

Key Points
Human CD3 copotentiation can enhance the clonal expansion of several classes of recall T cells responding to antigens.
Enhanced expansion follows a unique pattern based on the immunodominance or weakness of antigen and public or private TCR status.

Abstract
Human cytomegalovirus (HCMV) induces long-lasting T-cell immune responses that control but do not clear infection. Typical responses involve private T-cell clones, expressing T-cell antigen receptors (TCRs) unique to a person, and public T-cell clones with identical TCRs active in different people. Here, we report the development of a pretherapeutic immunostimulation modality against HCMV for human T cells, CD3 copotentiation, and the clonal analysis of its effects in recall assays at single-cell resolution. CD3 copotentiation of human T cells required identification of an intrinsically inert anti-CD3 Fab fragment that conditionally augmented signaling only when TCR was coengaged with antigen. When applied in recall assays, CD3 copotentiation enhanced the expansion of both public and private T-cell clones responding to autologous HLA-A2(+) antigen-presenting cells and immunodominant NLVPMVATV (NLV) peptide from HCMV pp65 protein. Interestingly, public vs private TCR expression was associated with distinct clonal expansion signatures in response to recall stimulus. This implied that besides possible differences in their generation and selection in an immune response, public and private T cells may respond differently to pharmacoimmunomodulation. Furthermore, a third clonal expansion profile was observed upon CD3 copotentiation of T-cell clones from HLA-A2(−) donors and 1 HLA-A2(+) presumed-uninfected donor, where NLV was of low intrinsic potency. We conclude that human T-cell copotentiation can increase the expansion of different classes of T-cell clones responding to recall antigens of different strengths, and this may be exploitable for therapeutic development against chronic, persistent infections such as HCMV.
Introduction
β-Herpes human cytomegalovirus (HCMV) infects the population at high incidence.1 Infection is often asymptomatic and controlled by long-lasting T-cell responses driving the virus to latency, although sterilizing immunity is not induced.2-4 Chronic inflammation or immune compromise can allow HCMV reactivation and life-threatening disease.5-7 Thus, there is great interest in developing new immune-boosting therapies to treat/prevent HCMV recurrence.8-10
In mice, we previously developed an immunostimulatory concept, CD3 copotentiation, that, if applicable to humans, might add a new boosting strategy against a range of diseases from cancer to chronic viral infections, including HCMV. We described an anti-mouse-CD3 mono-Fab fragment whose binding was functionally inert if T cells encountered no antigen and did not inhibit T cells stimulated by strong antigens, but if T cells were stimulated by weak antigens, then coincident Fab-CD3 engagement improved various responses elicited from naive CD8 T cells.11 In vivo, the Fab reduced tumor burden of B16-F10 melanoma by a mechanism dependent on CD4 and CD8 T cells and T-cell antigen receptor (TCR) antigen specificity.11 The anti-CD3 mono-Fab induces a stimulation-poised CD3 conformation thought to amplify signaling upon weak antigen engagement by TCR.12,13 Here, we report the development and characterization of an anti-human-CD3 mono-Fab with copotentiation function.
Whether CD3 copotentiation can boost responses from previously activated (not naive) T cells, and whether responses to strong antigens in addition to weak antigens might be enhanced, are outstanding questions. HCMV(+) peripheral blood samples contain clonally expanded recall T cells that can be classified regarding their response to immunodominant vs weak HCMV antigens and whether they bear public or private TCRs.14,15 Here, we focus on immunodominant pp65495-503 peptide NLVPMVATV (NLV) in HLA-A*02:01(+) individuals,16 which is reported to induce both public and private CD8 T-cell responses.17-25 Both bulk recall assays and single-cell assessment of TCR clonality (TRBV-CDR3 sequencing) from peripheral blood reveal that human CD3 copotentiation can amplify expansion of public and private T cell clones. This effect occurs in response to (1) immunodominant NLV:HLA-A2, (2) NLV as a weak antigen in HLA-A2(−) conditions and HLA-A2(+) presumed-uninfected conditions, and (3) undefined HLA-dependent antigens in autologous antigen-presenting cells (APCs). We propose that CD3 copotentiation can amplify clonal expansion of recall T cells under various conditions of antigen experience and stimulatory strength, making the CD3 copotentiation strategy potentially attractive for translation to HCMV and other disease treatments.
Methods
Cell lines
PBMCs
With Mayo Institutional Review Board approval, whole human blood was collected from healthy volunteers. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll gradient and used fresh or cryopreserved in fetal bovine serum, 10% dimethyl sulfoxide. Where indicated, T cells were isolated untouched using the magnetic human pan, CD4, or CD8-T-cell isolation kits following the manufacturer’s protocols (MACS, Miltenyi Biotec).
Mono-Fab preparation
Mono-Fabs were prepared as described previously.27 After papain digestion (Sigma-Aldrich), monoclonal antibody (mAb) digests were terminated with iodoacetamide (Sigma-Aldrich) and dialyzed in phosphate-buffered saline (PBS) with periodic buffer exchange. Fc was removed with Protein A Sepharose (GE Healthcare). Following isolation by size exclusion chromatography over 2 tandem Superdex200 10/300GL columns (GE Life Sciences) on NGC-Quest10 FPLC system (Bio-Rad), mono-Fabs were sterile-filtered in cold PBS + 2 M l-proline to preserve monovalency.28 Protein concentration was quantified on DeNovixDS-11 Spectrophotometer.
Peptides and antibodies
The peptide ovalbumin (pOVA, SIINFEKL) and the peptide FARL (pFARL, SSIEFARL) were purchased from Elim Biopharmaceuticals. NLV peptide (NLVPMVATV) was synthesized by the Mayo Clinic Rochester Proteomics Core. Culture supernatants of OKT3 hybridoma were purified in-house to obtain OKT3 mAb. The following antibodies were purchased: UCHT1 mAb (BioXCell), anti-CD8 DK25 mAb (Agilent), serum mouse immunoglobulin G (Ms-IgG) and Ms-IgG-Fab (Jackson ImmunoResearch), anti-mouse IgG (BioLegend, Poly4060), anti-human CD4 (BD Biosciences, RPA-T4, OKT4), anti-human CD8 (BD Biosciences, HIT8a, RPA-T8, SK1), anti-human CD69 (BD Biosciences, FN50), anti-human CD45 (BioLegend, UCHL1), anti-human CD56 (BD Biosciences, B159), anti-human CD19 (BioLegend, HIB19), anti-Vβ5 (BioLegend, MR9-4), and anti-Nur77 (Invitrogen, 12.14). H2-Kb/OVA-tetramer was made in-house as previously described.29 HLA-A*02:01/NLV (A2/NLV)-tetramer was purchased from MBL International. GhostDye discriminated live/dead cells (Tonbo).
Flow cytometry
Cells stained with indicated fluorophore-conjugated antibodies were collected on either Guava easyCyte HT Flow Cytometer (Luminex) or BD-Accuri C6 Flow Cytometer (BD Biosciences). For intracellular staining of Nur77, samples were fixed and permeabilized with Cytofix/Cytoperm Kit per the manufacturer’s protocol (BD Biosciences). Data analysis was performed using FlowJo (Tree Star) or guavaSoft software.
T-cell activation
A total of 50 000 OT-I.JRT3 T cells per well were stimulated with 0.2 nM indicated peptides presented by T2-Kb APCs and 10 µg/mL Ms-IgG-Fab control or specific mono-Fabs for 24 hours. Human PBMCs were rested overnight following fresh isolation or thaw from cryopreservation. Next, 0.2 × 106 PBMCs per well were stimulated with indicated control or specific immunoglobulins (10 µg/mL) for 4 to 6 hours.
Western blot
A total of 1 × 106 human PBMCs were stimulated with 10 µg/mL immunoglobulins as indicated in the presence or absence of pervanadate for 5 minutes, 37°C. Cells were washed twice in cold PBS and lysed for 10 minutes in 1% TritonX-100, 20 mM Tris/HCl pH 7.4, and 150 mM NaCl plus Halt-protease/phosphatase inhibitors (ThermoFisher). Equivalent cell lysates were subjected to SDS-PAGE (reducing, 10% gel), polyvinylidene difluoride membrane transfer, and western blot analysis with anti-phosphotyrosine (EMD Millipore, 4G10) and secondary anti-mouse IgG horseradish peroxidase (Cell Signaling).
CD3 pull-down (CD3-PD)
The CD3-PD assay was used to quantify CD3 conformational change (CD3Δc) by detection of CD3ε proline-rich sequence exposure.11-13,30 30 × 106 PBMCs per sample were lysed in isotonic ice-cold buffer containing 1% Brij58 (Sigma-Aldrich) and centrifuged to obtain postnuclear fractions. Samples were precleared with glutathione S-transferase (GST) beads (4°C, 1 hour) in the presence of indicated immunoglobulins (10 µg/mL), followed by specific CD3-PD with GST-SH3.1-NCK beads (4°C, 12 hours). CD3-PD samples were subjected to SDS-PAGE (reducing, 13% gel), nitrocellulose transfer, and western blot with rabbit serum 448 antibody, specific for CD3ζ (gift from Balbino Alarcón, Universidad Autónoma de Madrid, Madrid, Spain). The mAb APA 1/1 (GE Biosciences) set the assay background level.30 Protein acetone precipitates from a fraction of postnuclear lysates controlled for total CD3 content per sample. Quantification was performed as described previously.13
Recall T-cell expansion cultures
CD8 T cells were expanded as described previously,31 with minor modifications. 0.2 × 106 total PBMCs per well were seeded on day 0 with no exogenous peptide or 1 μM exogenous NLV peptide in RPMI, 10% fetal bovine serum. On day 2, 10 μg/mL mono-Fab and 20 U/mL interleukin-2 (IL-2) (Proleukin, Mayo Pharmacy) were added to culture. On days 4 and 7, half the media was replaced with fresh media containing 20 U/mL IL-2. Flow cytometry was run on day 9. For antigen-blocking experiments, 5 μg/mL DK25 antibody or Ms-IgG control was added on days 0, 2, and 4, and flow cytometry was run on day 7.
ELISA
Supernatants were collected from day 7 recall assays and stored at −20°C. Supernatants were thawed at room temperature and analyzed for human interferon-γ (IFN-γ) and granzyme B levels by sandwich enzyme-linked immunosorbent assay (ELISA) according to manufacturer’s protocol (R&D Systems).
Cytotoxic T lymphocyte (CTL) assay
Performed as described previously,32 CD8 T cells from NLV-expanded recall assays and target CD4 T cells from thawed, autologous PBMCs were isolated using negative-selection magnetic beads. CD4 T cells were loaded with 10 µM NLV and labeled with 0.01 µM carboxyfluorescein diacetate succinimidyl ester (specific targets) or not loaded but labeled with 0.1 µM carboxyfluorescein diacetate succinimidyl ester (nonspecific targets). Specific and nonspecific targets were mixed 1:1 for coculture with serially diluted recall CD8 T cells overnight and then analyzed by flow cytometry. Specific lysis was calculated based on the ratio of live NLV-loaded/nonloaded target cells.
TRBV-CDR3 sequencing and analysis
Genomic DNA was isolated from frozen cell pellets (QIAamp DNA mini kit). TRBV-CDR3 sequencing and preliminary analysis was completed using the immunoSEQ platform33 (Adaptive Biotechnologies, hsTCRβ kit). Per the manufacturer’s protocol, 1.6 µg genomic DNA per sample was subjected to polymerase chain reaction (PCR) to amplify all TRBV-CDR3 sequences in a bias-controlled manner using multiplexed V- and J-gene primers. Amplified TRBV-CDR3 underwent a second PCR to generate barcoded libraries. Sequencer-ready barcoded libraries were pooled and sequenced on an Illumina MiSeq. Raw sequencing data were sent to Adaptive Biotechnologies for processing to report the normalized, annotated TCR-β repertoire of each sample. Data analysis was performed using the provided immunoSEQ Analyzer program. The VDJdb database34 was accessed to identify TRBV-CDR3 clones in the data set matching those from previously published public TCRs associated with HLA-A2 and NLV peptide. “Public” TCR was operationally defined as 100% identity of TRBV-CDR3 amino acid sequence between individuals; it is possible that clones classified as private here could be found public upon deeper/broader population sequencing.
Statistical analysis
Statistics performed using GraphPad Prism included 2-tailed, 1-tailed, unpaired, and paired Student t tests and Fisher’s exact tests. Results showing central values represent mean ± standard deviation (SD) or standard error of the mean (SEM).
Results
Mono-OKT3-Fab binds to human CD3 without blocking TCR-antigen interactions
Binding distinct but overlapping epitopes of human CD3ε,35-37 mAbs OKT3 and UCHT1 were subjected to papain digestion to obtain mono-Fabs. For copotentiation, mono-Fabs must bind CD3 without sterically hindering TCR-antigen binding or signaling. OKT3 and UCHT1 mono-Fabs bound surface CD3 of OT-I.JRT3 cells, expressing mouse CD8αβ and OT-I-TCR-αβ in complex with human CD326 (Figure 1A, left). Both mono-Fabs also bound primary human CD4 and CD8 T cells (Figure 1A, middle and right). When bound, mono-OKT3-Fab did not block TCR-antigen interaction, as demonstrated by unaffected Kb/OVA-tetramer staining of OT-I.JRT3 cells (Figure 1B, top left) and uninterrupted A2/NLV-tetramer staining of HLA-A*02:01(+) CD8 T cells (Figure 1B, top right). Furthermore, binding of mono-OKT3-Fab to OT-I.JRT3 cells did not alter surface TCR downregulation or CD69 upregulation in response to SIINFEKL antigenic peptide (Figure 1C, pOVA). In contrast, mono-UCHT1-Fab inhibited TCR-antigen interaction in both OT-I.JRT3 and human A2/NLV-tetramer(+) CD8 T cells (Figure 1B, bottom) and inhibited surface TCR downregulation and CD69 upregulation of OT-I.JRT3 cells in response to SIINFEKL (Figure 1C, pOVA).

Mono-OKT3-Fab binds to human T cells and induces CD3Δc without blocking TCR-antigen interactions or initiating early T-cell signaling. (A) Mono-OKT3-Fab and mono-UCHT1-Fab bind T cells, detected by positive secondary anti-Ms-IgG staining by flow cytometry of OT-I.JRT3 cells and primary human CD4 and CD8 T cells from PBMCs. (B) Mono-OKT3-Fab does not block TCR-antigen binding, in contrast to mono-UCHT1-Fab. OT-I.JRT3 or CD8 T cells isolated from PBMCs that were previously expanded with NLV peptide were preincubated with indicated immunoglobulins and stained for binding of Kb/OVA-tetramer (B, left) or A2/NLV-tetramer (B, right), respectively. (C) Mono-OKT3-Fab does not impair the T-cell response to cognate antigen, unlike mono-UCHT1-Fab. OT-I.JRT3 cells were cultured with null peptide (pFARL) or antigenic peptide (pOVA) presented on T2-Kb APCs in the presence of indicated immunoglobulins and analyzed for CD69 upregulation and TCR downregulation. Frequencies of CD69(+) and Vβ5(+) cells are shown (mean ± SD from triplicate samples, 2-tailed unpaired Student t test). (D-E) Binding of mono-Fabs does not stimulate T cells in the absence of antigen. (D) PBMCs were incubated with indicated immunoglobulins, after which CD4 and CD8 T cells were analyzed for the induction of surface CD69 and intracellular Nur77 by flow cytometry. Frequencies of CD69(+) and Nur77(+) T cells are shown (mean ± SD from triplicate samples, 2-tailed unpaired Student t test). (E) PBMCs were incubated with indicated immunoglobulins in the presence or absence of pervanadate (PV). Phosphotyrosine was detected by western blot (WB) of equivalent cell lysates. (F) Mono-Fabs induce CD3Δc. PBMC lysates were incubated with APA1/1 (to block CD3 pull-down), Ms-IgG-Fab (to reveal basal level of CD3Δc), or mono-Fabs (test conditions) and assessed for induction of CD3Δc by the CD3 pull-down assay. Post-CD3Δc open conformation was detected with anti-CD3ζ by western blot. Inducible CD3Δc is measured by fold-increase over basal level. TL, total lysate before pull-down. Data are representative of ≥3 independent experiments. Ms-IgG-Fab, negative control.
Mono-OKT3-Fab binds to human T cells and induces CD3Δc without blocking TCR-antigen interactions or initiating early T-cell signaling. (A) Mono-OKT3-Fab and mono-UCHT1-Fab bind T cells, detected by positive secondary anti-Ms-IgG staining by flow cytometry of OT-I.JRT3 cells and primary human CD4 and CD8 T cells from PBMCs. (B) Mono-OKT3-Fab does not block TCR-antigen binding, in contrast to mono-UCHT1-Fab. OT-I.JRT3 or CD8 T cells isolated from PBMCs that were previously expanded with NLV peptide were preincubated with indicated immunoglobulins and stained for binding of Kb/OVA-tetramer (B, left) or A2/NLV-tetramer (B, right), respectively. (C) Mono-OKT3-Fab does not impair the T-cell response to cognate antigen, unlike mono-UCHT1-Fab. OT-I.JRT3 cells were cultured with null peptide (pFARL) or antigenic peptide (pOVA) presented on T2-Kb APCs in the presence of indicated immunoglobulins and analyzed for CD69 upregulation and TCR downregulation. Frequencies of CD69(+) and Vβ5(+) cells are shown (mean ± SD from triplicate samples, 2-tailed unpaired Student t test). (D-E) Binding of mono-Fabs does not stimulate T cells in the absence of antigen. (D) PBMCs were incubated with indicated immunoglobulins, after which CD4 and CD8 T cells were analyzed for the induction of surface CD69 and intracellular Nur77 by flow cytometry. Frequencies of CD69(+) and Nur77(+) T cells are shown (mean ± SD from triplicate samples, 2-tailed unpaired Student t test). (E) PBMCs were incubated with indicated immunoglobulins in the presence or absence of pervanadate (PV). Phosphotyrosine was detected by western blot (WB) of equivalent cell lysates. (F) Mono-Fabs induce CD3Δc. PBMC lysates were incubated with APA1/1 (to block CD3 pull-down), Ms-IgG-Fab (to reveal basal level of CD3Δc), or mono-Fabs (test conditions) and assessed for induction of CD3Δc by the CD3 pull-down assay. Post-CD3Δc open conformation was detected with anti-CD3ζ by western blot. Inducible CD3Δc is measured by fold-increase over basal level. TL, total lysate before pull-down. Data are representative of ≥3 independent experiments. Ms-IgG-Fab, negative control.
Mono-OKT3-Fab induces CD3Δc without initiating early signaling
In the absence of antigen recognition, neither mono-Fab induced signaling-dependent surface TCR downregulation or CD69 upregulation in OT-I.JRT3 cells, as expected for noncrosslinking species (Figure 1C, pFARL). Likewise, neither mono-Fab induced CD69 or Nur77 upregulation in primary human T cells, unlike their parent bivalent mAbs (Figure 1D). Furthermore, mono-Fabs did not induce accumulation of tyrosine-phosphorylated proteins following engagement of human PBMCs compared with positive control, pervanadate38 (Figure 1E). Despite their inability to trigger CD3 signaling, mono-Fab binding induced a signal-amplifying conformational change in CD3 (CD3Δc), as indicated by a CD3-PD assay (Figure 1F), where GST-SH3.1-Nck beads capture TCR/CD3 complexes displaying a CD3ε proline-rich sequence exposed upon optimal TCR engagement.11-13,30 Based on its ability to bind human CD3 and induce CD3Δc without blocking TCR-antigen interaction and without intrinsically initiating early signaling, mono-OKT3-Fab was selected to study copotentiation of human T cells.
Mono-OKT3-Fab enhances recall T-cell response to autologous APCs and NLV:HLA-A2
Seven healthy blood donors were classified by HLA-A*02:01 expression and the presence of CD8 T cells positive for binding A2/NLV-tetramer, an immunodominant antigen and marker of HCMV positivity. Four donors were HLA-A*02:01(+) and A2/NLV-tetramer(+) (72F, 53M, 28M, and 47M), 1 donor was HLA-A*02:01(+) but A2/NLV-tetramer(−) (74M), and 2 donors were HLA-A*02:01(−) and A2/NLV-tetramer(−) (78F and 59F; supplemental Table 1). Bulk PBMCs were tested in recall T-cell expansion assays driven by exogenous NLV peptide. On day 0, donors presented variable numbers of bulk CD8 T cells, including low but detectable A2/NLV-tetramer(+) CD8 T cells in the expected prescreened donors (supplemental Table 1). After culturing PBMCs for 9 days in the presence of exogenous NLV + irrelevant Ig + late IL-2 (see “Methods”), donors positive for A2/NLV tetramer presented higher CD8 T-cell counts than cultures without exogenous peptide (Figure 2A, gray). These donors were categorized as exogenous NLV responsive in bulk culture (exog-NLV-bulk-responsive). For these donors, NLV + mono-OKT3-Fab increased A2/NLV-tetramer(+) CD8 T-cell numbers even more (Figure 2A, red). NLV + mono-OKT3-Fab stimulation also induced significant increase in IFN-γ and granzyme B accumulation by day 7 in culture supernatants (Figure 2B-C). Bulk CD8 T cells isolated at day 7 from NLV recall cultures ± mono-OKT3-Fab were tested for CTL activity against autologous NLV-loaded CD4+ target cells. Greater specific lysis was observed in the mono-OKT3-Fab copotentiated cultures (Figure 2D, left panels) correlating with increased NLV-A2-tetramer(+) cells (Figure 2D, right panels), demonstrating anti-NLV functional specificity.
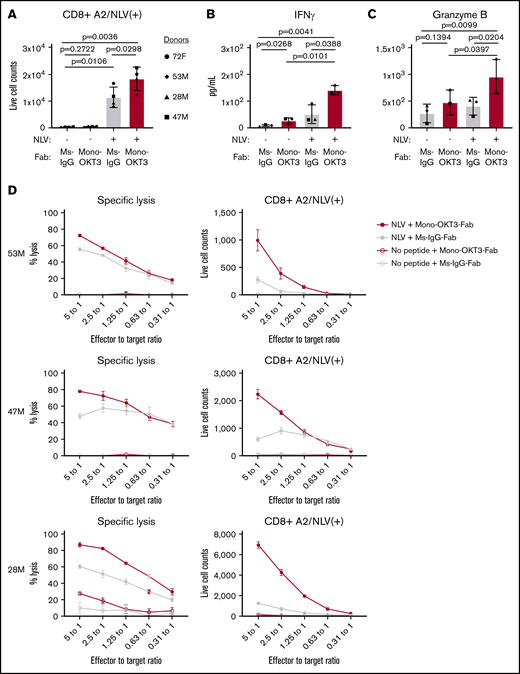
Mono-OKT3-Fab enhances recall T-cell expansion to NLV:HLA-A2. (A-D) PBMCs were cultured with or without exogenous NLV peptide in the presence of mono-OKT3-Fab or control Ms-IgG-Fab. (A) On day 9, cells were analyzed by flow cytometry for the number of A2/NLV-tetramer(+) CD8 T cells from exog-NLV-bulk-responsive donors (mean ± SD, 2-tailed paired Student t test). Mono-OKT3-Fab increased the production of IFN-γ (B) and granzyme B (C), as measured by ELISA of day 7 supernatants (mean ± SD, 1-tailed paired Student t test). Each symbol represents the average of ≥3 independent experiments per donor (A-C). (D) CD8 T-cell isolates (effectors) were cultured at the indicated effector to target ratios with NLV-loaded CD4 T cells (targets) overnight and analyzed for specific lysis of targets (mean ± SD of duplicates).
Mono-OKT3-Fab enhances recall T-cell expansion to NLV:HLA-A2. (A-D) PBMCs were cultured with or without exogenous NLV peptide in the presence of mono-OKT3-Fab or control Ms-IgG-Fab. (A) On day 9, cells were analyzed by flow cytometry for the number of A2/NLV-tetramer(+) CD8 T cells from exog-NLV-bulk-responsive donors (mean ± SD, 2-tailed paired Student t test). Mono-OKT3-Fab increased the production of IFN-γ (B) and granzyme B (C), as measured by ELISA of day 7 supernatants (mean ± SD, 1-tailed paired Student t test). Each symbol represents the average of ≥3 independent experiments per donor (A-C). (D) CD8 T-cell isolates (effectors) were cultured at the indicated effector to target ratios with NLV-loaded CD4 T cells (targets) overnight and analyzed for specific lysis of targets (mean ± SD of duplicates).
CD3 copotentiation is dependent on TCR-HLA and CD8 coreceptor engagement
Interestingly, A2/NLV-tetramer(−) CD8 T cells in the same recall cultures increased in the presence of mono-OKT3-Fab, but not exogenous NLV peptide (Figure 3A), and PBMCs from A2/NLV-tetramer(−) donors responded likewise (Figure 3B). Thus, mono-OKT3-Fab increased the numbers of both A2/NLV-tetramer(+) and A2/NLV-tetramer(−) CD8 T cells. To distinguish mono-OKT3-Fab intrinsic T-cell stimulation from TCR-HLA–dependent copotentiation, recall assays were performed in the presence or absence of blocking reagents to TCR-HLA or CD8 coreceptor. First, CD8 T cells from 1 exog-NLV-bulk-responsive donor were cultured in the presence of mono-OKT3-Fab or mono-UCHT1-Fab, the latter binding to CD3 and inducing CD3Δc but impairing antigen binding to T cells (Figure 1). Mono-UCHT1-Fab significantly reduced copotentiation when compared with mono-OKT3-Fab of CD8 T cells either positive (Figure 3C) or negative (Figure 3D) for A2/NLV-tetramer. These results indicate that with impaired TCR-antigen interactions, induction of CD3Δc by mono-Fabs is insufficient to mediate copotentiation. Second, parallel experiments showed that the anti-CD8 blocking antibody, DK25,39 inhibited both exogenous NLV–specific and mono-OKT3-Fab–specific responses in CD8 T cells either positive (Figure 3E) or negative (Figure 3F) for A2/NLV tetramer. Thus, mono-OKT3-Fab copotentiation is dependent on CD8-TCR-HLA engagement for both A2/NLV-tetramer(+) CD8 T cells driven by exogenous NLV and for A2/NLV-tetramer(−) CD8 T cells driven only by autologous APCs.

CD3 copotentiation is dependent on TCR-HLA and CD8 co-receptor engagement. (A-B) PBMCs were cultured with or without exogenous NLV peptide in the presence of mono-OKT3-Fab or control Ms-IgG-Fab for 9 days as in Figure 2A. Counts of A2/NLV-tetramer(−) CD8 T cells are shown for exog-NLV-bulk-responsive donors (A) and exog-NLV-bulk-nonresponsive donors (B). Each symbol represents the average of ≥3 independent experiments per donor (mean ± SD, 2-tailed paired Student t test). (C-D) PBMCs were cultured with or without exogenous NLV peptide in the presence of Ms-IgG-Fab (control), mono-OKT3-Fab, or mono-UCHT1-Fab for 9 days. Mono-UCHT1-Fab dampened the copotentiation effect as compared with mono-OKT3-Fab in both A2/NLV-tetramer(+) (C) and A2/NLV-tetramer(−) (D) CD8 T cells. (E-F) PBMCs were cultured with or without exogenous NLV peptide in the presence of Ms-IgG-Fab or mono-OKT3-Fab and with or without the CD8 blocking antibody, DK25, for 7 days. Blocking CD8 reduced the copotentiation effect of mono-OKT3-Fab for both A2/NLV-tetramer(+) (E) and A2/NLV-tetramer(−) (F) CD8 T cells. One representative experiment of donor 47M is shown for 3 replicates (C-F) (mean ± SD from triplicate samples, 2-tailed unpaired Student t test).
CD3 copotentiation is dependent on TCR-HLA and CD8 co-receptor engagement. (A-B) PBMCs were cultured with or without exogenous NLV peptide in the presence of mono-OKT3-Fab or control Ms-IgG-Fab for 9 days as in Figure 2A. Counts of A2/NLV-tetramer(−) CD8 T cells are shown for exog-NLV-bulk-responsive donors (A) and exog-NLV-bulk-nonresponsive donors (B). Each symbol represents the average of ≥3 independent experiments per donor (mean ± SD, 2-tailed paired Student t test). (C-D) PBMCs were cultured with or without exogenous NLV peptide in the presence of Ms-IgG-Fab (control), mono-OKT3-Fab, or mono-UCHT1-Fab for 9 days. Mono-UCHT1-Fab dampened the copotentiation effect as compared with mono-OKT3-Fab in both A2/NLV-tetramer(+) (C) and A2/NLV-tetramer(−) (D) CD8 T cells. (E-F) PBMCs were cultured with or without exogenous NLV peptide in the presence of Ms-IgG-Fab or mono-OKT3-Fab and with or without the CD8 blocking antibody, DK25, for 7 days. Blocking CD8 reduced the copotentiation effect of mono-OKT3-Fab for both A2/NLV-tetramer(+) (E) and A2/NLV-tetramer(−) (F) CD8 T cells. One representative experiment of donor 47M is shown for 3 replicates (C-F) (mean ± SD from triplicate samples, 2-tailed unpaired Student t test).
Mono-OKT3-Fab copotentiation primarily enhances expansion of top-ranked T-cell clones
To analyze the T-cell clonal dynamics of the copotentiation response, recall assays were followed by DNA extraction and TRBV-CDR3 analysis via immunoSEQ with single-cell resolution (Adaptive Technologies, see “Methods”). The number of clones sampled per donor per culture condition reached as high as ∼50 000. Clonal diversity was estimated by scaled Shannon entropy, a value whose range is 0 to 1, where 0 represents minimum diversity exhibited by a monoclonal T-cell population and 1 represents maximal repertoire diversity when all TRBV-CDR3 sequences are expressed equally.40 We observed that exogenous NLV decreased entropy compared with negative-control cultures for 4 out of 4 exog-NLV-bulk-responsive donors and likewise decreased the total clone number sampled from cultures (supplemental Figure 1A-H), an expected outcome when T-cell clones specific for a single peptide proliferate and increase their relative representation. In contrast, mono-OKT3-Fab did not reliably produce such an effect (observed in 2/4 exog-NLV-bulk-responsive donors), nor did mono-OKT3-Fab tend to further decrease entropy when administered in combination with exogenous NLV compared with NLV alone (supplemental Figure 1A-H). Thus, mono-OKT3-Fab was not producing a clonal effect identical to that of exogenous peptide.
To determine if mono-OKT3-Fab indiscriminately caused many clones to expand, we used single-cell clonal sequencing data to estimate total copy number of each clone in recall cultures and compared between conditions by rank analysis. Interestingly, we found that despite thousands of clones measured, substantial differences in cell numbers ranked according to abundance were heavily concentrated in the top 10 clones (Figure 4A-D; supplemental Figure 1I-L). Thus, copotentiation must have a mechanism of clonal specificity, which is more deeply analyzed here, discussing the HLA-A2(+) exog-NLV-bulk-responsive donor 72F as an example. Among the top 10 clones from negative-control cultures, none were NLV responsive (supplemental Table 2, donor 72F; no exogenous peptide + Ms-IgG-Fab, rows 4-13), and most scored as “zeros” in the other culture conditions. This likely indicates the individual clones were too infrequent for consistent sampling. In exogenous NLV cultures, the top clone reached ∼130 000 cells (supplemental Table 2, donor 72F; NLV + Ms-IgG-Fab, rows 16-25), having represented ∼1500 cells in negative-control culture. Several other top clones were absent in ≥1 other culture conditions, and thus, as above, some analysis had sampling limitations. However, ranks 3 and 5 were consistently sampled (supplemental Table 2, donor 72F; no exogenous peptide + mono-OKT3-Fab, rows 18 and 20) and showed matching clones that were also amplified in the mono-OKT3-Fab-only condition, with synergistic highest abundance in NLV + mono-OKT3-Fab cultures. Rank 8 was already high in negative-control culture and was neither exogenous NLV-responsive nor amplified by mono-OKT3-Fab (supplemental Table 2, donor 72F; no exogenous peptide + mono-OKT3-Fab, row 23). In mono-OKT3-Fab-only cultures (supplemental Table 2, donor 72F; no exogenous peptide + mono-OKT3-Fab, rows 28-37), 3 out of 10 top clones were independently NLV responsive in exogenous NLV cultures, while 4 out of 10 top clones responded to mono-OKT3-Fab, but not exogenous NLV. Finally, in NLV + mono-OKT3-Fab cultures (Figure 4E; supplemental Table 2, donor 72F; NLV + mono-OKT3-Fab, rows 40-49), 4 out of 10 top clones were NLV responsive and synergistically amplified, while 5 out of 10 top clones were not NLV responsive but were amplified to a similar extent as mono-OKT3-Fab-only cultures. Therefore, copotentiation amplified clonal abundance of certain top clones only, with some, but not others, also being responsive to exogenous NLV.

Mono-OKT3-Fab copotentiation primarily enhances expansion of top-ranked T-cell clones. (A-D) TCR clones for each exog-NLV-bulk-responsive donor were ranked from most abundant to least abundant for each condition. Differences in rank-vs-rank performance concentrated in the top 10 clones. (E-H) Among top-ranked clones, there was variability in the extent to which clones were amplified by exogenous NLV, mono-OKT3-Fab, or both in combination. The top 10 ranked clones from the NLV + mono-OKT3-Fab condition are shown with their corresponding live-cell number abundance in the other 3 conditions. Boxed amino acid sequences indicate NLV-specific clones (clones with greater abundance in NLV + Ms-IgG-Fab vs no exogenous peptide + Ms-IgG-Fab condition or, for public TCRs, observation of that pattern in ≥1 other donor or previously reported in the literature). Sequences in bold represent public TCR-bearing clones appearing in multiple donors in the present study or previously reported in the literature. Heatmaps visualize the increase in clonal cell number generated by exogenous NLV, mono-OKT3-Fab, or both in combination.
Mono-OKT3-Fab copotentiation primarily enhances expansion of top-ranked T-cell clones. (A-D) TCR clones for each exog-NLV-bulk-responsive donor were ranked from most abundant to least abundant for each condition. Differences in rank-vs-rank performance concentrated in the top 10 clones. (E-H) Among top-ranked clones, there was variability in the extent to which clones were amplified by exogenous NLV, mono-OKT3-Fab, or both in combination. The top 10 ranked clones from the NLV + mono-OKT3-Fab condition are shown with their corresponding live-cell number abundance in the other 3 conditions. Boxed amino acid sequences indicate NLV-specific clones (clones with greater abundance in NLV + Ms-IgG-Fab vs no exogenous peptide + Ms-IgG-Fab condition or, for public TCRs, observation of that pattern in ≥1 other donor or previously reported in the literature). Sequences in bold represent public TCR-bearing clones appearing in multiple donors in the present study or previously reported in the literature. Heatmaps visualize the increase in clonal cell number generated by exogenous NLV, mono-OKT3-Fab, or both in combination.
Different classes of T-cell clones respond to CD3 copotentiation with distinct clonal expansion signatures
The other 3 exog-NLV-bulk-responsive donors showed similar examples of clonal responses (Figure 4F-H), while unlike donor 72F, the others had exogenous NLV-responsive public TCRs (supplemental Table 3A) among top clones, which in 12 out of 14 occurrences responded to mono-OKT3-Fab (supplemental Table 3B). However, there was a curious pattern in their response: public clones tended to respond best to either mono-OKT3-Fab only or exogenous NLV but less optimally to the combination. Assigning first-place performance (“gold”) to conditions with highest clonal abundance and second/third-place (“silver-bronze”), we found that considering all top 10 clones, private much more than public TCRs showed synergy in combination treatment (Table 1).
We next examined the T-cell clonal dynamics of copotentiation in the A2/NLV-tetramer(−) donors, all of which amplified T-cell expansion by mono-OKT3-Fab but were exog-NLV-bulk-nonresponsive (Figure 3B). Exogenous NLV and mono-OKT3-Fab status did not correlate with predictable changes in entropy and total clones sampled, although rank differences remained largely concentrated in top-10 clones (supplemental Figure 2). Interestingly, the top clone for each donor appeared exogenous NLV responsive, as did several other top clones, with further enhancement combined with mono-OKT3-Fab (supplemental Table 4). To assess NLV + mono-OKT3-Fab combinatorial synergy, we applied gold/silver-bronze analysis to these 13 apparently NLV-responsive clones and found that the NLV + mono-OKT3-Fab condition produced the highest clonal abundance in all cases (Figure 5A). This was similar to the “private TCR” signature noted previously for exog-NLV-bulk-responsive donors, but there was also a distinct difference. The exogenous NLV-only condition always (but barely) increased the cell number of these clones above the negative-control culture condition (∼1- to 4-fold); in contrast, combination-treatment “gold-response” clones from exog-NLV-bulk-responsive donors were much more peptide responsive, (∼10- to 500-fold; Figure 5B; supplemental Table 5). Comparing clonal cell numbers from cultures with mono-OKT3-Fab ± exogenous NLV, combination-treatment gold-response clones from exog-NLV-bulk-responsive donors appeared in 2 clusters: one for which exogenous NLV peptide increased clonal abundance by ∼75- to 150-fold and another that only increased ∼1- to 11-fold; in contrast, NLV-responsive clones from exog-NLV-bulk-nonresponsive donors all appeared in the low-peptide-response cluster (Figure 5C). This pattern flipped when assessing the contribution of mono-OKT3-Fab to combinatorial synergy; here, exog-NLV-bulk-responsive donor gold-response clones increased ∼2-fold on average, while gold-response clones from exog-NLV-bulk-nonresponsive donors increased ∼30-fold (Figure 5D). Taken together, these data show that exog-NLV-bulk-responsive donors responded to CD3 copotentiation by amplifying potent NLV-focused clones, while exog-NLV-bulk-nonresponsive donors responded to combination treatment with synergy driven mostly by CD3 copotentiation and low-but-positive intrinsic potency toward exogenous NLV. We conclude that mono-OKT3-Fab provides antigen-specific CD3 copotentiation that can increase expansion of recall public and private clones against antigens that are immunodominant or of intrinsically weak potency (Figure 6).
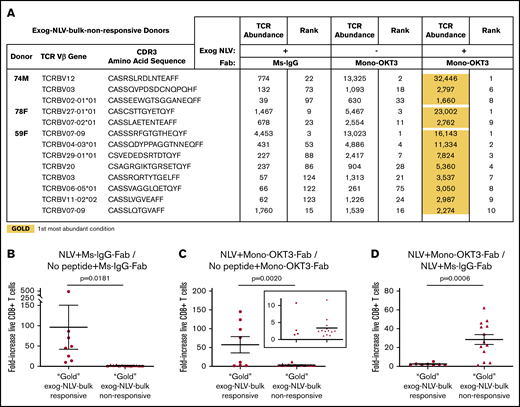
Although bulk cultures from A2/NLV-tetramer(−) donors appear nonresponsive to exogenous NLV, single-clone analysis reveals weak NLV-responsive clones amplified by CD3 copotentiation in a unique expansion signature. (A) Gold/silver-bronze analysis was applied to exog-NLV-bulk-nonresponsive donors for the top clones showing greater abundance in NLV + Ms-IgG-Fab vs no peptide + Ms-IgG-Fab conditions. It was observed that for each of these clones, the NLV + mono-OKT3-Fab combination condition yielded greatest abundance. (B) Among top NLV-specific clones, those from exog-NLV-bulk-responsive donors respond more than those from exog-NLV-bulk-nonresponsive donors to exogenous NLV. NLV-specific fold increase in TCR abundance was determined for gold-response clones from exog-NLV-bulk-responsive donors vs those from nonresponsive donors (supplemental Table 5). (C) NLV-specific fold-increase in TCR abundance was also assessed when gold-response clones from both types of donors were cultured in the presence of mono-OKT3-Fab. (D) Exog-NLV-bulk-nonresponsive donors respond more than exog-NLV-bulk-responsive donors to CD3 copotentiation when it is driven by exogenous NLV. Mono-OKT3-Fab-specific fold increase in TCR abundance was determined for gold-response clones from exog-NLV-bulk-responsive donors vs those from nonresponsive donors (supplemental Table 5). Data are included for gold-response clones in exog-NLV-bulk-responsive and nonresponsive donors. Each dot represents the fold-increase of a TRBV-CDR3-bearing clone (B-D) (mean ± SEM, 1-tailed unpaired Student t test).
Although bulk cultures from A2/NLV-tetramer(−) donors appear nonresponsive to exogenous NLV, single-clone analysis reveals weak NLV-responsive clones amplified by CD3 copotentiation in a unique expansion signature. (A) Gold/silver-bronze analysis was applied to exog-NLV-bulk-nonresponsive donors for the top clones showing greater abundance in NLV + Ms-IgG-Fab vs no peptide + Ms-IgG-Fab conditions. It was observed that for each of these clones, the NLV + mono-OKT3-Fab combination condition yielded greatest abundance. (B) Among top NLV-specific clones, those from exog-NLV-bulk-responsive donors respond more than those from exog-NLV-bulk-nonresponsive donors to exogenous NLV. NLV-specific fold increase in TCR abundance was determined for gold-response clones from exog-NLV-bulk-responsive donors vs those from nonresponsive donors (supplemental Table 5). (C) NLV-specific fold-increase in TCR abundance was also assessed when gold-response clones from both types of donors were cultured in the presence of mono-OKT3-Fab. (D) Exog-NLV-bulk-nonresponsive donors respond more than exog-NLV-bulk-responsive donors to CD3 copotentiation when it is driven by exogenous NLV. Mono-OKT3-Fab-specific fold increase in TCR abundance was determined for gold-response clones from exog-NLV-bulk-responsive donors vs those from nonresponsive donors (supplemental Table 5). Data are included for gold-response clones in exog-NLV-bulk-responsive and nonresponsive donors. Each dot represents the fold-increase of a TRBV-CDR3-bearing clone (B-D) (mean ± SEM, 1-tailed unpaired Student t test).
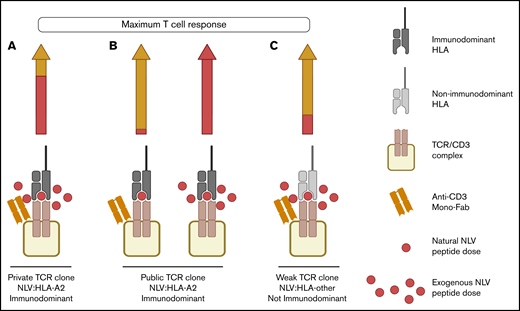
Different T-cell clonal signatures of maximal recall response to NLV when providing copotentiation with anti-CD3 mono-Fab. (A) Maximum recall response of private immunodominant TCR clones to exogenous NLV is mainly caused by the peptide (arrow, red segment), with a smaller contribution coming from copotentiation delivered by anti-CD3 mono-Fab (arrow, yellow segment). (B) Maximum recall immunodominant response of public TCR clones to NLV is driven by either (1) copotentiation (left arrow, yellow segment), with the smallest contribution from natural amounts of NLV presented in HCMV(+) APCs; or (2) exogenous NLV alone (right arrow). (C) NLV weak TCR clones reach their maximum recall response to exogenous NLV mainly by copotentiation (arrow, yellow segment), with a smaller contribution coming from exogenous NLV peptide (arrow, red segment). Created with BioRender.
Different T-cell clonal signatures of maximal recall response to NLV when providing copotentiation with anti-CD3 mono-Fab. (A) Maximum recall response of private immunodominant TCR clones to exogenous NLV is mainly caused by the peptide (arrow, red segment), with a smaller contribution coming from copotentiation delivered by anti-CD3 mono-Fab (arrow, yellow segment). (B) Maximum recall immunodominant response of public TCR clones to NLV is driven by either (1) copotentiation (left arrow, yellow segment), with the smallest contribution from natural amounts of NLV presented in HCMV(+) APCs; or (2) exogenous NLV alone (right arrow). (C) NLV weak TCR clones reach their maximum recall response to exogenous NLV mainly by copotentiation (arrow, yellow segment), with a smaller contribution coming from exogenous NLV peptide (arrow, red segment). Created with BioRender.
Discussion
We show that mono-OKT3-Fab provides human CD3 copotentiation to enhance expansion of several classes of recall CD8 T cells with relevance to HCMV. First, mono-OKT3-Fab fulfilled the biochemical requirements to deliver copotentiation: binding to CD3 and inducing CD3Δc without initiating intrinsic signaling or interfering with TCR-antigen binding (Figure 1). Functional copotentiation was observed in recall assays where PBMCs from healthy blood donors were cultured with or without exogenous NLV and/or mono-OKT3-Fab. Because enhanced expansion was observed both in A2/NLV-tetramer(+) and A2/NLV-tetramer(−) cells and in exog-NLV-bulk-nonresponsive donors (Figures 2A and 3A-B), the question arose whether mono-OKT3-Fab induced pan-T-cell activation vs HLA-restricted copotentiation. Expansion was impaired when using mono-UCHTI-Fab (Figure 3C-D), which inhibits TCR-antigen binding and signaling (Figure 1), suggesting that Fab-CD3 was insufficient for copotentiation without TCR-antigen interaction. Mono-OKT3-Fab–mediated copotentiation was inhibited by anti-CD8 blocking antibody, suggesting that copotentiation depends on the tripartite CD8-TCR-HLA antigenic interaction (Figure 3E-F). Furthermore, a pan-T-cell stimulatory effect was not supported by clonal analysis, where, despite thousands of clones that were identified and measured, both copotentiation and exogenous peptide effects were mainly concentrated in top clones. The observation of strong NLV-reactive clones from A2/NLV-tetramer(+) donors (Figures 4 and 5) and weak NLV-reactive clones from an HLA-A2+, A2/NLV-tetramer(−) donor (74M, Figure 5) suggests that a wide range of TCR-antigen affinities/avidities may be responsive to CD3 copotentiation, although future experiments are needed to directly test this hypothesis. Together, these results support a model in which mono-OKT3-Fab performs copotentiation by enhancing HLA-dependent responses (Figure 6). We speculate that autologous APCs present some antigens for which recall T-cell clones are specific, and these are copotentiated by mono-OKT3-Fab, while other recall T cells do not encounter their specific antigens on PBMC APCs, and these are not affected by mono-OKT3-Fab.
Previous mouse experiments showed the CD3 copotentiation principle in naive CD8 T cells in vitro, and antitumor effects in vivo, without ruling out the possibility that other T-cell classes and/or activation states could be responsive.11 In the current work, we observed copotentiation of peripheral blood CD8 T cells in classic recall assays with increased clonal expansion and effector function, suggesting that previously clonally expanded T cells are responsive to copotentiation (Figures 2 and 3). Among them, we observed public and private clones responding to immunodominant NLV:HLA-A2 antigen (Figure 4; Table 1) and clones for which NLV was a weak antigen (Figure 5).
Interestingly, these 3 classes of clonal response showed distinct clonal expansion signatures. A private signature against immunodominant NLV:HLA-A2 saw highest clonal abundance achieved upon combination stimulus of exogenous NLV + mono-OKT3-Fab (Table 1). This was also observed for exog-NLV-bulk-nonresponsive donors for whom NLV was an intrinsically weak antigen (Figure 5). The difference between these 2 response classes was in their opposite intrinsic potencies of NLV peptide (strong vs weak) and in a corresponding switch in contribution to the combinatorial synergistic response, peptide>mono-Fab for immunodominance vs mono-Fab>peptide for weak antigens (Figure 5). These patterns raise important questions. First, these responses can be confirmed as recall responses (not naive), because negative-control culture conditions contained multiple copies of these clones, consistent with prior clonal expansion. This is not surprising for responders to immunodominant antigen, but when NLV is a weak antigen, why would recall assays contain previously expanded T-cell clones for which intrinsic reactivity to exogenous NLV is minimal? Three nonexclusive possibilities are that (1) over time, in vivo, the weak response slowly accumulates numerous cells; (2) in vivo, the response to NLV is potent, although in recall assays, it is not; or (3) clones were expanded in vivo against other antigens but cross-react with NLV-HLA. This latter possibility is attractive, because it has been suggested that heteroclitic stimulation of T-cell clones might increase response to other cross-reactive antigens,41 which could recruit preexisting recall cells for other antigens to contribute to this otherwise nonsterilizing immune response.
The third expansion signature saw public T cells generating highest clonal abundance with either exogenous NLV or mono-OKT3-Fab, but not with both in combination. The rest of the data set reinforced the idea that the 2 modes of TCR/CD3 engagement were sterically and functionally compatible and synergistic, but apparently not in this case. One possible explanation is that the 2 modes of engagement remain synergistic but can cause hyperstimulation that favors activation-induced cell death42,43 or an antiproliferative signal,44,45 both of which decrease clonal expansion by incompletely understood mechanisms. Why these responses might be favored in public more than private T-cell clones is not clear, but it leads to the proposal of a second possible explanation. Perhaps public T cells possess a physical/functional distinction that has not been previously predicted, and their TCR-CD3 complexes engage or signal in response to cognate antigen and mono-OKT3-Fab differently than most private TCR clones. The nature of such a distinct property of the public T cell or its TCR-CD3 complex cannot be surmised at this time, but for now, we report that public T cells responded with a unique clonal expansion pattern to CD3 copotentiation.
Finally, the question of whether CD3 copotentiation may benefit patients merits further exploration. The present study provides compelling evidence that anti-human-CD3 Fabs can enhance expansion of several classes of recall T-cell clones responding to antigens. It is conceivable that these effects may find useful translation in vitro for adoptive cell therapies, in vivo for immunoboosting, or in other immunotherapeutic strategies against chronic/persistent antigens in cancer or infectious diseases like HCMV.
For original data, please contact gilpagesd@health.missouri.edu and markovic.svetomir@mayo.edu.
Acknowledgments
The authors thank Balbino Alarcón (Universidad Autónoma de Madrid, Madrid, Spain) and Ed Palmer (University of Basel, Switzerland) for kind gifts of reagents.
This work was funded by Mayo Graduate School of Biomedical Sciences (L.R.E.B.), the Mayo Foundation (S.N.M.), and the National Institutes of Health (National Cancer Institute grants U01 CA244314 [D.G.] and R33CA228979 [A.G.S.]; National Institute of Allergy and Infectious Diseases grant R01AI097187 [D.G.]; and National Institute of General Medical Sciences grant R01GM103841 [A.G.S.]).
Authorship
Contribution: L.R.E.B. designed, performed, and analyzed experiments and wrote the paper; W.K.N., S.L.S., M.A., M.M.H., C.A.P., and L.R.P. designed, performed, analyzed, and/or interpreted data and revised the paper; A.G.S. and D.G. designed experiments, analyzed data, and wrote the paper; and S.N.M. designed experiments, analyzed and interpreted data, wrote the paper, and conceived the project.
Conflict-of-interest disclosure: A.G.S. and D.G. declare a patent filed on the use of anti-CD3 mono-Fabs for immunotherapy. The remaining authors declare no competing financial interests.
The current affiliation for M.M.H. is Department of Immunology and Microbiology, University of Colorado Denver, Anschutz Medical Campus, Denver, CO.
The current affiliation for C.A.P. is Department of Medicine, Columbia University Medical Center, New York, NY.
Correspondence: Diana Gil, University of Missouri, 1 Hospital Dr, Columbia, MO 65212; e-mail: gilpagesd@health.missouri.edu; Adam G. Schrum, University of Missouri, 1 Hospital Dr, Columbia, MO 65212; e-mail: schruma@health.missouri.edu; and Svetomir N. Markovic, Mayo Clinic, 200 1st St SW, Rochester, MN 55905; e-mail: markovic.svetomir@mayo.edu.




