Key Points
The VWF p.D1472H variant does not affect VWF expression or cause bleeding in a murine model.

Abstract
The von Willebrand factor ristocetin cofactor activity assay (VWF:RCo) is used for diagnosis of von Willebrand disease (VWD) because of its ability to evaluate VWF binding to platelets. VWF sequence variant p.D1472H is associated with lower VWF:RCo levels in the absence of associated bleeding symptoms, indicating the VWF:RCo may not be accurate for characterizing VWF function in individuals with this variant. Thus, this study aimed to determine the implications of the variant on VWF functioning in vivo. Mice were engineered with humanized wild-type (WT*) VWF A1/A2 and VWF with the p.D1472H (1472H) variant along with humanized platelet GPIbα and bred to homozygosity. VWF antigen and VWF binding to GPIbα were measured using enzyme-linked immunosorbent assay. Gel electrophoresis was used for VWF multimer analysis. Tail bleeding assays were performed at a 3-mm defined length. Normal VWF multimers were preserved in both WT* and 1472H mice. VWF expression was normal in the WT* and 1472H mice, and VWF binding to GPIbα did not statistically differ between the groups. Additionally, tail bleeding times were similar for WT* and 1472H mice. These results show the p.D1472H variant does not impair hemostasis in mice, and support the conclusion that p.D1472H is a normal variant in humans.
Introduction
von Willebrand disease (VWD) is a bleeding disorder caused by defects in the von Willebrand factor (VWF) protein. VWF functions in coagulation initiation through linkage of collagen exposed in vascular injury to circulating platelets via binding of the VWF A1 domain to the platelet GP1b complex.1 Because of the crucial role that VWF plays in facilitating hemostasis, defects in VWF can lead to impaired coagulation and associated bleeding symptoms.2 Variants in the VWF gene can lead to disease through both quantitative and qualitative defects in expressed VWF protein.3 Type 1 VWD is the most prominent variation of disease and seen in patients with decreased levels in VWF multimers, whereas patients with type 3 VWD have very minimal, if any, expression of VWF. Type 2 VWD presents with qualitative defects in VWF, leading to impaired multimer formation, platelet adhesion, and affinity for factor VIII.4
VWD diagnosis relies on patient reports of bleeding symptoms and a diagnostic workup that includes measurement of total VWF protein expression, as well as a measure of VWF functionality through platelet binding via the VWF ristocetin cofactor activity assay (VWF:RCo). The VWF:RCo assay is a key diagnostic test used in identifying patients with VWD. Ristocetin is used to induce VWF binding to the platelet GPIb receptor, which allows for investigation of the VWF protein ability to form bonds with platelets and initiate hemostasis.5,6
Several sequence variants in the A1 domain of VWF have been identified to affect the VWF:RCo, indicating these variants reduce VWF functionality. Specifically, VWF variant p.D1472H has been shown to lower VWF:RCo levels because of impaired VWF binding to ristocetin.7,8 However, individuals with the p.D1472H variant do not present with an elevated bleeding score, indicating that the ristocetin assay may not accurately measure VWF function for these patients.7 In addition, VWF activity measured by the VWF:GPIbM assay, which uses a gain of function GPIbα, is normal in subjects with p.D1472H.7
The objective of this study was to determine if the p.D1472H variant affects VWF activity in vivo to better understand the implications of this sequence variant. Since mouse platelets do not respond to ristocetin,9 we used a mouse model using human GPIbα along with the human exon 28 containing the GPIbα binding site.10
Methods
A construct with human VWF exon 28 replacing the murine exon 28 sequence was used to create knock-in C57BL/6J mice with human exon 28 VWF expression in endothelial cells and platelets.10 The following mice were created: wild-type (WT*) mice with wild-type human exon 28 and mice with human exon 28 containing the 1472H variant. The mice were then crossed with a mouse containing humanized GPIbα and no murine GPIbα that was previously created and described by Ware and colleagues to create WT* and 1472H variant mice with humanized VWF exon 28 and GPIbα.11 The targeting vector and breeding protocols have been previously described in detail by Kanaji et al.10 The resulting chimeric mice were then used to analyze the effect of the human p.D1472H variant on hemostasis.
VWF antigen (VWF:Ag) was measured through enzyme-linked immunosorbent assay (ELISA) (n ≥ 12 per group). Murine capture antibody 344.2 (Versiti Blood Research Institute, Milwaukee, WI) diluted with carbonate coating buffer to 5 μg/mL was used to coat Immulon-1b ELISA plates. Blocking buffer was created using 1% bovine serum albumin in phosphate-buffered saline and was then used for diluting WT* and 1472H VWF samples. Samples were incubated for 1 hour at room temperature, at which point VWF was detected using a biotinylated polyclonal anti-VWF antibody (Dako, Carpinteria, CA). The detection antibody was diluted to 2 μg/mL in the described blocking buffer before use.7 Quantification of murine VWF used a calibrator composed of pooled plasma from C75Bl/6J mice collected via vena cava puncture.
VWF binding to human GPIbα (VWF:GPIbM/VWF:Ag) was measured by ELISA (n ≥ 12 per group) using the 142.16 monoclonal antibody against human GPIbα (Versiti Blood Research Institute) diluted to 5 μg/mL in carbonate coating buffer for capture and biotinylated monoclonal anti-VWF antibodies AVW-1 and AVW-15 (Versiti Blood Research Institute) diluted to 2 μg/mL in ELISA blocking buffer for detection of VWF.12 Multimer analysis was performed via gel electrophoresis using previously described protocols to ensure VWF structure was preserved in the 1472H variant mice.7 For the multimers, VWF detection was achieved using an anti-VWF polyclonal antibody (Dako) with a secondary goat anti-rabbit antibody (IRDye 800CW; Li-Cor, Lincoln, NE).
Collagen binding was tested using human collagen III (Southern Biotech, Birmingham, AL) and murine collagen IV (Southern Biotech) as previously described.13,14
Tail clip bleeding assay was performed at a defined length of 3 mm after mice were anesthetized (n ≥ 5 per group).15 Bleeding time and total blood lost were measured from initial tail clip to cessation of bleeding or until cautery was performed at 10 minutes.
Results and discussion
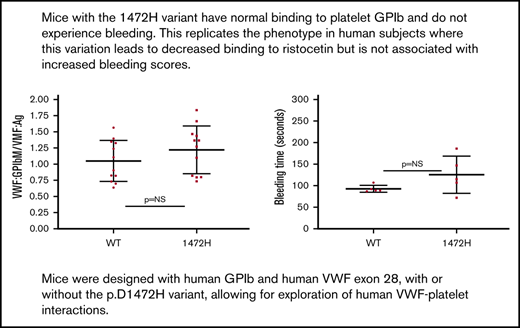
Normal VWF expression was seen for both groups of mice (Figure 1A), with no significant difference in VWF:Ag for the WT* and 1472H mice. The VWF:Ag for the WT* mice was 83 ± 45 IU/dL (mean ± 1 standard deviation) as compared with 111 ± 53 IU/dL for the 1472H mice (P = .11). This matches the normal VWF:Ag seen in human subjects with the D1472H variant. VWF binding to GPIbα using a human gain of function GPIbα (VWF:GPIbM) was similar for the WT* and 1472H mice (Figure 1B). VWF:GPIbM/VWF:Ag was 1.05 ± 0.32 for WT* mice vs 1.22 ± 0.37 for 1472H mice (P = .24). These results also parallel the results seen in human subjects, where those homozygous for the p.D1472H variant had significantly lower than normal VWF:RCo/VWF:Ag ratios but normal VWF:GPIbM/VWF:Ag ratios.12 In addition, normal binding was observed for both collagen III and collagen IV (P > .99 and P = .12, respectively) (Figure 1C). Tail bleeding times did not statistically differ between the WT* and 1472H mice (93 ± 8 vs 125 ± 43 seconds, P = .16) (Figure 1D). No other phenotypic bleeding was observed in the homozygous 1472H mice. Normal multimer distribution was observed for both the WT* and 1472H mice, confirming expression of the full VWF protein (Figure 1E).

Comparison of wild-type and 1472H mice. (A) VWF expression was normal for both the WT* (human exon 28, human GPIbα replacing the murine sequence in those regions) and 1472H mice, as indicated by the similar levels of VWF:Ag between the groups. The y-axis denotes the VWF:Ag in U/dL for wild-type (WT*, D1472) and 1472H mice. Error bars denote 1 standard deviation. Squares represent male mice; circles represent female mice. (B) VWF binding to platelet GPIbα (VWF:GPIbM/VWF:Ag) also did not differ for 1472H mice. The y-axis denotes VWF:GPIbM/VWF:Ag ratios for the same groups of mice. (C) Collagen binding with type III (CB3) and type IV (CB4) collagen did not differ. The y-axis shows the collagen binding to VWF (VWF:CB/VWF:Ag) ratios for collagen III on the left and collagen IV on the right. Triangles represent mice for whom sex was not known. (D) Tail bleeding times in seconds for WT* and 1472H mice. No statistical difference was seen. (E) Multimer distribution for the WT* and 1472H mice. Lane 1 has a C57Bl6 plasma standard, lane 2 has WT* plasma, and lane 4 has the 1472H mouse plasma. Full multimer distribution was seen for all mice. (Lane 3 contains an irrelevant plasma.)
Comparison of wild-type and 1472H mice. (A) VWF expression was normal for both the WT* (human exon 28, human GPIbα replacing the murine sequence in those regions) and 1472H mice, as indicated by the similar levels of VWF:Ag between the groups. The y-axis denotes the VWF:Ag in U/dL for wild-type (WT*, D1472) and 1472H mice. Error bars denote 1 standard deviation. Squares represent male mice; circles represent female mice. (B) VWF binding to platelet GPIbα (VWF:GPIbM/VWF:Ag) also did not differ for 1472H mice. The y-axis denotes VWF:GPIbM/VWF:Ag ratios for the same groups of mice. (C) Collagen binding with type III (CB3) and type IV (CB4) collagen did not differ. The y-axis shows the collagen binding to VWF (VWF:CB/VWF:Ag) ratios for collagen III on the left and collagen IV on the right. Triangles represent mice for whom sex was not known. (D) Tail bleeding times in seconds for WT* and 1472H mice. No statistical difference was seen. (E) Multimer distribution for the WT* and 1472H mice. Lane 1 has a C57Bl6 plasma standard, lane 2 has WT* plasma, and lane 4 has the 1472H mouse plasma. Full multimer distribution was seen for all mice. (Lane 3 contains an irrelevant plasma.)
Our 1472H mice, using the human GPIbα receptor and human A1 domain VWF, had normal VWF expression and normal tail bleeding times. These results suggest that the p.D1472H variant, even in homozygous form, does not result in impaired hemostasis. These results are consistent with the lack of bleeding seen in human subjects with the D1472H variant despite low VWF:RCo levels. Before this, the available data suggested that human subjects with the D1472H variant had an association with low VWF:RCo, but normal bleeding scores. Now for the first time, it is possible to state that presence of the D1472H variant does not result in increased bleeding. This model is also important because it replicates the human VWF-GPIb interaction. This will prove useful in future attempts to dissect the relationship between these 2 important ligands.
These findings, paired with previous findings showing lack of association between the 1472H sequence variant and bleeding, indicate that the VWF:RCo assay using ristocetin-induced platelet aggregation is not always the most accurate method in detection of clinically significant VWF dysfunction. In conclusion, use of the chimeric VWF human exon28 with human GPIbα mice served as a relevant model to confirm p.D1472H is a nonpathogenic variant in vivo.
For data sharing requests, send e-mails to the corresponding author, Veronica H. Flood, at vflood@mcw.edu.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute HL126810 (V.H.F.), HL081588 (R.R.M.), and HL144457 (R.R.M.), and by the MACC Fund (Midwest Athletes Against Childhood Cancer).
Authorship
Contribution: H.L.K. and T.L.S. performed the research; S.K. designed the mice. S.L.H., R.R.M., and V.H.F. conceived the study; H.L.K., S.L.H., and V.H.F. wrote the manuscript; and all authors edited and approved the manuscript.
Conflict-of-interest disclosure: R.R.M. holds a patent assigned to the Versiti BloodCenter of Wisconsin for a VWF platelet binding assay. The remaining authors declare no competing financial interests.
Correspondence: Veronica H. Flood, Pediatric Hematology/Oncology/BMT, Medical College of Wisconsin, 8701 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: vflood@mcw.edu.