Key Points
Represents the largest set of safety and efficacy data for tisagenlecleucel.
Outcomes in the real-world setting are similar to results in the pivotal trials.

Abstract
Tisagenlecleucel is a CD19 chimeric antigen receptor (CAR) T-cell therapy approved for treatment of pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia (ALL) and adults with non-Hodgkin lymphoma (NHL). The initial experience with tisagenlecleucel in a real-world setting from a cellular therapy registry is presented here. As of January 2020, 511 patients were enrolled from 73 centers, and 410 patients had follow-up data reported (ALL, n = 255; NHL, n = 155), with a median follow-up of 13.4 and 11.9 months for ALL and NHL, respectively. Among patients with ALL, the initial complete remission (CR) rate was 85.5%. Twelve-month duration of response (DOR), event-free survival, and overall survival (OS) rates were 60.9%, 52.4%, and 77.2%, respectively. Among adults with NHL, the best overall response rate was 61.8%, including an initial CR rate of 39.5%. Six-month DOR, progression-free survival, and OS rates were 55.3%, 38.7%, and 70.7%, respectively. Grade ≥3 cytokine release syndrome and neurotoxicity were reported in 11.6% and 7.5% of all patients, respectively. Similar outcomes were observed in patients with in-specification and out-of-specification products as a result of viability <80% (range, 61% to 79%). This first report of tisagenlecleucel in the real-world setting demonstrates outcomes with similar efficacy and improved safety compared with those seen in the pivotal trials.
Introduction
Tisagenlecleucel (Kymriah; Novartis) is a second-generation chimeric antigen receptor (CAR) T-cell therapy targeting the CD19 antigen expressed on the surface of cells, manufactured from autologous T cells transduced to express a 4-1BB costimulatory domain and a CD3ζ T-cell activation signaling domain.1 The approval of tisagenlecleucel in the United States was based on outcomes seen in children and young adults with acute lymphoblastic leukemia (ALL) and adults with non-Hodgkin lymphoma (NHL).2–4 A better understanding of the safety and efficacy outcomes of patients receiving tisagenlecleucel outside of a clinical trial remains crucial to the care of these patients.
The manufacturing cycle of tisagenlecleucel starts with cell collection by leukapheresis and cryopreservation at the treatment center, transportation to a central facility, activation of T cells using CD3/CD28 antibody–coated beads, transduction with a lentiviral vector containing the CAR construct, expansion of the cell population, product release according to a set of criteria, cryopreservation, and transportation back to centers.5 Before tisagenclecleucel release, required product specifications are assessed. These include product viability, cell dose, CAR expression, demonstration of CAR T-cell interferon-γ release assays for potency, and routine microbiologic clearance.6 Manufacturing CAR T cells is a challenging process, and although some products that do not meet specifications, termed out of specification (OOS), might be considered safe to infuse, it is unclear whether the outcomes resulting from their use are similar to those from in-specification products.
Transduction of T cells with a lentiviral vector could potentially result in long-term oncogenic effects, and this is the basis for follow-up of recipients for 15 years to assess the late effects and risk of subsequent primary malignancies (SPMs) that could be attributed to CAR T cells.7 In North America, long-term follow-up is a postmarket requirement (PMR), structured as a prospective, multicenter, observational study with a planned accrual goal of 2500 patients. The Center for International Blood and Marrow Transplant Research (CIBMTR) has a long-established outcomes database for hematopoietic cell transplantation (HCT), and it more recently established a robust non-HCT cellular therapy (CT) registry. This standardized approach for data collection on CAR T-cell recipients is now used for capturing outcomes of tisagenlecleucel in the real-world setting. This report provides data on 410 patients receiving commercial tisagenlecleucel in the real-world setting and an early analysis of safety and efficacy outcomes, with an assessment of manufacturing parameters on those outcomes.
Methods
Data sources
The CIBMTR is a research collaboration between the Medical College of Wisconsin and the National Marrow Donor Program/Be The Match that developed infrastructure for collection of data on non-HCT cellular therapies. Additionally, the CIBMTR operates the National Cancer Institute–funded Cellular Immunotherapy Data Resource, with the objective of collecting, processing, and sharing data on cellular therapies for treatment of cancer. Cellular therapy data for any CAR T-cell recipient are collected longitudinally from 130 participating centers in the United States and Canada. The CIBMTR assures data quality by a multistage error check, on-site data audits, and metrics for on-time data reporting by participating centers. Data use for research is defined according to a data sharing protocol review by local ethics committees, and all participants sign informed consent to share data with the CIBMTR. The tisagenlecleucel PMR study was reviewed and approved by the institutional review board of the National Marrow Donor Program/Be The Match.
For all tisagenlecleucel infusions with a valid batch identification number available within the CIBMTR, specification parameters, such as cell dose and viability for the final product, were obtained from the Novartis manufacturing database.6 The clinical outcomes and final product attributes were linked using a unique tisagenlecleucel batch number.
Patients and study design
In this noninterventional prospective study, patients who received tisagenlecleucel for an approved indication (ie, relapsed/refractory pediatric/young adult ALL or adult NHL) after 30 August 2017 (date of first approval of tisagenlecleucel) in the United States or Canada were eligible. Clinical data on tisagenlecleucel were reported by treatment centers to the CIBMTR, and manufacturing attributes of tisagenlecleucel were provided by Novartis. The data collection forms can be found on the CIBMTR Web site.8 The main outcomes analyzed included the incidence and severity of cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS), type and frequency of SPMs, hematologic recovery (neutrophils and platelets), overall response rate (ORR), duration of response (DOR), event-free survival (EFS), progression-free survival (PFS), and overall survival. CRS was determined by the reporting center using the American Society of Transplant and Cellular Therapy (ASTCT) grading criteria and based on the most severe manifestation occurring during the reporting period.9 ICANS was graded according to the ASTCT criteria and based on the neurologic manifestations related to tisagenlecleucel therapy. Among patients with ALL, complete remission (CR) was assessed based on achievement of a complete morphologic response (<5% blasts), and minimal residual disease (MRD) status was described when available. Among patients with NHL, disease response was assessed by the treating physician based on either computed tomography or positron emission tomography scan. If both were available, the positron emission tomography scan was used. DOR was defined as time from the date of first CR or partial remission (PR) to the date of progression, relapse, or death resulting from underlying disease. EFS among patients with ALL was defined from time from tisagenlecleucel infusion to death resulting from any cause, relapse, or treatment failure (failure to achieve remission, including death without remission), whichever occurred first. PFS in NHL patients was defined as time from tisagenlecleucel infusion to disease progression or death resulting from any cause. Patients were censored at the time of other anticancer therapy, including HCT, for both EFS and PFS. OS was defined as time from tisagenlecleucel infusion to death resulting from any cause. Neutrophil or platelet recovery was defined as achievement of absolute neutrophil count >500/mm3 or platelet count >20 × 109/L. Prolonged cytopenia was defined as the lack of recovery of blood counts above the defined threshold within 30 days from CAR T-cell infusion.
Cell viability and cell dose were the final product release parameters included in our analyses. Cell viability is the percentage of viable T cells in the final product, and the release specification is ≥80% in the United States. Cell dose is the total number of CAR+ viable T cells. Release specifications were as follows: for ALL, 0.2 × 106 to 5.0 × 106 CAR+ viable T cells per kilogram of body weight, if weight ≤50 kg, and 0.1 × 108 to 2.5 × 108 CAR+ viable T cells if weight >50 kg; for NHL, 0.6 × 108 to 6.0 × 108 CAR+ viable T cells. A batch was considered OOS if any of these release parameters did not meet the specifications.
Analysis set
Because study enrollment is ongoing, each data freeze includes patients with different follow-up times. The cellular registry captures data on efficacy and safety in separate forms, and the numbers of patients with complete information may differ. Figure 1 shows the CONSORT diagram with all patients enrolled by the time of the data freeze, but for the analysis, only patients infused and with completed 3-month post-CT follow-up forms were included (n = 410; 70% of total recipients of tisagenlecleucel). Additionally, for efficacy end points, only patients with information on disease outcomes were included (n = 401). Lastly, the manufacturing data set includes patients with available batch numbers in the registry database, allowing retrieval of tisagenlecleucel manufacturing parameters.

CONSORT diagram. 1, patients without the complete set of baseline forms were excluded; 2, infusion set is the cohort with complete baseline information at the time of data freeze; 3, identifiable tisagenlecleucel batch numbers with available product characteristics; 4, analysis set defined as patients with available follow-up forms after tisagenlecleucel with information related to safety and efficacy outcomes.
CONSORT diagram. 1, patients without the complete set of baseline forms were excluded; 2, infusion set is the cohort with complete baseline information at the time of data freeze; 3, identifiable tisagenlecleucel batch numbers with available product characteristics; 4, analysis set defined as patients with available follow-up forms after tisagenlecleucel with information related to safety and efficacy outcomes.
Statistical analysis
Data analysis was based upon a data freeze on 23 January 2020. Descriptive statistics were used for baseline data on all patients, including demographic and disease characteristics. Numbers and percentages of safety events after the first infusion were summarized descriptively. ORRs by indication were defined as rates of best disease response of CR for ALL and CR/PR for NHL, as recorded after tisagenlecleucel infusion until progressive disease or start of new anticancer therapy, whichever occurred first. ORRs were summarized along with 2-sided exact Clopper-Pearson confidence intervals (CIs). OS, PFS, EFS, and DOR were summarized using Kaplan-Meier estimates. The event-free probabilities at 12 and 6 months with 95% CIs were determined for ALL and NHL, respectively, based on the number of patients at risk. The frequencies and types of SPMs were summarized, and the incidence rates per patient-year were calculated.
The relationship of cell viability and cell dose with clinical end points of safety (any-grade CRS or ICANS within 100 days) and best overall response (BOR) was assessed using logistic regression for ALL (separately for weight ≤50 and >50 kg) and NHL patients, respectively. ORs with 95% CIs were determined for 10% increase in cell viability and doubling of cell dose, respectively. Potential confounding effects of covariate fields, namely bone marrow blasts before tisagenlecleucel infusion (for ALL) and disease status at tisagenlecleucel infusion, were also studied. In addition, selected clinical efficacy and safety end points were compared between patients who received products with cell viabilities <80% and ≥80%. SAS software package was used for data analyses.
Results
Demographics
At the time of data freeze, a total of 511 appropriately enrolled patients in the PMR study (Figure 1) and 410 patients treated in 73 centers in North America had available safety follow-up data to be included in the analysis. Characteristics of tisagenlecleucel recipients, along with numbers and percentages of patients comprising relevant subgroups for ALL and NHL, are listed in Table 1, respectively. Median time from ALL diagnosis to CAR T-cell infusion was 32 months. At the time of tisagenlecleucel infusion, 81.3% had detectable disease morphologically or by flow cytometry, and of the patients in CR (n = 95), 50 (53%) were MRD+, 44 (46%) were MRD−, and 1 had no MRD assessment reported. Forty-six patients (18%) had poor cytogenetics before tisagenlecleucel infusion, including Philadelphia chromosome positivity, t(4:11), t(8:14), mixed lineage leukemia, intrachromosomal amplification of chromosome 21, hypodiploidy, and complex cytogenetics. Median blast percentage in bone marrow before infusion was 2% (range, 0% to 100%); 28% of patients had no marrow blasts, 20% had 0% to <5% marrow blasts, and 33% had ≥5% marrow blasts before tisagenlecleucel. Of the patients with >5% marrow blasts at time of infusion, median blast percentage was 48% (range, 6% to 100%). Twenty-eight percent of patients had undergone prior allogeneic HCT, and 14.9% and 10.6% had received prior blinatumomab or inotuzumab, respectively. Median time from NHL diagnosis to CAR T cells was 16 months; 94.8% of patients had primary refractory or relapsed disease, and 68% had no response to the last line of therapy. Median size of the largest nodal mass before CAR T-cell infusion was 9.5 cm2. Eleven percent of patients had double- or triple-hit lymphoma, and 29% had undergone prior HCT, primarily with autologous stem cells. The most common lymphodepleting chemotherapy regimen was fludarabine and cyclophosphamide for both indications, and 9% of patients with NHL had received a bendamustine-based regimen. Median follow-up of patients with ALL and NHL was 13.4 and 11.9 months, respectively.
Relapsed/refractory ALL
CRS and ICANS.
Overall, 55% of patients had CRS (Table 2). Grade ≥3 CRS was reported for 16.1% of patients, including 1 death resulting from CRS. Overall, 25% of patients received tocilizumab and 6% of patients received corticosteroids alone or in combination with tocilizumab (Figure 2A). Among patients with CRS, 45% received tocilizumab and 10.7% received corticosteroids. Supportive care for patients with CRS included fluid boluses in 37.9%, vasopressor treatment in 22.1%, supplemental oxygen in 30%, and positive pressure ventilatory support in 9.3%. Overall, 27% of patients had ICANS (Figure 2D). Grade ≥3 ICANS was reported in 9.0% of patients. Among patients with ICANS, the most common symptoms of neurotoxicity included depressed consciousness (47.8%), tremors (21.7%), seizure (18.8%), hallucinations (17.4%), and dysphasia/aphasia (15.9%). Overall, ICANS was treated with corticosteroids in 6% of patients (Figure 2C). Among patients with ICANS, corticosteroids were used in 21.7%. Fifty-five patients (22%) underwent allogeneic HCT after CAR T-cell therapy; 34 of these transplantations were performed in remission as a consolidation approach and 21 were performed as treatment for disease relapse. Subgroup analyses of children <3 years of age, patients who had prior therapy with blinatumomab or inotuzumab, and patients with prior central nervous system involvement are summarized in supplemental Table 2A.
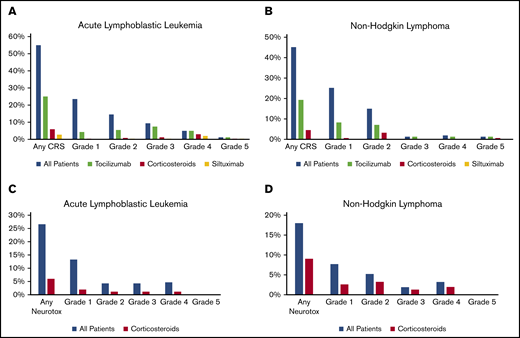
Safety outcomes among patients treated with tisagenlecleucel. CRS (A-B) and ICANS (C-D) by disease: ALL (A,C) and NHL (B,D).
Safety outcomes among patients treated with tisagenlecleucel. CRS (A-B) and ICANS (C-D) by disease: ALL (A,C) and NHL (B,D).
The cumulative incidences of neutrophil recovery at 28 and 100 days were 75% and 91%, respectively; corresponding cumulative incidences for platelet recovery were 79% and 89%, respectively. At 30 days after CAR T-cell infusion, 22% and 18% of patients experience prolonged neutropenia and thrombocytopenia. Regarding long-term safety outcomes, subsequent neoplasms were reported in 6 patients (2.4%), including acute myeloid leukemia and myelodysplastic syndrome (MDS) in 4 and 2 patients, respectively (supplemental Table 3). The cases of acute myeloid leukemia were confirmed as relapsed ALL with lineage switch, and 3 of these patients had confirmed mixed lineage leukemia rearrangement detected before tisagenelecleucel. One of the patients with MDS had reported Li-Fraumeni as a baseline genetic abnormality. No pregnancies were reported among female patients, nor among partners of male patients. Additional safety end points are included in the data supplement (supplemental Table 3).
ALL efficacy outcomes.
The complete remission rate was 85.5% (Table 3). Among the 116 patients in CR evaluated for MRD, 115 (99.1%) were MRD−. In subgroups of patients age <3 years, receiving prior treatment with blinatumomab, and with prior central nervous system involvement, the complete remission rates were similar at 86.7%, 78.4%, and 82.6%, respectively (supplemental Table 2A). Twelve-month DOR, EFS, and OS rates were 60.9%, 52.4%, and 77.2%, respectively (Figure 3A-C). Among patients in CR, 34 (16.1%) went on to undergo HCT while in remission.
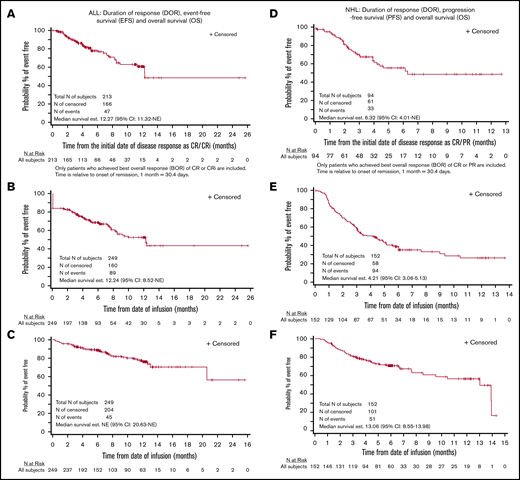
Efficacy outcomes for recipients of tisagenlecleucel. DOR (A), EFS (B) and OS (C) in patients with ALL, and DOR (D), PFS (E), and OS (F) in patients with NHL. CRi, CR with incomplete hematologic recovery; NE, not evaluable.
Efficacy outcomes for recipients of tisagenlecleucel. DOR (A), EFS (B) and OS (C) in patients with ALL, and DOR (D), PFS (E), and OS (F) in patients with NHL. CRi, CR with incomplete hematologic recovery; NE, not evaluable.
Relapsed/refractory NHL
CRS and ICANS.
Overall, 45% of patients had CRS. Severe grade ≥3 CRS was infrequent (4.5%; Table 2). Two patients died as a result of disease progression, with CRS as a contributing factor. Overall, tocilizumab treatment was used in 19% of patients and corticosteroids in 5% (Figure 2B). Among patients with CRS, tocilizumab was used in 42.9% and corticosteroids in 10%. Supportive care for patients with CRS included fluid boluses in 32.9%, vasopressor treatment in 4.3%, supplemental oxygen in 18.6%, and positive pressure ventilatory support in 5.7%. Overall, 18% of patients had ICANS (Figure 2D). Grade ≥3 ICANS was uncommon (5.1%). Overall, corticosteroids were used in 9% of patients for ICANS (Figure 2D). Among patients with ICANS, corticosteroids were used in 50%. The most common symptoms of neurotoxicity included depressed consciousness (39.2%) and dysphasia/aphasia (25%). A similar safety profile was noted for patients with double- or triple-hit lymphoma and patients ≥age 65 years (supplemental Table 2B).
Cumulative incidences of neutrophil recovery at 28 and 100 days were 93% and 97%, respectively; corresponding cumulative incidences for platelet recovery were 86% and 89%, respectively. At day 30 after CAR T-cell therapy, 5% and 13% of patients had prolonged neutropenia and thrombocytopenia. Subsequent neoplasms were reported in 6 patients (3.9%), with an exposure-adjusted event rate of 0.08 events per 100 person-years (supplemental Table 3). These malignancies included basal cell carcinoma (n = 1), genitourinary malignancy (n = 2), MDS (n = 1), B-cell ALL (n = 1), and cholangiocarcinoma (n = 1). No pregnancies were reported among female patients, nor among partners of male patients. Additional safety end points are included in the data supplement (supplemental Table 3).
NHL efficacy outcomes.
The ORR was 61.8%, including CR in 39.5% and PR in 22.4% of patients (Table 3). Among patients with double- or triple-hit lymphoma and patients age ≥65 years, response rates were similar to those of the overall population at 70.6% and 61.7%, respectively (supplemental Table 2B). The 6-month DOR, PFS, and OS rates were 55.3%, 38.7%, and 70.7%, respectively (Figure 3D-F).
Tisagenlecleucel final product attributes
Among the analysis set, a batch number was available for 383 patients (Figure 1). Median time from leukapheresis acceptance to infusion was 32 days (range, 21–130 days). Median tisagenlecleucel doses for ALL (<50 kg) were 2.0 × 106 CAR+ viable T cells per kilogram; for ALL (>50 kg), 0.9 × 108 CAR+ viable T cells; and for NHL, 1.8 × 108 CAR+ viable T cells. Median cell viabilities were 87.1% (range, 66.7% to 96.8%) and 83.8% (range, 61.4% to 94.9%) for patients with ALL and NHL, respectively. The commercial products in the registry had lower median doses and viabilities compared with the products in the pivotal trials (supplemental Figure 1).
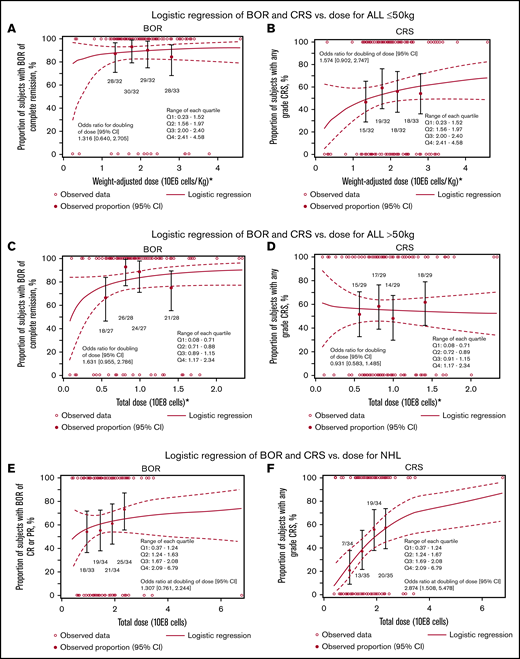
Logistic regression for BOR and CRS vs CAR T-cell dose infused in patients with ALL and NHL is shown in Figure 4A-F. For patients with ALL and weight ≤50 kg, the unadjusted cell dose response logistic regression analyses indicated no apparent impact of dose on BOR (OR, 1.32; 95% CI, 0.64-2.7) or CRS (OR, 1.57; 95% CI, 0.90-2.74; Figure 4A-B). Similarly, for patients with ALL and weight >50 kg, the OR of BOR was 1.63 (95% CI, 0.96-2.79); for CRS, the OR was 0.93 (95% CI, 0.58-1.49; Figure 4C-D). Adjusted analyses for BOR according to bone marrow blasts (<5% vs ≥5%; OR, 1.62; 95% CI, 0.87-3.00) and disease status (CR vs non-CR) at infusion (OR, 1.62; 95% CI, 0.91-2.87) demonstrated similar results (data not shown).

Correlation of cell product release specifications with clinical outcome. Logistic regression of BOR (A,C,E) and CRS (B,D,F) vs dose for ALL ≤50 kg (A-B), ALL >50 kg (C-D), and NHL (E-F).
Correlation of cell product release specifications with clinical outcome. Logistic regression of BOR (A,C,E) and CRS (B,D,F) vs dose for ALL ≤50 kg (A-B), ALL >50 kg (C-D), and NHL (E-F).
For NHL, the logistic regression analyses showed that by doubling the dose, the OR for BOR was 1.31 (95% CI, 0.761-2.24); for CRS, the OR was 2.874 (95% CI, 1.508-5.478; nominal P = .001; Figure 4C-D). Adjusting for disease status (CR/PR vs resistant) at infusion showed similar results (data not shown).
For ALL, 13.5% of patients received products with cell viability <80% (1% had <70%); all products had cell counts within the approved dosing range (supplemental Figure 1). For NHL, 31% patients received products with cell viability <80% (1.9% had <70%). One product had both viability <80% and CAR T-cell dose below the approved range. An additional 2 products had cell viability ≥80% but did not meet release criteria, because cell counts were not within the approved dosing range (1 above and 1 below; supplemental Figure 1). The BORs for patients with ALL who received a batch with cell viability <80% vs ≥80% were 94% and 84%, respectively (Table 4). The ORRs for patients with NHL who received a batch with cell viability <80% vs ≥80% were 52% and 65%, respectively (Table 4).
Discussion
This report represents the largest published series of patients with ALL and NHL treated with tisagenlecleucel and is the only commercial CAR T-cell therapy analysis incorporating product specification data. The CT registry already provides data on more than twice the number of patients as the registration trials. Although median follow-up is shorter in the PMR study compared with the pivotal trials, there now exists substantial experience in the short-term follow-up safety and efficacy outcomes of tisagenlecleucel.
This report demonstrates that in the real-world setting, efficacy of tisagenlecleucel for ALL and NHL are comparable to that in the pivotal ELIANA and JULIET trials. The short-term efficacy noted in response rates and 6-month estimates of time-to-event end points (ie, DOR, EFS/PFS, and OS) shows similarity to that in the pivotal trials (Tables 2 and 3). Admittedly, long-term results of time-to-event end points need more time to mature.
The patient population in the PMR study reflects the real-time use of CAR T-cell therapy, and important differences were observed compared with the cohort of patients enrolled in the pivotal trials (supplemental Table 1A-B). Previous reports have suggested that eligibility criteria for organ function, age, and prior therapy are less restrictive in the real-world setting than in the tisagenlecleucel registration trials.10 Children <3 years of age were not included in the pivotal tisagenlecleucel trial; however, almost 6% of the ALL cohort in our study were age <3 years. Prior treatment with allogeneic HCT was less frequent among patients in this study compared with the pivotal ALL trial (28% vs 61%). Primary refractory patients were more common in the registry than the pivotal trials (15% vs 8%), although median number of prior therapies was comparable among patients in the registry and pivotal trial (median, 3). Among adults with NHL, median age was older (65 vs 56 years) in the registry compared with the pivotal trial. Additionally, fewer patients underwent autologous stem cell transplantation befor tisagenlecleucel in the registry (26% vs 49%). Differences in baseline tumor burden were difficult to compare because of the disparity in collection timing (before and after bridging and lymphodepleting chemotherapy). Finally, the use of bendamustine as lymphodepleting chemotherapy was less frequent in the registry compared with the pivotal trial (9% vs 20%).4 In summary, there were notable differences in age groups and number of patients undergoing HCT before tisagenlecleucel among patients entered into the registry compared with pivotal trials for both ALL and NHL.
The regulatory approval of tisagenlecleucel in the United States, similar to other CAR T cells, required additional safeguards in addition to the PMR study, including the implementation of a Risk Evaluation and Mitigation Strategy (REMS) program to track and mitigate the key effects of CRS and ICANS.2 REMS, different than the PMR study, is mandatory for any prescriber or treatment center. A REMS program provides a framework to mitigate the risks of CRS and ICANS by certifying hospitals, clinics, prescribers, and ancillary staff with training for these adverse effects and requires immediate access to tocilizumab. The CIBMTR CT registry collects the necessary information to fulfill REMS requirements. Because a majority of cases of CRS and neurotoxicity occur within the first 1 to 2 weeks, the current data on >400 patients, with a median follow-up of ∼1 year, represent a substantial snapshot of the utility of the REMS program. Although grading scales are not identical between the registry and pivotal trials, the ASTCT grading scale is similar to the University of Pennsylvania CRS grading scale used in the pivotal trials, allowing rough comparison.11 The overall rate and incidence of grade ≥3 CRS seemed lower among patients in the real-world setting captured by the registry compared with the pivotal trials (Table 2). However, it is notable that for ALL, 95 patients were in CR (37.2%), including 44 who were MRD−, highlighting an overall lower disease burden that might have contributed to the differences in rates of toxicity. The use of tocilizumab seemed less frequent in pediatric ALL patients (24% vs 37%) and comparable in NHL patients (19% vs 14%) compared with the pivotal trials.3,4 Similarly, the overall rate and incidence of grade ≥3 ICANS seemed lower among registry patients than the pivotal trial patients (Figure 2D). The registry data indicate that toxicity is tolerable when tisagenlecleucel is administered with a REMS program.
CAR T-cell manufacturing is technically challenging. Most manufactured products fall within a narrow specification for cell count, viability, potency, and sterility. Some manufactured products do not meet full specifications but can and have been administered to patients with limited treatment options. The CIBMTR CT registry collects the same data for these patients, providing the opportunity to evaluate critically the utility of OOS products. Interestingly, a majority of OOS products are designated as such because of viability <80%. The product viability in the pivotal trials of commercial tisagenlecleucel was ≥70%; however, in the United States, it must be ≥80% per US Food and Drug Administration regulations. The viability of OOS products within the PMR study ranged from 61% to 79%. Supplemental Figure 1 highlights the differences in dose and viability between commercial tisagenlecleucel and products used in the pivotal trials, with commercial products having lower doses and viabilities (supplemental Data). For patients with ALL and NHL, there has not seemed to be an association between viability and inferior survival in prior studies.12,13 This PMR study of tisagenlecleucel also showed similar efficacy as well as toxicity between products with viabilities of <80% and ≥80% for both ALL and NHL (Table 4).
The dose range of tisagenlecleucel was approved based on the pivotal trials, in the absence of formal dose finding first-in-human studies. Preclinical data suggest a threshold dose of tisagenlecleucel for efficacy.14 In the pivotal and supportive trials of tisagenlecleucel in patients with ALL, logistic regression analysis showed an increasing probability of response with dose.15 Modeling predicted a lower response rate for patients treated with the lowest doses compared with the overall population.16 No dose-response or dose-expansion relationship was observed among patients with NHL in the tisagenlecleucel pivotal trial.4,17 In this study, there was no significant association between cell dose and disease response among patients with ALL. In patients weighing >50 kg, there was a possible association, which will need to be further investigated with a larger number of patients. Similar to the pivotal clinical study in NHL patients, the logistic regression analyses suggested an increasing rate of CRS with increases in the dose. The baseline tumor burden data were not available in this study to evaluate as a confounder; however, previous analyses from the pivotal clinical study demonstrated a relationship between high tumor burden and worsened severity of CRS.18
Established CIBMTR collaboration with hematopoietic stem cell transplantation centers, where most of these therapies are currently used, allowed the rapid implementation of this PMR study. This regulatory requirement is directed to the manufacturer. Data collection is voluntary, and patients need to sign an informed consent for data sharing with the CIBMTR. At time of the data freeze for this analysis, it was estimated that a little >50% of all tisagenlecleucel shipped to US centers was included in this study. Some of the limitations of the registry include: differences in data collection compared with the pivotal trials (eg, timing of efficacy assessments), lack of inclusion of patients considered for tisagenlecleucel therapy but not receiving infusion (eg, adverse event precluding treatment or death), difficult ascertainment of bridging chemotherapy, and lack of B-cell aplasia data or information related to CD19− relapses. As the field of CAR T cells evolves, optimizing data collection will fill some of these initial identified gaps. Balancing the collection of enough information to understand important efficacy and safety outcomes without increasing burden at the treatment sites is fundamental for building a CT registry to be a resource to the biomedical community.
This report provides the largest set of data for patients treated with tisagenlecleucel. The differences in the real-world patient population treated in the registry compared with the patient selection in the pivotal trials highlight the need for more generalizable real-world data in addition to those from clinical trials. As accrual continues up to 2500 patients followed for 15 years, this PMR study is well positioned to continue to yield important insights into the use of tisagenlecleucel.
Send data sharing requests to Marcelo C. Pasquini (mpasquini@mcw.edu).
Acknowledgments
The CIBMTR is supported primarily by Public Health Service grant U24CA076518 from the National Cancer Institute (NCI), the National Heart, Lung, and Blood Institute (NHLBI), and the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH); U24HL138660 from NHLBI and NCI; U24CA233032 from the NCI; R21HL140314 and U01HL128568 from the NHLBI; HHSH250201700006C, SC1MC31881-01-00, and HHSH250201700007C from the Health Resources and Services Administration; and N00014-18-1-2850, N00014-18-1-2888, and N00014-20-1-2705 from the Office of Naval Research. Additional federal support is provided by NIH grants P01CA111412, R01CA152108, R01CA215134, R01CA218285, and R01CA231141 from NCI, R01AI128775, U01AI069197, and U01AI126612 from NIAID, R01HL129472, R01HL130388, and R01HL131731 from NHLBI; and BARDA from the US Department of Health and Human Services. Support is also provided by Be the Match Foundation, Boston Children’s Hospital, Dana-Farber, Japan Hematopoietic Cell Transplantation Data Center, St Baldrick’s Foundation, the National Marrow Donor Program, the Medical College of Wisconsin, and the following commercial entities: AbbVie, Actinium Pharmaceuticals, Inc., Adaptive Biotechnologies, Adienne SA, Allovir, Inc., Amgen, Inc., Angiocrine Bioscience, Anthem, Inc., Astellas Pharma US, AstraZeneca, Atara Biotherapeutics, Inc., bluebird bio, Inc., Bristol-Myers Squibb Co., Celgene Corp., CSL Behring, CytoSen Therapeutics, Inc., Daiichi Sankyo Co., Ltd, Gamida-Cell, Ltd, Genzyme, HistoGenetics, Inc., Incyte Corporation, Janssen Biotech, Inc., Janssen/Johnson & Johnson, Jazz Pharmaceuticals, Inc., Kiadis Pharma, Kite, a Gilead Company, Kyowa Kirin, Legend Biotech, Magenta Therapeutics, Mallinckrodt LLC, Medac GmbH, Merck & Co., Inc., Merck Sharp & Dohme Corp., Millennium, the Takeda Oncology Co., Miltenyi Biotec, Inc., Novartis Oncology, Novartis Pharmaceuticals Corp., Omeros Corp., Oncoimmune, Inc., OptumHealth, Orca Biosystems, Inc., Pfizer, Inc., Pharmacyclics, LLC, REGiMMUNE Corp., Sanofi Genzyme, Shire, Sobi, Inc., Takeda Pharma, Terumo BCT, Viracor Eurofins, and Xenikos BV. M.J.F. acknowledges support from NIH, NCI grant K12CA087723.
The views expressed in this article do not reflect the official policy or position of the National Institutes of Health, Department of the Navy, Department of Defense, Health Resources and Services Administration, or any other agency of the US Government.
Authorship
Contribution: M.C.P., Z.-H.H., L.Y., R.C., and E.B. designed the research, analyzed the data, and wrote the paper; S.G., K.C., T.L., F.L., R.R., M.A.P., C.L.P., A.K., M.F., D.S., S.J., J.P.S., J.R., M.G., D.L., S.M., P.L.M., and M.K.K. enrolled patients, interpreted results, and edited/reviewed the paper; P.H., S.N., C.T., M.-A.P., P.S., M.M.H., A.M., and L.P. interpreted results and wrote/edited/reviewed the manuscript.
Conflict-of-interest disclosure: M.C.P. has received research funding from Novartis, Kite Pharma, and Bristol-Myers Squibb, and consulted for Amgen and Bristol-Myers Squibb. K.C. has received research support from Juno Therapeutics and Novartis; has consulted, served on advisory boards, and participated in educational seminars for Juno Therapeutics, Novartis, and Mesoblast. F.L. served as a scientific advisor for Kite, Novartis, BMS/Celgene, Allogene, Amgen, Wugen, Calibr, and GammaDelta Therapeutics; as a consultant for Cellular BioMedicine Group Inc; received research support from Kite; and discloses patents to improve cellular immunotherapy held by his institution. T.L. has consulted for Novartis, Cellectis, and Bayer and received research funding from Novartis, Bayer, and Pfizer. R.R. has received honoraria from Novartis and KTE/Gilead for CAR T advisory boards, and research funding from Tessa Therapeutics. M.A.P. has received honoraria from AbbVie, Bellicum, Bristol-Myers Squibb, Incyte, Kite (Gilead), Merck, Novartis, Nektar Therapeutics, and Takeda; served on data and safety monitoring boards for Servier, Medigene, and Cidara Therapeutics and on scientific advisory boards for MolMed and NexImmune; and received research support for clinical trials from Incyte, Kite (Gilead), and Miltenyi Biotec. C.L.P. has served on an advisory board for Novartis. M.J.F. reports consulting with Novartis, Arcellx, Kite/Gilead, and Bristol-Myers Squibb. S.J. has received research funding from Kite, Novartis, and Unum Therapeutics and advisory board funding from Kite, Novartis, Juno, and CRISPR Therapeutics. J.P.S. has participated in a speaker program for Kite when its X19 product was approved for MCL. D.L. served on a board or advisory committee for Celgene, Curis, Inc, Karyopharm, and Morphosys; was a consultant for Curis, Inc; served on a speaker’s bureau for Seattle Genetics; and received research funding from Curis, Inc, Takeda, and Triphase. S.M. served on an advisory board for Novartis. M.K.K. served on a speaker’s bureau for Seattle Genetics; and was a consultant or participated on advisory boards for Celgene, Beigene, AZD, Pharmacyclics, Curio Science, and Adaptive. S.N. has served on ad hoc advisory boards for Novartis, Kite, and NKarta. C.T. has received research funding from Juno Therapeutics, Nektar Therapeutics, AstraZeneca, and TCR2 Therapeutics; served on scientific advisory boards for Precision Biosciences, Eureka Therapeutics, Caribou Biosciences, T-CURX, Myeloid Therapeutics, ArsenalBio, and Century Therapeutics; served on ad hoc advisory boards (last 12 months) for Nektar Therapeutics, Allogene, PACT Pharma, and AstraZeneca; discloses equity options with Precision Biosciences, Eureka Therapeutics, Caribou Biosciences, Myeloid Therapeutics, and ArsenalBio; and a patent licensed to Juno Therapeutics. M.-A.P. reports steering committee participation for the Eliana/Ensign trial, advisory boards, and education support from Novartis; education and study support from Miltenyi; study support from Adaptive; advisory board participation for Jasper and Mesoblast; and education support from Bellicum. S.A.G. reports consultancy for Allogene, CBMG, Cellectis, GSK, Janssen/J&J, Jazz Pharma, Novartis, and Roche; board or advisory committee membership for Allogene, Cabaletta, Cellectis, CRISPR/Vertex, Juno/BMS, Novartis, and TCR2; and research funding from Kite/Gilead, Novartis, and Servier. L.P., L.Y., R.C., and E.B. are employees of Novartis Pharmaceutical Corp. The remaining authors declare no competing financial interests.
Correspondence: Marcelo C. Pasquini, Medical College of Wisconsin, 9200 W Wisconsin Ave, CC 5500, Milwaukee, WI 53226; e-mail: mpasquini@mcw.edu.