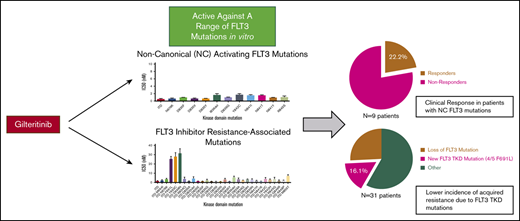
Key Points
Gilteritinib is active against a wide range of clinically relevant activating FLT3 mutations.
Resistance-conferring FLT3 kinase domain mutations are responsible for a minority of clinical resistance to gilteritinib.

Abstract
Gilteritinib is the first FMS-like tyrosine kinase 3 (FLT3) tyrosine kinase inhibitor (TKI) approved as monotherapy in acute myeloid leukemia with FLT3 internal tandem duplication and D835/I836 tyrosine kinase domain (TKD) mutations. Sequencing studies in patients have uncovered less common, noncanonical (NC) mutations in FLT3 and have implicated secondary TKD mutations in FLT3 TKI resistance. We report that gilteritinib is active against FLT3 NC and TKI resistance-causing mutations in vitro. A mutagenesis screen identified FLT3 F691L, Y693C/N, and G697S as mutations that confer moderate resistance to gilteritinib in vitro. Analysis of patients treated with gilteritinib revealed that 2/9 patients with preexisting NC FLT3 mutations responded and that secondary TKD mutations are acquired in a minority (5/31) of patients treated with gilteritinib. Four of 5 patients developed F691L mutations (all treated at <200 mg). These studies suggest that gilteritinib has broad activity against FLT3 mutations and limited vulnerability to resistance-causing FLT3 TKD mutations, particularly when used at higher doses.
Introduction
Mutations in the FMS-like tyrosine kinase 3 (FLT3) gene are the most common mutations in acute myeloid leukemia (AML).1,2 Activating internal tandem duplication (ITD) mutations are the most frequently identified FLT3 mutations. ITD mutations occur in ∼20% to 25% of AML and confer poor prognosis.3,4 Five to 10% of AML is associated with activating point mutations in the FLT3 tyrosine kinase domain (TKD), particularly at the residue D835.5,6 In recent years, FLT3 tyrosine kinase inhibitors (TKIs) have entered clinical development in AML with variable success. Midostaurin, a multitargeted inhibitor, demonstrated little activity as monotherapy,7 but prolonged survival when added to induction chemotherapy.8 This led to approval of midostaurin in newly diagnosed FLT3-mutated AML. In contrast to midostaurin, quizartinib, a more potent and selective inhibitor, demonstrated a high response rate as monotherapy in relapsed/refractory (R/R) patients with FLT3-ITD mutations.9,10 A randomized study of quizartinib vs chemotherapy in R/R FLT3-ITD+ AML (QuANTUM-R: An Open-label Study of Quizartinib Monotherapy vs. Salvage Chemotherapy in Acute Myeloid Leukemia [AML] Subjects who are FLT3-ITD Positive) demonstrated superior survival for patients treated with quizartinib.11 However, duration of response on quizartinib is short9,10 and is limited by acquired resistance-conferring FLT3 TKD mutations. These mutations occur frequently at the activation loop (AL) residue D835 and less commonly at the kinase “gatekeeper” residue F691.12 Because of limited resistance studies in patients treated on FLT3 TKI clinical trials, the true frequency of resistance-causing FLT3 TKD mutations is unknown. Nonetheless, TKD mutations, particularly at the D835 residue, are a commonly reported mechanism of clinical resistance to type II FLT3 inhibitors, which bind only the inactive kinase conformation. In addition to quizartinib, FLT3 TKD mutations have been associated with resistance to sorafenib,13,14 PLX3397 (pexidartinib),15 and sunitinib.13 In a small case series, 4/6, 14/15, and 6/9 assessed patients developed new FLT3 TKD mutations at the time of progression on sorafenib,14 quizartinib,16 and PLX3397,15 respectively.
Type I FLT3 inhibitors, which bind the active kinase conformation, have been developed to combat resistance caused by FLT3 D835 mutations. Of these, crenolanib17 and gilteritinib18 demonstrated preclinical activity against type II FLT3 inhibitor resistance-conferring D835 mutations and clinical activity as monotherapy in R/R FLT3-mutated AML.19,20 Gilteritinib (ASP2215), an oral inhibitor of FLT3 and AXL, has demonstrated significant single-agent activity in R/R FLT3-mutated AML, achieving an overall response rate of 40% in a phase 1/2 clinical trial (CHRYSALIS).19 A randomized phase 3 study of gilteritinib (ADMIRAL) compared with salvage chemotherapy (NCT02421939) in AML demonstrated a significant overall survival benefit in the gilteritinib arm (9.3 months) compared with chemotherapy (5.6 months).21 Event-free survival in the gilteritinib arm was also superior.21 Based on findings from this study, the US Food and Drug Administration (FDA) approved gilteritinib as the first FLT3 inhibitor indicated for use as monotherapy in FLT3-mutated R/R AML.
The activity of gilteritinib against a limited panel of FLT3 TKD mutations, including equipotent activity against FLT3 D835H/V/Y mutations found in native FLT3 (compared with FLT3-ITD), has been previously demonstrated in vitro18 ; however, the full activity of gilteritinib against other clinically relevant interchanges at D835 and other TKD mutations in native FLT3 is unknown. Moreover, the clinical activity of gilteritinib against FLT3 mutations beyond canonical ITD and D835 mutations has not been reported. These noncanonical (NC) FLT3 mutations make up ∼5% to 10% of FLT3 mutations identified in AML patients overall (though not all are confirmed to be kinase-activating).1,22 Knowledge of the ability of gilteritinib to inhibit a wider array of clinically relevant FLT3 mutations is critical to the appropriate selection of patients for gilteritinib treatment, especially as next-generation sequencing (NGS) technologies uncover patients with less common FLT3 substitutions.1,22 Notably, NC FLT3 TKD mutations have also been shown to cause resistance to midostaurin.23,24 As midostaurin becomes more commonly used in the upfront setting,8 resistance-associated FLT3 TKD mutations may be more frequently observed in the R/R setting. Previous studies identified that the FLT3 gatekeeper mutation F691L, in the context of FLT3-ITD, confers relative resistance to gilteritinib in vitro,18 but the role of this and other secondary FLT3 TKD mutations in clinical resistance to gilteritinib has not been systematically assessed in a complete clinical trial population. We aimed to test the activity of gilteritinib against a range of clinically relevant activating FLT3 TKD point mutations and secondary FLT3-ITD TKD mutations associated with FLT3 TKI resistance. Using a well-validated in vitro mutagenesis assay,12,25 we sought to prospectively identify novel FLT3-ITD TKD mutations that may confer clinical resistance to gilteritinib. Finally, we report the in vitro and clinical activity of gilteritinib against FLT3 mutations identified in patients treated in the phase 1/2 trial of gilteritinib in R/R FLT3-mutated AML.
Methods
Mutagenesis and resistance screen
We used a strategy for random mutagenesis of FLT3-ITD or FLT3-ITD/D835V, as previously described.12 Ba/F3 cells were selected with 20 or 40 nM gilteritinib (Astellas Pharma, Inc.) in soft agar. Whole genomic DNA was isolated from resistant Ba/F3 cells and the FLT3 TKD was amplified and sequenced as described.12
Generation of mutant Ba/F3 cell lines
FLT3 mutations were engineered into pMSCVpuroFLT3-ITD by QuikChange mutagenesis (Stratagene) and stable Ba/F3 lines were generated by retroviral infection as previously described.12
Cell-viability assay
Ba/F3 cells were plated in 0.1 to 320 nM gilteritinib in triplicate and expanded for 2 days. Viability was assessed and 50% inhibitory concentration (IC50) values were calculated as previously described.12
Immunoblotting
Immunoblotting using Ba/F3 cells stably expressing FLT3-mutated isoforms was performed as previously described,26 using anti-phospho-FLT3 (Cell Signaling Technology #3461) and anti-FLT3 antibody (Cell Signaling Technology #3462).
FLT3 sequencing of gilteritinib-treated patients
Blood or bone marrow samples from patients from the CHRYSALIS phase 1/2 open-label, randomized, dose-escalation/dose-expansion study of once-daily oral gilteritinib in patients with R/R AML were sequenced. Full details of the trial have been previously published.19 The entire coding sequence of the FLT3 gene in baseline and relapse samples was sequenced using a capture-based NGS assay on an Illumina MiSeq platform. Any non-silent variant detected at ≥0.6% variant allelic frequency (VAF) in the FLT3 TKD was reported.
Plasma inhibitory assay
Plasma inhibitory assay was performed as previously described.26 Plasma was obtained from healthy controls or from patients treated on the phase 1/2 study of gilteritinib (NCT00660920) at University of California San Francisco or University of Pennsylvania. All samples were collected under institutional review board–approved cell banking protocols. Informed consent was obtained in accordance with the Declaration of Helsinki.
Modeling of gilteritinib-FLT3 interaction
Docking simulation of gilteritinib with FLT3 was performed as previously described.27 The modeling software MOE (Chemical Computing Group Inc., Montreal, QC, Canada) was used to visualize the molecules.
Results
Gilteritinib is active against oncogenic and resistance-causing FLT3 mutations in vitro
Sequencing studies have identified a variety of activating NC FLT3 TKD mutations in AML patients.1,22,28,29 We engineered a subset of diverse clinically identified FLT3 TKD mutations into Ba/F3 cells and confirmed ability of these mutations to transform Ba/F3 cells to cytokine independence. We next assessed the ability of gilteritinib to impair proliferation of these cell lines in the absence of cytokine. Gilteritinib demonstrated potent antiproliferative activity against all assessed mutations, including all FLT3 AL mutations, which have been previously implicated in resistance to Type II FLT3 inhibitors such as quizartinib16 (Figure 1A; supplemental Table 1). The activity of gilteritinib against these less common FLT3 TKD mutations was equivalent to the activity of gilteritinib against FLT3-ITD, implying gilteritinib has potential for clinical activity in patients with a variety of activating FLT3 mutations.
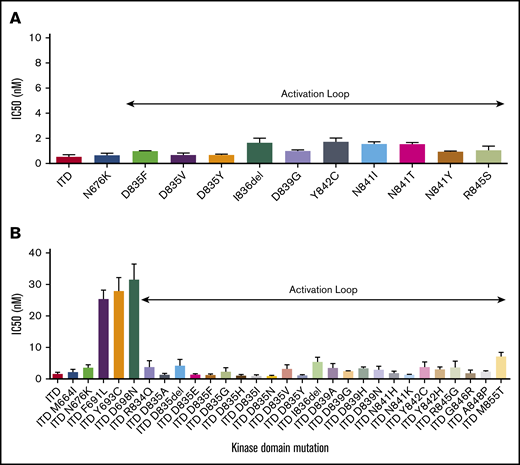
Gilteritinib activity against activating and TKI resistance-causing FLT3 mutations. (A) Gilteritinib IC50 levels for proliferation of Ba/F3 cells expressing activating FLT3 TKD mutations. Error bars represent standard deviation (SD) of 3 or more independent experiments. (B) Gilteritinib IC50 levels for proliferation of Ba/F3 cells expressing FLT3-ITD TKD mutations previously associated with FLT3-TKI resistance. Error bars represent SD of 3 or more independent experiments.
Gilteritinib activity against activating and TKI resistance-causing FLT3 mutations. (A) Gilteritinib IC50 levels for proliferation of Ba/F3 cells expressing activating FLT3 TKD mutations. Error bars represent standard deviation (SD) of 3 or more independent experiments. (B) Gilteritinib IC50 levels for proliferation of Ba/F3 cells expressing FLT3-ITD TKD mutations previously associated with FLT3-TKI resistance. Error bars represent SD of 3 or more independent experiments.
Many activating FLT3 TKD mutations identified de novo in AML patients, most notably FLT3 AL mutations, have also been implicated in FLT3 TKI resistance when identified in cis with FLT3-ITD. We assessed the activity of gilteritinib against FLT3-ITD TKD substitutions implicated in in vitro and clinical resistance to a variety of type I and type II FLT3 TKIs.12,15,17,24,26,30,31 Gilteritinib demonstrated potent activity against all TKD mutations assessed in cis with FLT3-ITD, with the exception of ITD/F691L, ITD/Y693C, and ITD/D698N mutations (Figure 1B; supplemental Table 1), all of which were identified in a screen for crenolanib resistance.17 These data indicate that the structurally diverse type I FLT3 inhibitors crenolanib and gilteritinib exhibit overlapping resistance profiles.17 Of note, a FLT3 extracellular domain mutation, K429E, identified to cause clinical crenolanib resistance32 was also tested but did not confer resistance to gilteritinib (supplemental Table 1). We also assessed the ability of gilteritinib to inhibit proliferation of a panel of FLT3-ITD+ human AML Molm14 clones that developed FLT3 TKD mutations after selection in quizartinib. Gilteritinib demonstrated equipotent activity in all assessed quizartinib-resistant cell lines, except in the case of the F691L line, which demonstrated resistance similar to that observed in Ba/F3 cells (supplemental Figure 1). Overall, these data suggested that gilteritinib has activity against the majority of known FLT3 TKI resistance-conferring mutations.
Mutagenesis screen identifies a limited number of gilteritinib resistance-conferring mutations
With the goal of identifying novel mutations that may confer resistance to gilteritinib, we used a well-validated mutagenesis screen which has previously successfully predicted clinically relevant TKI resistance-causing TKD mutations for both BCR-ABL25,33 and FLT3 TKIs12,15 (supplemental Table 2). Using this technique, we randomly mutagenized FLT3-ITD and FLT3-ITD/D835V and selected for drug-resistant clones in the presence of 20 and 40 nM of gilteritinib (∼20-40× the IC50 for FLT3-ITD). We elected to mutagenize FLT3-ITD/D835V in addition to FLT3-ITD in anticipation that some patients who develop secondary D835 mutations on treatment with quizartinib or sorafenib could receive gilteritinib as a second-line agent. Using this strategy, we identified 15 unique secondary FLT3-ITD TKD mutations in 64 colonies in the presence of 20 nM gilteritinib and no mutations in the presence of 40 nM gilteritinib (Figure 2A). On the background of FLT3-ITD/D835V, we identified 7 unique secondary TKD mutations in 59 colonies in 20 nM gilteritinib and 4 unique mutations in 13 colonies in 40 nM gilteritinib (Figure 2B). We recreated the mutations and confirmed that, in addition to the crenolanib-resistant F691L, Y693C, and D698N mutations, an additional substitution at residue Y693 (Y693N) and a G697S mutation conferred >10-fold resistance to gilteritinib in proliferation assays (Figure 2C; supplemental Table 1). All identified individual gilteritinib resistance-conferring mutations (F691L, Y693C/N, G697S, and D698N) resulted in comparable degrees of resistance in either the ITD or ITD/D835V background. The G697S mutation conferred the greatest degree of resistance in either background. Biochemical assays confirmed maintenance of FLT3 phosphorylation and downstream signaling in keeping with the degree of resistance observed in proliferation assays (Figure 2D; supplemental Figure 2; supplemental Figure 3). The G697S mutation has been previously implicated in a screen for resistance to midostaurin,23 but has never been identified clinically. Of the 5 identified gilteritinib resistance-conferring mutations, only the F691L mutation has been previously associated with clinical resistance to FLT3 TKI therapy, including in a recent report of clinical crenolanib resistance.12,32,34
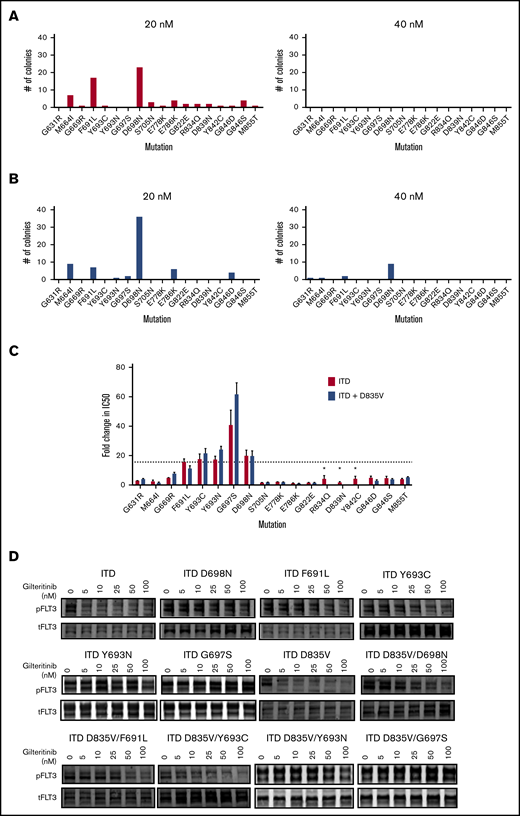
Mutation screen of FLT3-ITD and FLT3-ITD/D835 reveals kinase domain mutations that cause resistance to gilteritinib. Numbers of independently derived gilteritinib-resistant Ba/F3/FLT3-ITD subpopulations with amino acid substitution at the indicated residue obtained from a saturation mutagenesis assay for FLT3-ITD (A) and FLT3-ITD/D835V at the indicated gilteritinib concentration (B). (C) Relative resistance of Ba/F3 cells expressing indicated FLT3 TKD mutations in FLT3-ITD (red) and FLT3-ITD/D835V (blue) backgrounds. Resistance is expressed as fold-change compared with FLT3-ITD (ratio of mutant cell line IC50 over IC50 of FLT3-ITD cells). Error bars represent SD of 3 or more independent experiments. *Mutation was only assessed in FLT3-ITD background. Dotted line indicates fold-change in IC50 for FLT3-ITD/F691L cell line. (D) Western blot analysis using anti–phospho-FLT3 and anti-FLT3 antibody performed on lysates from interleukin-3-independent Ba/F3 populations expressing the FLT3-ITD–mutated isoforms indicated. Cells were exposed to gilteritinib at the indicated concentrations for 90 minutes.
Mutation screen of FLT3-ITD and FLT3-ITD/D835 reveals kinase domain mutations that cause resistance to gilteritinib. Numbers of independently derived gilteritinib-resistant Ba/F3/FLT3-ITD subpopulations with amino acid substitution at the indicated residue obtained from a saturation mutagenesis assay for FLT3-ITD (A) and FLT3-ITD/D835V at the indicated gilteritinib concentration (B). (C) Relative resistance of Ba/F3 cells expressing indicated FLT3 TKD mutations in FLT3-ITD (red) and FLT3-ITD/D835V (blue) backgrounds. Resistance is expressed as fold-change compared with FLT3-ITD (ratio of mutant cell line IC50 over IC50 of FLT3-ITD cells). Error bars represent SD of 3 or more independent experiments. *Mutation was only assessed in FLT3-ITD background. Dotted line indicates fold-change in IC50 for FLT3-ITD/F691L cell line. (D) Western blot analysis using anti–phospho-FLT3 and anti-FLT3 antibody performed on lysates from interleukin-3-independent Ba/F3 populations expressing the FLT3-ITD–mutated isoforms indicated. Cells were exposed to gilteritinib at the indicated concentrations for 90 minutes.
The F691L mutation is identified in a minority of patients at the time of gilteritinib resistance
We sought to ascertain the role of specific FLT3 TKD mutations in clinical resistance to gilteritinib. We sequenced the FLT3 coding sequence in 240 patients enrolled in the CHRYSALIS phase 1/2 study of gilteritinib in R/R AML.19 Before gilteritinib treatment, 10 patients treated at clinically efficacious doses of gilteritinib (≥80 mg) had detectable NC FLT3 TKD mutations (defined as non-ITD, non-D835 mutations). These mutations were found alone or in combination with an ITD or D835 mutation (Table 1): 5/10 patients had both an ITD mutation and a NC FLT3 TKD mutation and 5/10 had a NC FLT3 TKD mutation without a cooccurring ITD. Two of 9 evaluable patients with preexisting NC FLT3 mutations had a clinical response to gilteritinib (partial response); the remaining 7 patients were nonresponders (Table 1). One responding patient expressed a N841Y35 TKD mutation predicted to be sensitive to gilteritinib in vitro (Figure 1A). This mutation was detectable at high VAF (33%) before gilteritinib treatment, suggesting that patients with activating NC FLT3 mutations can respond clinically to gilteritinib. The other responder expressed a variant of uncertain significance in the FLT3 kinase insert region (H721Y) at a similar VAF as FLT3-ITD. No mutations at FLT3 residues implicated in gilteritinib resistance in vitro were identified in any patient before gilteritinib treatment, including in nonresponders. Given the small number of patients with NC FLT3 mutations assessed in this study and the overall low response rate (2/9 patients, 22%), larger clinical experience will be required to determine the activity of gilteritinib in patients with NC FLT3 mutations.
We next sought to determine if acquired FLT3 mutations were associated with acquired gilteritinib resistance. Thirty-one patients in this cohort had a FLT3 mutation at baseline and had an available relapse sample for sequencing (supplemental Table 3). Of these, 23 patients had FLT3 mutations detectable at both baseline and relapse. Five patients developed newly detectable FLT3 mutations at relapse (Table 2; supplemental Table 3). One patient acquired a new FLT3 mutation of uncertain functional significance (N609T) at the time of relapse. The mutation was identified at extremely low VAF (<1%) at the time of relapse, making its significance as a driver of drug resistance unclear. Four of 5 patients who developed new FLT3 mutations at relapse acquired a new F691L mutation at the time of gilteritinib resistance (Table 2). All patients who developed secondary F691L mutations were FLT3-ITD+ at the time of relapse. Before gilteritinib treatment, 3 of 4 patients had both ITD and D835 mutations and the remaining patient had only an ITD mutation detectable. At the time of resistance, 1 patient had detectable ITD, D835Y, and F691L mutations. The remaining 3 patients, including 2 who had pretreatment D835 mutations, exhibited only ITD and F691L mutations at the time of drug resistance. In 2 patients, multiple distinct F691 codon mutations were noted at relapse (c.2071T>C and c.2073T>G), indicative of polyclonal resistance. All 4 patients who developed F691L mutations were treated at drug doses <200 mg. No patients treated at ≥200 mg developed new FLT3 mutations at the time of gilteritinib resistance in our cohort. Given the moderate degree of resistance conferred by the F691L mutation in vitro (Figures 2C; supplemental Figure 1), occurrence of F691L in a minority (4/31, 12.9%) of gilteritinib-resistant patients and only at drug doses <200 mg, we hypothesized that resistance because of F691L could potentially be overcome by higher drug exposure levels.
Plasma inhibitory assay (PIA) indicates activity of gilteritinib against the FLT3 F691L mutation
To assess the potential ability of a higher dose of gilteritinib to suppress outgrowth of F691L-mediated resistance, we sought to assess if gilteritinib plasma levels in patients treated at ≥200 mg were sufficient to biochemically inhibit FLT3-ITD/F691L to a degree comparable to FLT3-ITD in vitro. Using pretreatment and steady-state plasma obtained from 2 patients treated with 200 mg of gilteritinib, we demonstrated that FLT3 auto-phosphorylation in FLT3-ITD+ Molm14 parental and F691L expressing cells were suppressed to similar levels (normalized to pretreatment and normal control plasma) (Figure 3). We also demonstrated that steady-state plasma from 1 patient treated at 300 mg also potently suppressed FLT3 autophosphorylation compared with normal control, though a pretreatment sample was unavailable from this patient. This finding suggests that potent inhibition of FLT3 with 200 mg of gilteritinib or higher has potential to overcome the moderate degree of gilteritinib resistance induced by the F691L mutation.
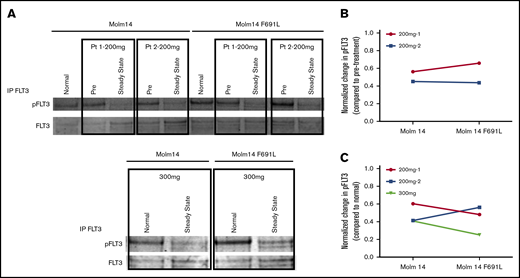
PIA shows gilteritinib is active against FLT3-ITD/F691L at clinically achievable plasma concentrations. (A) Western blot analysis for phosphotyrosine and total FLT3 performed after immunoprecipitation using anti-FLT3 antibody on lysates prepared from parental Molm14 cells and Molm14 cells expressing the FLT3-ITD/F691L mutation. Cells were exposed for 120 minutes to healthy normal control or pretreatment (pre) and steady-state plasma obtained from patients treated with gilteritinib at the 200 or 300 mg daily. Quantification of PIA data shown in panel A indicating reduction in normalized phospho-FLT3 (pFLT3) levels at steady-state timepoint compared with pretreatment (B) or normal (C) control plasma. Phosphotyrosine and total FLT3 band was quantified on a Licor Oddyssey imager and phosphotyrosine value was normalized to total FLT3 to derive normalized pFLT3 level. Data shown are ratios of steady-state to pretreatment normalized pFLT3 levels. Data represent a single experiment.
PIA shows gilteritinib is active against FLT3-ITD/F691L at clinically achievable plasma concentrations. (A) Western blot analysis for phosphotyrosine and total FLT3 performed after immunoprecipitation using anti-FLT3 antibody on lysates prepared from parental Molm14 cells and Molm14 cells expressing the FLT3-ITD/F691L mutation. Cells were exposed for 120 minutes to healthy normal control or pretreatment (pre) and steady-state plasma obtained from patients treated with gilteritinib at the 200 or 300 mg daily. Quantification of PIA data shown in panel A indicating reduction in normalized phospho-FLT3 (pFLT3) levels at steady-state timepoint compared with pretreatment (B) or normal (C) control plasma. Phosphotyrosine and total FLT3 band was quantified on a Licor Oddyssey imager and phosphotyrosine value was normalized to total FLT3 to derive normalized pFLT3 level. Data shown are ratios of steady-state to pretreatment normalized pFLT3 levels. Data represent a single experiment.
Molecular interaction of FLT3 with gilteritinib
To understand the structural basis of gilteritinib resistance induced by mutations at F691, Y693, G697, and D698, we modeled the binding of FLT3 to gilteritinib (Figure 4). As shown in Figure 4, gilteritinib interacts with the side chain atoms of F691 via van der Waals or C–H donor π-acceptor (CH−π) interactions. Therefore, the reduction of the inhibitory activity of gilteritinib for the F691L mutation would be attributed to the smaller side chain or the lack of aromaticity. The phenyl ring of gilteritinib forms π−π and CH−π interactions with Y693 and G697, respectively. Therefore, it is reasonable that gilteritinib has reduced affinity for Y693 mutations due to the lack of aromatic rings in Y693C/N. Because G697S lacks 1 Cα-H atom contributing to the CH−π interaction, the decreased activity of gilteritinib for G697S is reasonable. In addition to the loss of this interaction, the side chain of G697S would have a steric clash with the phenyl ring of gilteritinib, which can also contribute to significant reduction of gilteritinib inhibitory activity.
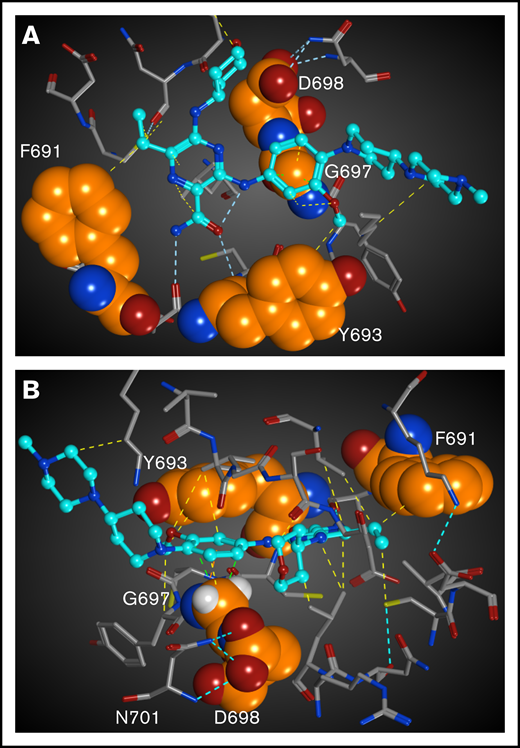
Structural mapping and interaction network of the gilteritinib-resistant mutations in FLT3. The residues for which mutations have been identified from the in vitro saturation mutagenesis screen are represented as spheres. Gilteritinib is shown as a ball-and-stick model. All of atoms are colored by the type of element (white: hydrogen; cyan, gray, and orange: carbon; blue: nitrogen; red: oxygen, yellow: sulfur). For clarity, the hydrogen atoms are omitted. Molecular interactions are shown as dotted lines and colored by the type of interaction (cyan: hydrogen bond; yellow: via van der Waals interaction; light green: CH−π interaction). (A) Gilteritinib bound to FLT3. The residues in front of the gatekeeper residue F691 is hidden. (B) Another view of gilteritinib bound to FLT3. The hydrogen atoms of G697 are shown.
Structural mapping and interaction network of the gilteritinib-resistant mutations in FLT3. The residues for which mutations have been identified from the in vitro saturation mutagenesis screen are represented as spheres. Gilteritinib is shown as a ball-and-stick model. All of atoms are colored by the type of element (white: hydrogen; cyan, gray, and orange: carbon; blue: nitrogen; red: oxygen, yellow: sulfur). For clarity, the hydrogen atoms are omitted. Molecular interactions are shown as dotted lines and colored by the type of interaction (cyan: hydrogen bond; yellow: via van der Waals interaction; light green: CH−π interaction). (A) Gilteritinib bound to FLT3. The residues in front of the gatekeeper residue F691 is hidden. (B) Another view of gilteritinib bound to FLT3. The hydrogen atoms of G697 are shown.
Gilteritinib does not contact directly with D698 in the docking model, though the inhibitory activity of gilteritinib is reduced for D698N. Analysis of the D698 interaction network shows that D698 forms an interaction network with N701 via hydrogen bonds. D698N mutation would cause loss of this hydrogen bonds, which would lead to steric clash with N701 and flip of the side chain to gilteritinib because there is no space around D698N to be accommodated other than the space for gilteritinib. This side chain flip would lead to steric clash with the tetrahydropyran ring of gilteritinib, which would result in reduced inhibitory activity of gilteritinib.
Discussion
As the first FLT3 inhibitor approved for use as monotherapy in R/R FLT3-mutated AML, gilteritinib represents an important clinical advance in this poor prognosis subtype of AML. The high single-agent response rate of gilteritinib19 coupled with in vitro activity against commonly identified FLT3 D835H/V/Y mutations18 potentially provides significant advantages over earlier inhibitors, including quizartinib. However, appropriate identification of patients likely to benefit from gilteritinib is vital to optimize clinical outcomes. The current FDA label for gilteritinib is based on identification of patients using a polymerase chain reaction-based companion diagnostic that distinguishes only FLT3-ITD or D835/I836 TKD mutations. In this study, we demonstrate that gilteritinib has potent in vitro activity against a wide range of TKD mutations, including less common but clinically relevant NC activating FLT3 TKD mutations. Modern large-scale genomic sequencing assays have demonstrated that the diversity of FLT3 mutations in AML extends beyond hotspot FLT3-ITD and D835 mutations.1,22 Widespread use of clinical NGS assays in AML ensures that patients with less common NC FLT3 mutations will be increasingly identified. The potent in vitro activity of gilteritinib against all clinically identified activating FLT3 TKD mutations assessed in this study suggests that some patients with NC FLT3 mutations may also clinically benefit from gilteritinib, though clinical trials will be required to confirm this possibility.
It is important to acknowledge that the clinical activity of most FLT3 inhibitors, either approved or in late-stage clinical development, was established in clinical trials in which the majority of patients had FLT3-ITD mutations. Overall, the clinical experience with gilteritinib in non-ITD patients is small and the antileukemic activity of gilteritinib in the published clinical report of the CHRYSALIS phase 1/2 study in FLT3-mutated AML is relatively low in non-ITD patients compared with patients with ITD mutations.19 In this study, only 2 of 12 (17%) patients with D835 point mutations without cooccurring ITD mutation responded (compared with 43% in the overall study population treated at active doses of gilteritinib ≥80 mg). In contrast, the complete remission rates of patients with FLT3-ITD mutations and FLT3 TKD mutations were similar in the phase 3 randomized trial of gilteritinib vs salvage chemotherapy (ADMIRAL). In the ADMIRAL study, 19% of patients with FLT3 TKD mutations only achieved complete remission compared with 20.5% in patients with FLT3 ITD mutations only, though the number of patients with TKD only mutations remained small.21 Clinical response data from CHYRSALIS (or ADMIRAL) patients with NC FLT3 mutations other than ITD and D835 substitutions have not been previously reported. In the subset of patients assayed for this report, just 1 of 5 patients with a NC FLT3 TKD mutation only responded, though the 1 responding patient did exhibit a known activating N841Y substitution. Given the relatively equipotent activity of gilteritinib against ITD and TKD point mutations in vitro, it is possible that the lower clinical response rates in patients with only TKD activating point mutations is because these mutations were present in a minor subclone or may be attributable to the nature of the mutations themselves. TKD mutations may be less likely to be “drivers” in patients than ITD mutations, may be more dependent on cooccurring genetic lesions for oncogenic activity, and may thereby be less likely to respond to single-agent FLT3 inhibitor treatment. More widespread experience with gilteritinib in patients with non-ITD FLT3 mutations will be required to establish the true clinical utility of this drug in patients with canonical and NC activating TKD mutations.
The development of acquired resistance remains the most critical barrier to improved survival in AML patients treated with targeted therapy. Secondary on-target FLT3-ITD TKD mutations have been a commonly identified resistance mechanism in patients treated with FLT3 inhibitors. Gilteritinib’s potent activity against FLT3-ITD/D835 and other mutations known to confer resistance to type II FLT3 inhibitors such as quizartinib and sorafenib suggest a potential advantage over these earlier drugs. In this study, we demonstrate that gilteritinib has limited vulnerability to resistance-causing TKD mutations in FLT3. In a prospective screen, we identified a small number of FLT3-ITD TKD mutations (F691L, Y693C/N, G697S, and D698N) that confer a moderate degree of resistance to gilteritinib in vitro. The IC50 of the most highly resistant mutation identified (G697S) is ∼40× higher than FLT3-ITD. In comparison, the most highly resistant FLT3-ITD/D835 substitutions confer >200 to 2000× resistance to type II FLT3 inhibitors in vitro.36 We selected very few resistant colonies at 40 nM gilteritinib, a concentration below steady-state concentrations achieved in patients at the FDA-approved dose of 120 mg.19 The paucity of resistance-causing mutations identified at 40 nM gilteritinib in either the ITD or ITD/D835V background is in keeping with the fact that the mean IC50 of the most highly gilteritinib-resistant G697S mutation is only ∼100 nM. All other mutations, including the clinically identified F691L mutation, exhibit mean gilteritinib IC50 values <40 nM, suggesting that further attempts to identify highly resistant FLT3 TKD mutations in vitro are unlikely to be fruitful.
Ultimately, examination of resistance mechanisms in patients who relapse on gilteritinib therapy is required to define clinically relevant mechanisms of resistance. To this end, we sequenced the FLT3 coding sequence from 240 of the 252 patients enrolled on the CHRYSALIS trial and identified FLT3 TKD mutations associated with clinical resistance in only a small minority of patients. Of 31 patients with paired pretreatment and relapse samples, only 4 patients (12.9%) acquired new FLT3 TKD mutations known to cause gilteritinib resistance; all 4 patients acquired the F691L “gatekeeper” mutation in the context of FLT3-ITD. In our study, all patients who acquired an F691L mutation were treated at doses of gilteritinib below 200 mg. Incubation of FLT3-ITD+ Molm14 cells with and without F691L mutation with plasma from patients treated at 200 mg or higher of gilteritinib suggests that this dose can achieve equipotent inhibition of FLT3 phosphorylation in F691L cells. These data suggest that clinical resistance resulting from F691L (and the other moderately resistant FLT3-ITD TKD mutations identified in this study) is likely to be uncommon and may potentially be addressed by use of higher doses of gilteritinib. It is important to note, however, although 200 mg and 300 mg were considered safe doses in the phase 1/2 study, the dose assessed in the randomized phase 3 study (and the FDA-approved dose) was 120 mg. Most patients treated now with gilteritinib will be treated at this lower dose without escalation, as this is the recommendation of the US label. In Japan and Europe, the approved dose of gilteritinib is also 120 mg, though dose escalation to 200 mg is allowed for suboptimal response. It is possible that with more widespread use of gilteritinib, resistance-causing FLT3-ITD TKD mutations may be responsible for a larger proportion of resistance than in the small number of patients assessed in our study. Future studies will be required to define the true frequency of FLT3 TKD mutations as a mechanism of clinical gilteritinib resistance. It is notable, however, that in a study of clinical resistance to the type I inhibitor crenolanib, resistance-causing FLT3 TKD mutations were similarly identified in only a small proportion of patients.32 This finding is congruent with results of our study and reinforces that type I inhibitors may be overall less susceptible to on-target resistance, as is suggested by prior mutagenesis studies which often identify less resistance mutations associated with type I inhibitors17,33 than type II inhibitors12,15,25,26 (supplemental Table 2) as well as by our recent report of non-FLT3 dependent gilteritinib resistance mechanisms.37 The increased vulnerability of type II inhibitors to TKD mutations is likely attributable to the fact that type II inhibitors bind to allosteric sites in the inactive kinase conformation. Type II inhibitors are therefore susceptible both to mutations which change kinase conformation as well as mutations at drug contact sites. In contrast, type I inhibitors are vulnerable largely to mutations at sites of direct drug–protein interaction.
This study was focused on the identification of on-target gilteritinib resistance-causing FLT3 TKD mutations. However, it is clear that alternative resistance mechanisms are responsible for a larger proportion of clinical resistance. Further work will be needed to fully define these off-target resistance mechanisms. In a recent report from our group, treatment-emergent Ras/MAPK pathway mutations were acquired in 15 of 41 (36.6%) patients treated with gilteritinib, including activating mutations in NRAS (13/41 patients; 31.7%), KRAS (3/41 patients; 7.3%), PTPN11 (3/41 patients; 7.3%), CBL (2/41 patients; 4.9%), and BRAF (1/41 patients; 2.4%).37 We also identified 2 patients with new BCR-ABL1 fusions, consistent with prior reports.37,38 These results suggest that acquisition of new or parallel oncogenic driver mutations will be common, as has been reported with crenolanib.32,37 Ultimately, although gilteritinib has activity against a wide variety of FLT3 mutations and appears less clinically vulnerable to on-target resistance, rational combination therapies will be required to suppress outgrowth of heterogeneous resistant leukemic clones.
Acknowledgments
The authors acknowledge Lu Wang from IQVIA for assistance with statistical analyses.
This work was supported by National Institutes of Health, National Cancer Institute grants R21CA198621 (A.E.P.) and K08 CA187577 (C.C.S.). C.C.S. is the Damon Runyon-Richard Lumsden Foundation Clinical Investigator supported (in part) by the Damon Runyon Cancer Research Foundation (CI-99-18).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: C.C.S. conceived of the study and analyzed data; T.C.T., J.E.H., K.M., E.B., L.R., and A.E.P. performed research and analyzed data; and C.C.S., T.C.T., and J.E.H. coauthored the manuscript with input from all authors.
Conflict-of-interest disclosure: A.E.P. has served as a consultant and/or advisory board member for Astellas Pharma, Arog Pharmaceuticals, Agios, Daiichi Sankyo, Novartis, and Takeda and has received research funding from Astellas, Daiichi Sankyo, FujiFilm, and Novartis. C.C.S. has received research support from Astellas Pharma and FujiFilm and has served as an advisory board member for Astellas Pharma and Daiichi Sankyo. J.E.H., E.B., and K.M. are employees of Astellas Pharma. The remaining authors declare no competing financial interests.
Correspondence: Catherine C. Smith, University of California, San Francisco, 505 Parnassus Ave, Suite M1286, Box 1270, San Francisco, CA 94143; e-mail: catherine.smith@ucsf.edu.