

Key Points
Cyclin-dependent kinase 9 and bromodomain and extraterminal inhibitors are synergistic in MLL-rearranged leukemia.
Multiple AML driver genes are downregulated by the combined therapy suggesting broad applicability for this subtype.
Introduction
Chromosomal rearrangements of the lysine methyltransferase 2A (KMT2A or MLL) gene are observed in ∼10% of all acute leukemias, with particularly high frequency (∼80%) in infant acute lymphoblastic leukemia (ALL),1 where, despite aggressive chemotherapy, patients still experience poor outcome and long-term side effects.2 Mixed lineage leukemia (MLL) rearrangements (MLL-r) also indicate particularly poor outcomes for patients with acute myeloid leukemia (AML).3 Mechanistically, MLL-r frequently generates fusion proteins involving partners that function in the super elongation complex,4 the result of which is aberrant recruitment to MLL target genes of the positive transcription elongation factor b (PTEFb), composed of cyclin-dependent kinase 9 (CDK9) as the catalytic subunit.5 CDK9 positively regulates transcription elongation through phosphorylation of serine 2 of RNA polymerase II (RNAPII).6 Given the central role of CDK9 in the leukemic MLL-r gene-expression program,7 and the well-described ability of CDK9 inhibitors to reduce levels of the short-lived prosurvival protein MCL1,8 a number of CDK9 inhibitors have been selected for clinical trials focusing on acute leukemias, including those with MLL-r.8,9 In MLL-r leukemia, the bromodomain and extraterminal (BET) family member bromodomain-containing 4 (BRD4)10 acts to recruit PTEFb to superenhancers and together with CDK9 drives increased expression of many oncogenes including MYC.11,12 The roles of CDK9 and BRD4 in MLL-r leukemias present a strong case for testing inhibitors of these proteins in combination as a potential treatment of MLL-r acute leukemias.
Methods
The patient-derived xenograft (PDX) model was established by engrafting infant MLL-r ALL or adult MLL-r AML cells into female NSG mice. Recipient mice were treated with either vehicle, iBET-151 (15 mg/kg; intraperitoneally; 5 days for 2 weeks), CDKI-73 (25 mg/kg; orally; 14 days), or the combination. These doses do not show hematological toxicity (supplemental Table 1). Detailed methods including methods for in vitro assays are provided in the supplemental Methods.
Results and discussion
We first investigated the combination of a CDK9 and BET inhibitor in vitro using cells derived from a panel of established infant MLL-r ALL PDX,13 and AML PDX cells carrying the MLL-MLLT3 rearrangement (AML-18) (supplemental Table 2).14 Combination treatment using CDKI-7315 and JQ116 resulted in further reduction of viable cells compared with either single drug treatment of all samples (Figure 1), with the Bliss-additive curve indicating synergy. We treated 3 MLL-r AML cell lines (MV4;11, MOLM-13, THP1) and a non–MLL-r ALL cell line (697; supplemental Table 2) with a combination of CDKI-73 and the BET inhibitor iBET-151, which has improved pharmacokinetic properties over JQ1. For all cell lines, we observed synergistic cell killing (supplemental Figure 1), consistent with the synergy not being limited to MLL-r.17
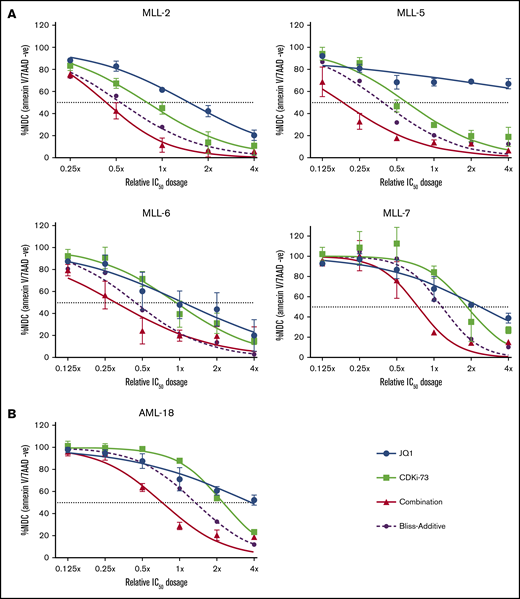
Synergistic activity of combined CDK9/BET inhibition in MLL-r PDXs in vitro. PDX cells from 4 MLL-ALL (n = 2) (A) and 1 MLL-AML (n = 3) (B) were treated for 48 hours, with single treatment of JQ1 (blue), CDKI-73 (light green), or the combination (red). Viability was measured by annexin V and 7-aminoactinomycin D (7AAD) staining and flow cytometry analysis. The double-negative (annexin V and 7-AAD negative) population (viable cells) of treated samples is plotted as a percentage of the no drug control (NDC). Bliss-additive effect is indicated by the purple dashed lines. Viability below this curve indicates synergy. IC50, 50% inhibitory concentration.
Synergistic activity of combined CDK9/BET inhibition in MLL-r PDXs in vitro. PDX cells from 4 MLL-ALL (n = 2) (A) and 1 MLL-AML (n = 3) (B) were treated for 48 hours, with single treatment of JQ1 (blue), CDKI-73 (light green), or the combination (red). Viability was measured by annexin V and 7-aminoactinomycin D (7AAD) staining and flow cytometry analysis. The double-negative (annexin V and 7-AAD negative) population (viable cells) of treated samples is plotted as a percentage of the no drug control (NDC). Bliss-additive effect is indicated by the purple dashed lines. Viability below this curve indicates synergy. IC50, 50% inhibitory concentration.
We next investigated the efficacy of this CDKI-73/iBET-151 combination treatment in vivo following engraftment of infant ALL and adult AML MLL-r PDXs (MLL-2 and AML-18, respectively) in NSG mice. For MLL-2, single treatments delayed disease progression with respect to event-free survival (EFS), however, improvement in median EFS was ≤7 days (Figure 2A-C). Neither single agent significantly delayed the progression of AML-18 (Figure 2D-F). For both PDX models, the combination treatment resulted in a significant increase in EFS compared with vehicle and single treatments (Figure 2B,E) and led to progression delays of 26.2 and 24.3 days for MLL-2 and AML-18, respectively (Figure 2C,F). At the end of the treatment period (day 14), a profound reduction of leukemic burden in the bone marrow, spleen, and peripheral blood compartments was observed in mice engrafted with AML-18 and treated with the combination, compared with mice treated with vehicle control or each single agent (supplemental Figure 2).
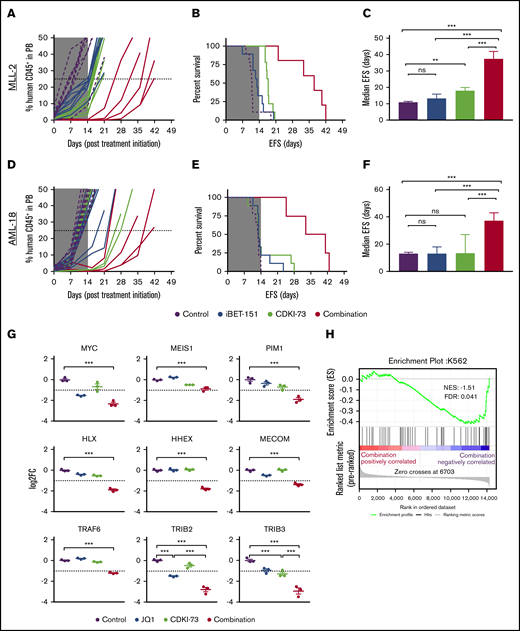
In vivo efficacy of combined CDK9/BET inhibition against MLL-r ALL and AML PDXs. Human CD45+ cells (percentage of huCD45+) in the peripheral blood (PB) of engrafted mice (left panels; A,D), EFS curves (middle panels; B,E), and median EFS of engrafted recipients (right panels; C,F, error bars depict 95% confidence interval) for MLL-ALL (A-C, n = 9 per treatment group) and MLL-AML (D-F, n = 9 per treatment group). Mice were treated with vehicle (purple), iBET-151 (blue), CDKI-73 (light green), or the combination (red) for 14 days as indicated (gray shading indicates treatment duration). (G) Messenger RNA expression of selected genes measured by RNA-seq in AML-18 treated with CDKI-73, JQ1, or in combination for 4 hours (error bars depict standard error, Tukey multiple comparison test; **P < .01, ***P < .001; dashed line indicates log fold change (FC) less than or equal to −1 cutoff for negatively differentially expressed genes). (H) Gene-set enrichment analysis using the ranked gene-expression list as determined by RNA-seq for the comparison of combination treatment vs vehicle in AML-18 cells. Plot shows significant negative enrichment for transcription factor genes associated with superenhancers in K562 cells.22 No significant enrichment was observed for this gene set with ranked gene expression from single treatments (supplemental Table 4). FDR, false discovery rate; NES, normalized enrichment score; ns, not significant.
In vivo efficacy of combined CDK9/BET inhibition against MLL-r ALL and AML PDXs. Human CD45+ cells (percentage of huCD45+) in the peripheral blood (PB) of engrafted mice (left panels; A,D), EFS curves (middle panels; B,E), and median EFS of engrafted recipients (right panels; C,F, error bars depict 95% confidence interval) for MLL-ALL (A-C, n = 9 per treatment group) and MLL-AML (D-F, n = 9 per treatment group). Mice were treated with vehicle (purple), iBET-151 (blue), CDKI-73 (light green), or the combination (red) for 14 days as indicated (gray shading indicates treatment duration). (G) Messenger RNA expression of selected genes measured by RNA-seq in AML-18 treated with CDKI-73, JQ1, or in combination for 4 hours (error bars depict standard error, Tukey multiple comparison test; **P < .01, ***P < .001; dashed line indicates log fold change (FC) less than or equal to −1 cutoff for negatively differentially expressed genes). (H) Gene-set enrichment analysis using the ranked gene-expression list as determined by RNA-seq for the comparison of combination treatment vs vehicle in AML-18 cells. Plot shows significant negative enrichment for transcription factor genes associated with superenhancers in K562 cells.22 No significant enrichment was observed for this gene set with ranked gene expression from single treatments (supplemental Table 4). FDR, false discovery rate; NES, normalized enrichment score; ns, not significant.
Immunoblots of lysates from the spleens of MLL-2–engrafted mice showed that all treatments resulted in depletion of MYC and BCL2 (supplemental Figure 3) as in previous reports.18 Consistent with on-target activity of CDKI-73, we observed reduced RNAPII-Ser2 phosphorylation with the single treatment (29%), which was further reduced (49%) in the combination. Most strikingly, combination treatment resulted in further reduction of the antiapoptotic protein MCL1, and increased levels of cleaved caspase 3, compared with either single agent alone, consistent with increased cell killing. The decrease of MCL1 is a potential mechanism for the enhanced MLL-2 in vivo response with the combination treatment, and is consistent with targeting of the MCL1 superenhancer.11
Downregulation of BCL2 family prosurvival proteins was not observed for the AML-18 PDX (data not shown). To investigate the mechanism for synergy in AML-18, gene-expression changes induced by treatment of the PDX cells in vitro for 4 hours with CDKI-73, JQ1, or the combination were determined by RNA sequencing (RNA-seq). Combination treatment resulted in significant downregulation of the hallmark MLL target genes MYC and MEIS1 (Figure 2G). The reduction in MYC expression is likely to be a significant contributor to the synergy observed in the MLL-r AML given that a BRD4- and CDK9-dependent MYC superenhancer is essential for maintenance of MLL-MLLT3–driven AML in mouse models.11,19,20 Another report also shows that combining a BET inhibitor with alternative CDK9 inhibitors synergistically repressed MYC in an MLL-AML cell line; however, this was not investigated in primary AML.21 To define other key myeloid oncogenic drivers, downregulation of which may contribute to the synergistic response, we determined genes that were uniquely downregulated in the combination treatment relative to control, or were downregulated by either single agent and in the combination (supplemental Figure 4A; supplemental Table 3). Through comparison with the DisGeNet AML database (supplemental Figure 4B-C; supplemental Table 3), multiple genes known to induce or promote AML were identified displaying enhanced downregulation in the combination treatment (Figure 2G). Of these, PIM1, HLX, TRAF6, and TRIB3 show similar responses in MV4;11 and MOLM-13 cell lines to that observed in AML-18 (supplemental Figure 5). Gene-set enrichment analysis showed, in combination treatment only, significant negative enrichment of transcription factor genes associated with superenhancers in the K562 myeloid leukemia cell line, but not in CD34+ cells, CD14+ cells, or nonhematopoietic tissue (Figure 2H; supplemental Table 4).22 Although it has been suggested that the CDK9/BET inhibitor combination may act via a global effect on transcriptional elongation,21 our results are most consistent with reduced expression by the combination treatment of multiple AML driver genes through targeting of myeloid leukemia superenhancers. Indeed, PIM1, HLX, and TRIB3 are linked to myeloid superenhancers and TRIB3 in particular has a very high superenhancer ranking in K562 cells.11,22
Our results highlight the potential of CDK9 inhibitors to act synergistically with transcriptional targeted therapies applicable to MLL-r acute leukemia, for example, BET, DOT1L, and Menin inhibitors.23 The synergy observed here for 2 PDX models of acute leukemia supports testing of the CDK9/BET inhibitor combination in MLL-r leukemia in the relapsed refractory setting or as an alternative to chemotherapy in high-risk cases. Such a tailored strategy has been successful in acute promyelocytic AML, where combination treatment with all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) has dramatically improved outcome and replaced chemotherapy.24 A key question will be whether this therapeutic approach reduces disease relapse, which is the major cause of poor survival outcomes in aggressive AML subtypes. However, it is very difficult to model clinical relapse in PDX models, as clonal evolution can differ compared with that in the patient.25 Thus, clinical testing will be required to establish whether this combination therapy is effective in improving survival for MLL-r acute leukemia patients.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (NHMRC; fellowships APP1059804 and APP1157871 [R.B.L.], and NHMRC program grant APP1091261 [R.B.L.]), a Tour de Cure research grant (17-UNSW-RS-01) (R.B.L. and R.J.D.), grants from the Ray and Shirl Norman Foundation, the Royal Adelaide Hospital Research Foundation, the Contributing Hematologist Committee (R.J.D., I.D.L., and S.W.), and an Industry Development grant from Bioinnovation SA (R.J.D. and S.W.).
Authorship
Contribution: H.M., K.L.L, L.J., S.C.B., D.A.C., S.E.S., I.D.L., R.B.L., and R.J.D. designed experiments and analyzed data; H.M., K.L.L., and L.J. performed the experiments; C.M., J.T., and K.L.L. performed bioinformatics analysis; N.S. and R.K.P. reviewed data and edited the manuscript; S.W. provided CDKI-73 and reviewed the manuscript; and R.J.D. and R.B.L. supervised the research and prepared the manuscript.
Conflict-of-interest disclosure: S.W. is shareholder of, holds patents with, and receives royalties from Le Sun Pharm Ltd. R.K.P. and N.S. are employees and shareholders of GlaxoSmithKline, which is carrying out clinical development of epigenetic inhibitors. The remaining authors declare no competing financial interests.
Correspondence: Richard J. D’Andrea, Acute Leukaemia Laboratory, Centre for Cancer Biology, University of South Australia, GPO Box 2471, Adelaide, SA 5001, Australia; e-mail: richard.dandrea@unisa.edu.au; and Richard B. Lock, Children’s Cancer Institute, PO Box 81, Randwick, NSW 2031, Australia; e-mail: rlock@ccia.org.au.

