

Key Points
P-selectin–deficient SCD mice are protected from lung vaso-occlusion.
P-selectin–deficient SCD mice will be useful in assessing the benefits of anti–P-selectin therapy in diverse complications of SCD.
Introduction
Sickle cell disease (SCD) is an autosomal-recessive genetic disorder that affects millions of people worldwide.1,2 Vaso-occlusion and hemolysis are the 2 predominant vascular events that contribute to the pathogenesis of SCD.3 Vaso-occlusion is believed to trigger acute systemic painful vaso-occlusive episode, which is the primary reason for emergency medical treatment of SCD patients.1,4 A role for P-selectin in promoting vaso-occlusion in the cremaster venules of SCD mice was first demonstrated by Kaul and Hebbel.5 Later, sickle erythrocytes were shown to adhere to P-selectin in vitro and undergo P-selectin–mediated rolling adhesion in postcapillary venules of mice in vivo, and P-selectin deletion or inhibition was shown to prevent adhesion of adoptively transferred sickle erythrocytes and vaso-occlusion in nonsickle mice in vivo.6-9 Also, platelet P-selectin–dependent neutrophil-platelet-erythrocyte heterocellular aggregates were shown to be significantly elevated in SCD patient blood.10
Epidemiological evidence suggests that a vaso-occlusive episode is often an antecedent to acute chest syndrome, a type of acute lung injury and one of the leading causes of mortality among SCD patients.1,11-13 Recently, we found that vaso-occlusive episode in transgenic humanized SCD mice triggered microembolism of precapillary pulmonary arterioles by platelet-neutrophil aggregates, which led to loss of blood flow in the lung microvasculature.13 Remarkably, platelet-neutrophil aggregates were attenuated, lung vaso-occlusion was prevented, and pulmonary blood flow was rescued in SCD mice following therapeutic blockade of P-selectin.13 A role for P-selectin in vaso-occlusion was further supported by a recent phase 2 study that reported a significant reduction in painful vaso-occlusive episodes among SCD patients receiving the P-selectin–blocking antibody crizanlizumab.14 Altogether, these findings suggest that SCD mice genetically deficient in P-selectin would be protected from vaso-occlusion. Such a mouse would also be useful in identifying the role of P-selectin in SCD-associated morbidities other than painful vaso-occlusive episode or acute chest syndrome.15 A role for P-selectin in systemic vaso-occlusion has been investigated using chimeric SCD mice lacking P-selectin only in the endothelium (intact in platelets),6,16 because SCD mice with global deletion of P-selectin did not exist. Here, we introduce the first SCD mice genetically lacking P-selectin in hematopoietic and nonhematopoietic compartments. Using our recently developed quantitative fluorescence intravital lung microscopy (qFILM)13,17 technique, we show that P-selectin deficiency protects SCD mice from lung vaso-occlusion.
Methods
Reagents
Violet 450 (V450) Rat anti-mouse CD49b monoclonal antibody (mAb; clone DX5) was purchased from BD Biosciences (San Jose, CA). Alexa Fluor 546 (AF546) rat anti-mouse Ly6G mAb (clone 1A8) was purchased from BioLegend (San Diego, CA). FITC-Dextran (molecular weight 70 000) was purchased from Molecular Probes (Eugene, OR). Gram-negative bacterial lipopolysaccharide (LPS) from Escherichia coli 0111:B4 was from Sigma-Aldrich (St. Louis, MO). Recombinant murine P-selectin (CD62P) Fc chimera was purchased from R&D Systems (Minneapolis, MN). DC Protein Assay Reagent A, B, and S were purchased from Bio-Rad (Hercules, CA). Bolt LDS sample buffer (4X), Bolt MES SDS Running Buffer (20X), Bolt transfer buffer (20X), Bolt 4% to 12% Bis-Tris plus gel, nitrocellulose membrane filter paper sandwich, and Novex sharp prestained protein standard were purchased from Life Technologies (Carlsbad, CA). HyBlot CL Autoradiography Film was purchased from Denville Scientific (Holliston, MA). Phosphate-buffered saline (without Ca2+ and Mg2+), M-PER Mammalian Protein Extraction Reagent, and SuperSignal West Pico Chemiluminescent Substrate were purchased from Thermo Fisher Scientific (Rockford, IL).
Generation of P-selectin–deficient SCD mice
Male and female (∼12- to 16-week-old) Townes SCD mice [SS mice; homozygous for Hbatm1(HBA)Tow, homozygous for Hbbtm2(HBG1,HBB*)Tow] and nonsickle control mice [AS mice; homozygous for Hbatm1(HBA)Tow, compound heterozygous for Hbbtm2(HBG1,HBB*)Tow/Hbbtm3(HBG1,HBB)Tow] were used in this study.18 Townes SS mice have human α-sickle and β-sickle globin (βS) genes knocked into the locus where mouse α and β genes were knocked out. Townes AS mice are sickle trait mice and, thus, do not develop SCD. Townes SS and AS mice have been used previously as SCD and control nonsickle mice, respectively.19,20 Townes SS and AS mice were bred and genotyped in-house. Breeding pairs of P-selectin–deficient (Selp−/−) mice (B6.129S7-Selptm1Bay/J; stock number 002289) were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in-house.21 Townes SS mice were bred to Selp−/− mice to generate P-selectin–deficient SS (SS-Selp−/−) mice using the breeding strategy described in supplemental Figure 1. Mice used in experiments were 12 to 16 weeks old and between the second and sixth generation. Mice were euthanized as per the guidelines of the American Veterinary Medical Association and the Department of Laboratory Animal Research at the University of Pittsburgh. Mice were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International–accredited pathogen-free animal facility at the University of Pittsburgh. The study was approved by Animal Care and Use Committee of the University of Pittsburgh.
Genotyping of SS-Selp−/− mice
DNA was isolated from crude mouse tail lysate, and polymerase chain reactions (PCR) were conducted on a thermocycler (Applied Biosystems, Foster City, CA) using a combination of protocols provided by The Jackson Laboratory for genotyping Townes SS and Selp−/− mice. The PCR products were used in 1.5% agarose gel electrophoresis, followed by visualization under UV light to determine the amplicon size. The following primers were used.
α-chain.
The following primers were used for α-chain: human α reverse (5′-TCC TGC AGG GTG AGG AAG GAA GG-3′); mouse α reverse (5′-CCC CAA GGC ACT CCA GGG ACA TAG-3′); common (5′-TCT ATG CAC ATC AAT TAG CAG AGG C-3′).
β-chain.
The following primers were used for β-chain: human-βA reverse (5′-GTT TAG CCA GGG ACC GTT TCA G-3′); human-βS reverse (5′-AAT TCT GGC TTA TCG GAG GCA AG-3′); mouse-β reverse (5′- ATG TCA GAA GCA AAT GTG AGG AGC A-3′); common (5′-TTG AGC AAT GTG GAC AGA GAA GG-3′).
P-selectin.
The following primers were used for P-selectin: mouse Selp−/−forward (5′-CTG AAT GAA CTG CAG GAC GA-3′); mouse Selp−/− reverse (5′-ATA CTT TCT CGG CAG GAG CA-3′); mouse wild type Selp forward (5′-TTG TAA ATC AGA AGG AAG TGG-3′); mouse wild type Selp reverse (5′-AGA GTT ACT CTT GAT GTA GAT CTC C-3′).
Real-time reverse-transcriptase quantitative PCR for P-selectin
Aortas were isolated from 3 SS-Selp−/− mice and 3 SS mice. Total RNA isolation was performed using Invitrogen TRIzol Reagent (Invitrogen, Carlsbad, CA), as per the vendor’s instructions. Reverse transcription was performed to synthesize complementary DNA using a High-Capacity cDNA Reserve Transcription Kit (Applied Biosystems). Real-time amplification of complementary DNA was conducted using PowerUp SYBR Green Master Mix on a StepOnePlus Real-Time PCR system (Applied Biosystems). β-Actin was used as a housekeeping gene. The following primers were used: mouse P-selectin forward primer, 5′-TCCAGGAAGCTCTGACGTACTTG-3′; mouse P-selectin reverse primer, 5′-GCAGCGTTAGTGAAGACTCCGTAT-3′; β-actin forward primer, 5′-ACGGCCAGGTCATCACTATTC-3′; and β-actin reverse primer, 5′-AGGAAGGCTGGAAAAGAGCC-3′.
Western blot analysis for P-selectin
Blood was drawn from the inferior vena cava of SS and SS-Selp−/− mice into trisodium citrate and supplemented with an equal volume of washing buffer (10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM glucose, and 1 mM pyruvate; pH 7.4) and centrifuged at 100g (22°C) for 10 minutes. The supernatant was treated with 0.5 µM prostaglandin I2 and centrifuged at 1100g (22°C) for 15 minutes. The platelet pellet was resuspended in washing buffer containing 0.5 µM prostaglandin I2 and centrifuged at 1100g (22°C) for 15 minutes. The platelet pellet was resuspended in ice-cold radioimmunoprecipitation assay buffer, supplemented with protease and phosphatase inhibitor cocktail, incubated for 5 minutes on ice, and centrifuged at 3000g (4°C) for 15 minutes to remove cell debris. Lysate (supernatant) was snap-frozen in liquid nitrogen and stored at −80°C. Protein concentrations in platelet lysates were measured using a Bio-Rad DC Protein Assay. Total protein (50 μg) was separated and blotted using a Mini Gel System and Mini Blot Module (both from Invitrogen), as described elsewhere.22 In addition, recombinant mouse P-selectin–Fc chimera protein (R&D Systems, Minneapolis, MN) was used as a standard positive control. CD62P protein was detected using goat–anti-mouse P-Selectin Antibody (R&D Systems). Chemiluminescent detection of P-selectin was done using horseradish peroxidase–conjugated Anti-goat IgG (R&D Systems) and SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA).
Histology and IHC
Immunohistochemistry (IHC) of paraffin-embedded liver tissue sections was performed as described elsewhere.23,24 Tissue sections (4 μm) were stained with hematoxylin and eosin (H&E) and Sirius Red. An Olympus Provis microscope was used to capture images, and Nikon NIS-Elements software was used for image analysis. Quantification of Sirius Red staining was done using ImageJ software.
qFILM
Recently, qFILM has been used by our group to study platelet-neutrophil aggregate–mediated pulmonary vaso-occlusion in transgenic humanized SCD mice.13 In the current study, qFILM was used to assess pulmonary vaso-occlusion in SS and SS-Selp−/− mice following IV challenge with saline or LPS. The qFILM experimental setup has been described in detail previously.13,17,22 Briefly, SS and SS-Selp−/− mice were injected IV with saline or 0.1 µg/kg LPS via the tail vein. Approximately 2 to 2.5 hours later, mice were anesthetized with an intraperitoneal injection of 100 mg/kg ketamine HCl (Henry Shein Animal Health, Dublin, OH) and 20 mg/kg xylazine (LLOYD Laboratories, Shenandoah, IA). A cannula was inserted into the right carotid artery, and a tracheotomy was performed to facilitate mechanical ventilation with 95% O2 and supply maintenance anesthesia (1%-2% isoflurane). The left lung was surgically exposed, and a small portion of the lung was immobilized against a coverslip using a vacuum enabled micro-machined device, as described elsewhere.13,17,22 Just prior to qFILM, ∼125 μg of FITC-Dextran, 12 μg of AF546-conjugated Ly6G mAb, and 7 μg of V450-conjugated CD49b mAb were injected into the carotid artery catheter for visualization of the pulmonary microcirculation and in vivo staining of neutrophils and platelets, respectively. qFILM was performed on a mouse for a total of 30 minutes, and the presence or absence of vaso-occlusion was assessed for 30 seconds in each FOV. The resulting series of qFILM images was processed using image subtraction, a median filter, a noise-reduction algorithm, and adjustment of intensity histograms, as described previously.13,22 Some of the channels were pseudo-colored to enhance contrast. Pulmonary vaso-occlusions were assessed in ∼15 to 20 fields of view (FOVs) in each mouse and across multiple mice per test group (n = 3 mice), as previously described.13,22 Pulmonary vaso-occlusion was quantified and compared between treatment groups using the following parameters, as described previously13 : average number of pulmonary vaso-occlusions per FOV, percentage of FOVs with pulmonary vaso-occlusions, and the average number of large pulmonary vaso-occlusions with an area >1000 µm2 per FOV.
Statistical analysis
Means were compared between groups using the unpaired Student t test. Percentages were compared using fourfold table analysis with χ2 statistics.25,26 Unless otherwise stated, error bars represent standard error (SE). P < .05 was considered significant.
Results and discussion
Townes SS male mice were bred to Selp−/− female mice (Figure 1A), using the breeding steps described in supplemental Figure 1, to generate SS-Selp−/− mice. Identical to SS mice, SS-Selp−/− mice lacked murine α- and β-globin genes but expressed human α and βS globin genes (representative gel shown in Figure 1B). SS-Selp−/− mice also lacked the P-selectin gene expressed in WT mice, but they expressed the mutant P-selectin gene expressed in Selp−/− mice (Figure 1B). P-selectin is stored preformed in Weibel-Palade bodies and α-granules of endothelial cells and platelets, respectively.27-29 Additionally, P-selectin is known to be transcriptionally upregulated in inflamed endothelial cells30,31 and chronically expressed on endothelium in SCD mice in vivo.32 Therefore, P-selectin protein levels were detected in isolated mouse platelets, whereas P-selectin messenger RNA (mRNA) levels were assessed in harvested mouse aortas. Real-time reverse-transcriptase quantitative PCR (RT2qPCR) of aortic tissue revealed significantly reduced P-selectin mRNA levels in aortas of SS-Selp−/− mice compared with SS mice (Figure 1C). Western blots confirmed that P-selectin protein was absent in platelets isolated from SS-Selp−/− mice but was present in normal SS mice platelets (Figure 1D). Because SS-Selp−/− females were unable to breed, the mice colony was maintained by breeding SS-Selp−/− males to littermate female mice heterozygous for murine β and human βS globin genes. P-selectin deficiency also led to significantly elevated counts of circulating neutrophils and monocytes in SS-Selp−/− mice compared with SS mice (Table 1), most likely as a result of the lack of P-selectin–dependent rolling along the vascular endothelium, leading to impaired recruitment of these cells to sites of inflammation or bone marrow. However, the hemoglobin and hematocrit values in SS-Selp−/− mice were comparable to those in SS mice; these values were below the normal range, suggestive of hemolytic anemia (Table 1). Next, the histology of isolated liver sections was assessed using the approach described previously.23,24 H&E staining (Figure 1E; supplemental Figure 2A) and Sirius Red staining (Figure 1F; supplemental Figure 2A) revealed sinusoidal congestion, liver injury (Figure 1G; supplemental Figure 2B), and liver fibrosis (Figure 1H; supplemental Figure 2C), respectively in SS mice, which was significantly ameliorated in SS-Selp−/− mice.
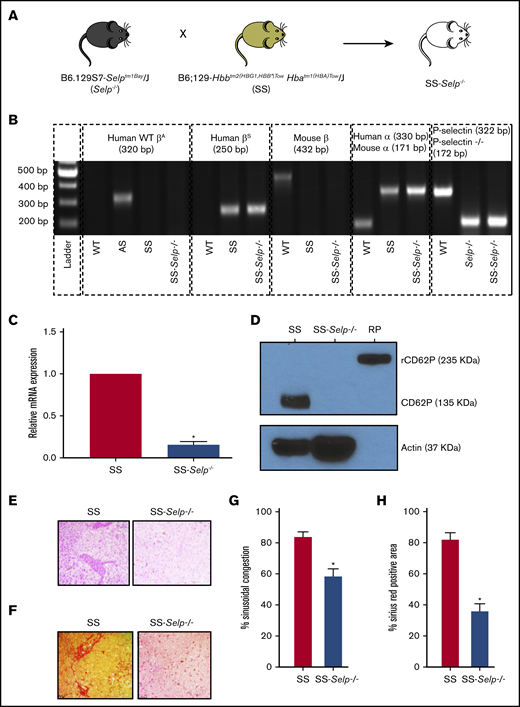
Generation and characterization of SS-Selp−/−mice. (A) Selp−/− mice were bred to Townes SS mice to generate SS-Selp−/− mice. Refer to supplemental Figure 1 for the breeding strategy. (B) Genomic PCR gel image showing the presence or absence of different alleles in C57BL/6 (WT) mice, Townes AS (AS) mice, Townes SS (SS) mice, Selp−/− mice, and SS-Selp−/− mice. Human WT β-globin (WT βA), mouse β-globin, mouse α-globin, and mouse WT P-selectin alleles were absent in SS-Selp−/− mice, but human βS, human α-globin, and mouse mutant P-selectin (P-selectin−/−) alleles were present in SS-Selp−/− mice. (C) RT2qPCR analysis revealed a significant reduction in the mRNA levels of P-selectin in the aortas of SS-Selp−/− mice compared with SS mice. Data are mean relative mRNA expression ± standard deviation (SD). (D) Western blot analysis of CD62P protein levels in platelets isolated from SS and SS-Selp−/− mice. P-selectin (135 kDa) was expressed in platelets of SS mice but was absent in platelets of SS-Selp−/− mice. Actin (37 kDa) was used as a loading control. Recombinant mouse P-selectin–Fc fusion protein (RP; 235 kDa; R&D Systems) was used as a positive control. Data in panels C-D are representative of 3 female SS mice and 3 female SS-Selp−/− mice. Representative IHC images showing sinusoidal congestion by H&E staining (E) and fibrosis and collagen deposition by Sirius Red staining (F) in the liver sections of SS and SS-Selp−/− mice. Original magnification ×40. (G) Percentage of sinusoidal congestion based on H&E staining. (H) Quantification of liver fibrosis as the percentage of the area positive for Sirius Red staining. Data in panels G-H are mean ± SE based on 3 male SS mice and 3 male SS-Selp−/− mice. *P < .05.
Generation and characterization of SS-Selp−/−mice. (A) Selp−/− mice were bred to Townes SS mice to generate SS-Selp−/− mice. Refer to supplemental Figure 1 for the breeding strategy. (B) Genomic PCR gel image showing the presence or absence of different alleles in C57BL/6 (WT) mice, Townes AS (AS) mice, Townes SS (SS) mice, Selp−/− mice, and SS-Selp−/− mice. Human WT β-globin (WT βA), mouse β-globin, mouse α-globin, and mouse WT P-selectin alleles were absent in SS-Selp−/− mice, but human βS, human α-globin, and mouse mutant P-selectin (P-selectin−/−) alleles were present in SS-Selp−/− mice. (C) RT2qPCR analysis revealed a significant reduction in the mRNA levels of P-selectin in the aortas of SS-Selp−/− mice compared with SS mice. Data are mean relative mRNA expression ± standard deviation (SD). (D) Western blot analysis of CD62P protein levels in platelets isolated from SS and SS-Selp−/− mice. P-selectin (135 kDa) was expressed in platelets of SS mice but was absent in platelets of SS-Selp−/− mice. Actin (37 kDa) was used as a loading control. Recombinant mouse P-selectin–Fc fusion protein (RP; 235 kDa; R&D Systems) was used as a positive control. Data in panels C-D are representative of 3 female SS mice and 3 female SS-Selp−/− mice. Representative IHC images showing sinusoidal congestion by H&E staining (E) and fibrosis and collagen deposition by Sirius Red staining (F) in the liver sections of SS and SS-Selp−/− mice. Original magnification ×40. (G) Percentage of sinusoidal congestion based on H&E staining. (H) Quantification of liver fibrosis as the percentage of the area positive for Sirius Red staining. Data in panels G-H are mean ± SE based on 3 male SS mice and 3 male SS-Selp−/− mice. *P < .05.
Previously,13 we have shown that IV administration of a few nanograms (0.1 μg/kg) of LPS triggered lung vaso-occlusion in SCD mice, which was facilitated by entrapment of large neutrophil-platelet aggregates in precapillary pulmonary arteriolar bottlenecks located at the junction of pulmonary arterioles and capillaries. Therefore, SS-Selp−/− mice and SS mice were administered LPS (IV, 0.1 μg/kg), and lung vaso-occlusion was compared using qFILM (Figure 2A). Identical to our previous findings, IV LPS triggered occlusion of pulmonary arterioles by large neutrophil-platelet aggregates in SS mice (Figure 2B; supplemental Figure 3). A large neutrophil-platelet aggregate (dotted oval) is shown in Figure 2B and supplemental Video 1. Three more representative qFILM images of large neutrophil-platelet aggregates occluding pulmonary arterioles in SS mice are shown in supplemental Figure 3. In contrast, lung vaso-occlusion was absent, and the majority of FOVs were free of neutrophil-platelet aggregates, in the lungs of SS-Selp−/− mice administered IV LPS (representative FOVs are shown in Figure 2C and supplemental Video 2). Analysis of the time-series of qFILM images revealed that the average number of pulmonary vaso-occlusions per FOV (Figure 2D), the percentage of FOVs with pulmonary vaso-occlusions (Figure 2E), and the average number of pulmonary vaso-occlusions with area >1000 μm2 (Figure 2F) were significantly smaller in SS-Selp−/− mice compared with SS mice. Taken together, these findings suggest that SS-Selp−/− mice lack the P-selectin gene, mRNA, and protein in hematopoietic and nonhematopoietic tissues, which lead to attenuation of vaso-occlusion and injury in the lung and liver. Our findings also highlight the need to study the effect of P-selectin antibody (crizanlizumab) treatment on circulating leukocyte counts in SCD patients.14
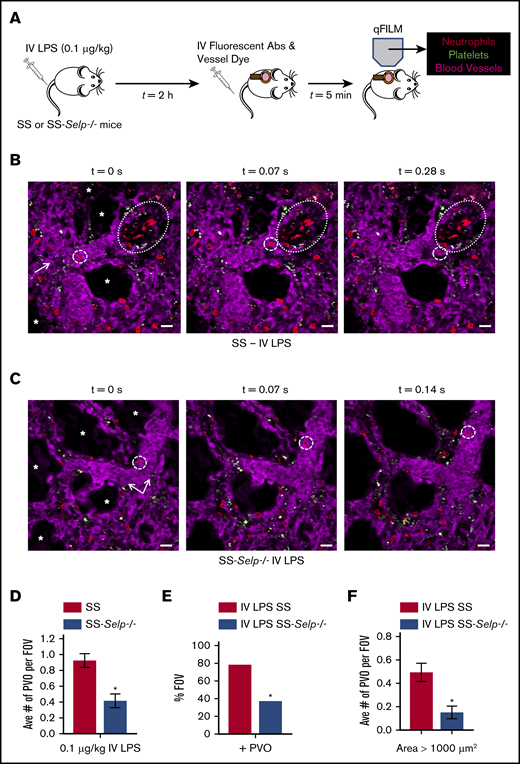
Genetic deletion of P-selectin attenuates pulmonary vaso-occlusion in SCD mice. (A) SS and SS-Selp−/− mice were challenged IV with 0.1 µg/kg LPS, and qFILM was used 2 to 2.5 hours later to visualize the pulmonary microcirculation. (B) IV LPS (0.1 µg/kg) triggered occlusion of pulmonary arteriolar bottlenecks (large dotted ovals) in SS mice by large aggregates of neutrophils (red) and platelets (green). The same FOV is shown over 3 time points. A neutrophil (red; small dashed circles) flows toward the occlusion but cannot pass through it and is forced to flow toward another open vessel to the side of the occlusion. Supplemental Video 1 shows the complete time series for the FOV in panel B. (C) In contrast to SS mice, the majority of FOVs in SS-Selp−/− mice were free of pulmonary vaso-occlusions. The same FOV is shown over 3 time points. A neutrophil (red; small dashed circles) is seen trafficking up the pulmonary arteriole that has no aggregates present. Supplemental Video 2 shows the complete time series for the FOV in panel C. The pulmonary microcirculation was labeled with FITC-Dextran and pseudo-colored purple. Neutrophils and platelets were labeled by IV administration of AF546-conjugated anti-Ly6G mAb and V450-conjugated anti-CD49b mAb, respectively. Neutrophils are shown in red, and platelets are pseudo-colored green. The arrows denote the direction of blood flow, and the asterisks (*) denote alveoli. Scale bars, 20 μm. The diameters of the vessels are 34 μm and 40 μm in panels B-C, respectively. (D-F) The neutrophil-platelet aggregates blocking pulmonary arterioles were quantified as described in Methods. After IV LPS administration, SS-Selp−/− mice had a significantly decreased average number of pulmonary vaso-occlusions per FOV (D), percentage of FOVs with pulmonary vaso-occlusions (E), and large pulmonary vaso-occlusions (area >1000 μm2) (F) compared with SS mice. The average number of pulmonary vaso-occlusions per FOV and large pulmonary vaso-occlusions (area >1000 µm2) were compared between groups using the unpaired Student t test, and the percentage of FOVs with pulmonary vaso-occlusions were compared between groups using fourfold table with χ2 analyses. SS mice: n = 1 male mice and 2 female mice, FOV = 45. SS-Selp−/− mice: n = 1 male mice and 2 female mice, FOV = 46. Error bars are mean ± SE. *P < .05.
Genetic deletion of P-selectin attenuates pulmonary vaso-occlusion in SCD mice. (A) SS and SS-Selp−/− mice were challenged IV with 0.1 µg/kg LPS, and qFILM was used 2 to 2.5 hours later to visualize the pulmonary microcirculation. (B) IV LPS (0.1 µg/kg) triggered occlusion of pulmonary arteriolar bottlenecks (large dotted ovals) in SS mice by large aggregates of neutrophils (red) and platelets (green). The same FOV is shown over 3 time points. A neutrophil (red; small dashed circles) flows toward the occlusion but cannot pass through it and is forced to flow toward another open vessel to the side of the occlusion. Supplemental Video 1 shows the complete time series for the FOV in panel B. (C) In contrast to SS mice, the majority of FOVs in SS-Selp−/− mice were free of pulmonary vaso-occlusions. The same FOV is shown over 3 time points. A neutrophil (red; small dashed circles) is seen trafficking up the pulmonary arteriole that has no aggregates present. Supplemental Video 2 shows the complete time series for the FOV in panel C. The pulmonary microcirculation was labeled with FITC-Dextran and pseudo-colored purple. Neutrophils and platelets were labeled by IV administration of AF546-conjugated anti-Ly6G mAb and V450-conjugated anti-CD49b mAb, respectively. Neutrophils are shown in red, and platelets are pseudo-colored green. The arrows denote the direction of blood flow, and the asterisks (*) denote alveoli. Scale bars, 20 μm. The diameters of the vessels are 34 μm and 40 μm in panels B-C, respectively. (D-F) The neutrophil-platelet aggregates blocking pulmonary arterioles were quantified as described in Methods. After IV LPS administration, SS-Selp−/− mice had a significantly decreased average number of pulmonary vaso-occlusions per FOV (D), percentage of FOVs with pulmonary vaso-occlusions (E), and large pulmonary vaso-occlusions (area >1000 μm2) (F) compared with SS mice. The average number of pulmonary vaso-occlusions per FOV and large pulmonary vaso-occlusions (area >1000 µm2) were compared between groups using the unpaired Student t test, and the percentage of FOVs with pulmonary vaso-occlusions were compared between groups using fourfold table with χ2 analyses. SS mice: n = 1 male mice and 2 female mice, FOV = 45. SS-Selp−/− mice: n = 1 male mice and 2 female mice, FOV = 46. Error bars are mean ± SE. *P < .05.
In addition to painful vaso-occlusive episode and acute chest syndrome, the recent improvement in the life expectancy of SCD patients has led to the increased incidence of other SCD-associated complications, such as pulmonary hypertension, sickle hepatopathy, renal dysfunction, retinopathy, cardiomyopathy, and priapism.15 The molecular mechanisms of these pathologies are incompletely understood, and current treatments are primarily supportive.15 The efficacy of P-selectin inhibition in preventing vaso-occlusive episode3,13,14,16 warrants basic science and clinical studies to determine whether P-selectin inhibition would show benefit in these other complications as well. Interestingly, P-selectin is constitutively expressed on SCD mice and patient platelets,33,34 and it is expressed on the vascular endothelium in the skin, lung, brain, liver, and kidney of SCD mice.20 Recently,35,36 P-selectin inhibition was also shown to prevent hypoxia-induced pulmonary hypertension in WT mice, suggesting that P-selectin may also be playing a role in SCD-associated pulmonary hypertension.37 P-selectin inhibition was also shown to attenuate blood-brain barrier permeability in SCD mice.38 Regardless of the difference in P-selectin expression between murine and human endothelial cells, SS-Selp−/− mice would be useful in future studies to assess the role of P-selectin and the benefits of anti–P-selectin therapy in diverse pathologies of SCD.
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung and Blood Institute grants 1R01HL128297 and 1R01HL141080, American Heart Association grants 18TPA34170588 and 11SDG7340005, and funds from the Hemophilia Center of Western Pennsylvania and Vitalant (all to P.S.). M.F.B. was supported by National Institutes of Health, National Heart, Lung and Blood Institute training grant T32HL110849 and National Institutes of Health National Research Service Award grant 1F32HL131216-01. T.B. was supported by a Postdoctoral Scholar Award from the American Society of Hematology. T.P.-S. was supported by a Community Liver Alliance award and by a Pittsburgh Liver Research Center pilot and feasibility grant. The Nikon multiphoton excitation microscope was funded by National Institutes of Health grant 1S10RR028478-01 (S.C.W.).
Authorship
Contribution: M.F.B. performed qFILM studies and analyses; E.T. generated SS-Selp−/− mice and performed genotyping and western blots; S.G. contributed to mice genotyping; T.B. isolated platelets from mice; E.T. and T.P.-S. performed RT2qPCR experiments; S.C.W. was involved in qFILM studies; T.P.-S. and S.P.M. performed histology and IHC of liver sections; and P.S. designed the experiment, supervised the project, and wrote the manuscript with contributions from all coauthors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Prithu Sundd, Division of Pulmonary, Allergy and Critical Care Medicine, Heart, Lung and Blood Vascular Medicine Institute, University of Pittsburgh School of Medicine, BST E1255, 200 Lothrop St, Pittsburgh, PA 15261; e-mail: prs51@pitt.edu.


