different cooperating mutations, preferring more frequently their own company (somatic RUNX1 mutation) and that of different chromatin remodelers and transcriptional regulators, such as GATA2, BCOR, PHF6, and WT1 (Figure 3). We also observed that in FPD-MM, somatic DNMT3A mutations and somatic RUNX1 mutations did not co-occur (Figure 3; supplemental Figure 11), and we speculate that an alteration of epigenetic states in combination with a single mutation in RUNX1 may generate an environment with access to a range of genes that alter the cellular state sufficiently that a second mutation in RUNX1 is not necessary, a pathway that would predominate in sporadic disease where RUNX1 is not an early mutational event. Interestingly, a recent study in ageing stem cells has demonstrated altered epigenetics that favor access of RUNX1 to key leukemia-associated genes, a process that could be recapitulated by direct mutation of epigenetic regulators.83 How this plays out functionally in terms of effects on pathways that require different genetic/epigenetic interdependencies and the triggers that determine secondary mutations that lead to progression of leukemia requires further investigation and generation of specific models.
Therefore, although it may be tempting to use somatically mutated RUNX1 sporadic malignancies as a proxy for germline RUNX1 predisposed malignancies when performing functional tumor modeling, therapy development, and clinical testing, our analysis indicates that there are real and important differences in both the germline and somatic mutation spectrum in FPD-MM. In particular, as the current curative therapy of choice, when to consider stem cell transplantation in germline-mutated individuals to maximize benefit and minimize risk is a topic of much recent discussion in the field and the answer is unclear in the case of germline RUNX1 mutations where penetrance and age of onset of malignancy have high variability.84 Therefore, although this is the biggest aggregation and analysis of FPD-MM cases to date, it is clear that further longitudinal studies of FPD-MM are warranted to conclusively answer questions about molecular progression to malignancy in germline RUNX1 mutation carriers and to provide rational targets for alternative therapies to potentially prevent leukemia development. Because of the relative rarity of this genetic disorder, further significant progress will continue to depend on collaborative efforts including international genomics and clinical trial programs.
The data reported in this article have been deposited in the European Genome-Phenome Archive (accession number EGAS00001004273).
Original data may be obtained by e-mail request to the corresponding author. Access to deidentified genomics data is available on request.
Acknowledgments
The authors thank the patients and their family members for their willingness to participate in this research and the SA Cancer Research Biobank for access to samples.
This work was supported by project grants APP1145278 and APP1164601 from the National Health and Medical Research Council of Australia, Principal Research Fellowship APP1023059 (H.S.S.), and the Royal Adelaide Hospital Research Foundation. This project is proudly supported by the Leukaemia Foundation of Australia, the Beat Cancer Project of the Cancer Council SA on behalf of its donors, and the State Government of South Australia through the Department of Health (project grant APP1125849). P.A. is supported by a fellowship from The Hospital Research Foundation.
Authorship
Contribution: A.L.B., P.A., and C.N.H. designed the research, analyzed and interpreted the data, and wrote the manuscript; M.B., C.-E.C., P.B., E.W., X.-C.L., J.M., and P.C. performed the research; J.D. performed the research, analyzed and interpreted the data, and contributed significantly to the manuscript; A.W.S., J.F., P.P.S.W., and T.H. performed the bioinformatic analyses; K.P., M.A., L.J., M.F., and C.B. collected the data and samples; C.C.H. and S.L.K.-S. analyzed and interpreted the data; L.R., C.V., A.D., J.B., G.M., D.H., A.W., and B.M. provided clinical molecular data and interpretation; S. Moore, M.N., and J.S. provided clinical cytogenetic data and interpretation; R.J.D. assisted with designing the study; I.D.L., D.K.H., L.A.G., and N.K.P. assisted with designing the study and provided clinical insight; E.P. provided data; M.S.H., S.I., A.K., and S.F. provided data and clinical and scientific insight; G.N. and H.Y.R. provided data and scientific insight; N.P., S. Morgan, R.S., S. Mapp, J.C., M.C., U.P., T.B., K.B., A.H.W., C.F., and H.M.F. provided data and clinical insight; H.S.S, and C.L.C. designed the research, analyzed and interpreted the data, and contributed significantly to the manuscript; and all authors critically reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for J.M. is Gene Predictis SA, Lausanne, Switzerland.
Correspondence: Anna L. Brown, Department of Genetics and Molecular Pathology, Centre for Cancer Biology, SA Pathology, PO Box 14, Rundle Mall, Adelaide, SA 5000, Australia; e-mail: anna.brown@sa.gov.au; Hamish S. Scott, Department of Genetics and Molecular Pathology, Centre for Cancer Biology, SA Pathology, PO Box 14, Rundle Mall, Adelaide, SA 5000, Australia; e-mail: hamish.scott@sa.gov.au; and Christopher N. Hahn, Department of Genetics and Molecular Pathology, Centre for Cancer Biology, SA Pathology, PO Box 14, Rundle Mall, Adelaide, SA 5000, Australia; e-mail: hahn@sa.gov.au.