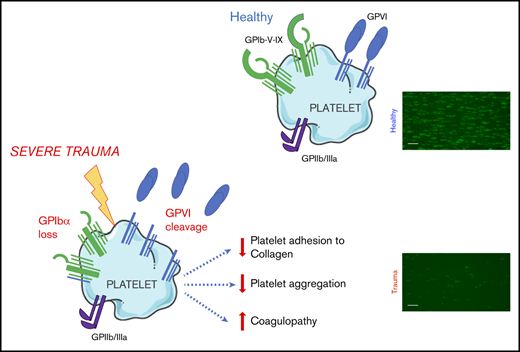
Key Points
GPVI and GPIbα are lost from the surface of circulating platelets in major trauma and contribute to acquired platelet dysfunction.
The targeting of proteolytic shedding of platelet receptors, particularly GPVI, is identified as a potential therapeutic strategy.

Abstract
Trauma-induced coagulopathy (TIC) is a complex, multifactorial failure of hemostasis that occurs in 25% of severely injured patients and results in a fourfold higher mortality. However, the role of platelets in this state remains poorly understood. We set out to identify molecular changes that may underpin platelet dysfunction after major injury and to determine how they relate to coagulopathy and outcome. We performed a range of hemostatic and platelet-specific studies in blood samples obtained from critically injured patients within 2 hours of injury and collected prospective data on patient characteristics and clinical outcomes. We observed that, although platelet counts were preserved above critical levels, circulating platelets sampled from trauma patients exhibited a profoundly reduced response to both collagen and the selective glycoprotein VI (GPVI) agonist collagen-related peptide, compared with those from healthy volunteers. These responses correlated closely with overall clot strength and mortality. Surface expression of the collagen receptors GPIbα and GPVI was reduced on circulating platelets in trauma patients, with increased levels of the shed ectodomain fragment of GPVI detectable in plasma. Levels of shed GPVI were highest in patients with more severe injuries and TIC. Collectively, these observations demonstrate that platelets experience a loss of GPVI and GPIbα after severe injury and translate into a reduction in the responsiveness of platelets during active hemorrhage. In turn, they are associated with reduced hemostatic competence and increased mortality. Targeting proteolytic shedding of platelet receptors is a potential therapeutic strategy for maintaining hemostatic competence in bleeding and improving the efficacy of platelet transfusions.
Introduction
Trauma-induced coagulopathy (TIC) is an acquired impairment of hemostatic competence that occurs in 25% of severely injured patients and results in a fourfold higher mortality.1 Circulating platelets develop a state of reduced responsiveness in the acute phase after major injury that is associated with poor outcomes and may contribute to the pathogenesis of TIC.2-5 Transfusion of allogeneic platelets during traumatic hemorrhage is a key component of modern resuscitation strategies and is associated with improved outcomes,6,7 but does not appear to restore platelet function during active bleeding.8,9 The contribution of platelets to TIC and the mechanisms underlying this state remain unclear, and therefore an improved understanding of the manifestations of trauma-induced changes in platelet behavior is essential for developing therapeutics to protect or restore platelet hemostatic function in bleeding patients.
The primary platelet response to vascular injury principally involves the coassociated10 glycoprotein (GP) Ib-IX-V receptor complex, for initial adhesion to exposed subendothelial collagen and von Willebrand factor11-13 and the coassociated platelet-specific collagen receptor GPVI, for subsequent platelet activation.14 The ectodomains of GPIbα15 and GPVI16 can undergo metalloproteolytic shedding. Loss of surface GPVI, in particular, has been identified as a mechanism of agonist-induced desensitization, because it can be triggered by collagen,17 fibrin,18 and factor Xa,19 resulting in a state of reduced responsiveness to collagen and other GPVI ligands. Given that widespread collagen exposure and massive activation of coagulation occur after major injury,20 GPVI loss has been proposed as a potential mediator of platelet dysfunction in TIC.21 Indeed, increased levels of shed GPVI fragments have been identified in other diseases18 ; however, to date, no studies have addressed the question of whether GPVI shedding occurs during bleeding in severely injured patients.
We therefore examined circulating platelets and platelet responses in blood samples obtained from critically injured patients on arrival at the trauma center. Our primary aim was to evaluate changes in platelet responses to collagen in severely injured patients, compared with healthy controls, and to investigate the involvement of platelet collagen receptors in altered responses. Our secondary aim was to determine how platelet-function parameters relate to coagulopathy and outcome after major injury.
Methods
Study design and setting
Patients were those who were recruited into the Activation of Coagulation and Inflammation in Trauma (ACIT) study approved by the East London and City regional ethics committee (07/Q0603/29).
Patients who met the criteria for advanced trauma team and/or major hemorrhage protocol activation were screened for inclusion. Exclusion criteria were presentation to the hospital >2 hours after injury, preadmission administration of >2000 mL crystalloid, burns involving >5% of the body surface area, known bleeding diathesis, and preinjury use of anticoagulants or antiplatelet agents, as described previously.1 Blood samples were obtained upon arrival in the emergency department, no more than 2 hours after injury, and processed immediately after collection. Because of the logistical challenges in studying platelet function in this patient population, experiments were performed in separate but overlapping patient cohorts with identical inclusion criteria and comparable clinical characteristics. Healthy volunteers who had been recruited into a separate study of platelet function testing acted as the control group (07/Q702/24).
Thromboelastometry
Rotational thromboelastometry (ROTEM delta instrument; Rotem, Leipzig, Germany) was performed with citrated whole blood after addition of tissue factor in the presence (FIBTEM) and absence (EXTEM) of the actin polymerization inhibitor cytochalasin D. The platelet contribution to each parameter was calculated by subtracting the FIBTEM value from the EXTEM value, as previously described.22 TIC was defined as a clot amplitude at 5 minutes (CA5) of <40 mm on EXTEM.23
Flow adhesion
Citrated whole blood, preincubated for 10 minutes with mepacrine (10 μM; quinacrine dihydrochloride; Sigma-Aldrich, Poole, United Kingdom), was passed through flow chambers (Ibidi, Gräfelfing, Germany) coated with type 1 (Horm) collagen (1 mg/mL; Takeda, Linz, Austria) at a shear rate of 1000/s, to mimic conditions in the human arterial circulation.24 Images were acquired with an inverted Nikon Eclipse TE-2000S microscope at 20× original magnification and analyzed with Image J software.
Platelet aggregometry
Platelet aggregation was measured in platelet-rich plasma (PRP) using the Optimul method, as described previously.25 Samples were stimulated with type 1 (Horm) collagen (0.1-30 μg/mL), collagen-related peptide (CRP-XL; 1-1000 ng/mL) purchased from Richard Farndale (Cambridge University, United Kingdom), or adenosine diphosphate (ADP; 0.1-30 μM; Labmedics, Salford, United Kingdom) for 5 minutes, mixing at 1200 rpm (Bioshake IQ, Q Instruments, Jena, Germany).
Flow cytometry
PRP, immediately or after 5 minutes of agonist stimulation, was diluted 1:1 with acid-citrate-dextrose (5 mM dextrose, 6.8 mM trisodium citrate, and 3.8 mM citric acid) before staining with anti-CD61-allophycocyanin (clone VI-PL2; eBioscience, Altrincham, United Kingdom), anti-CD62P-phycoerythrin (PE; clone AK4; Becton Dickinson [BD], Franklin Lakes, NJ), anti-CD42b-PE (clone HIP1; BD), anti-GPVI-PE (clone HY101; BD) or anti-IgG1k-PE (BD). Samples were fixed with 0.1% formalin and 10 000 platelets acquired with an LSRII flow cytometer (BD) before analysis with FlowJo v10 (TreeStar, OR).
Quantification of soluble GPVI
Soluble GPVI (sGPVI) levels in platelet-poor plasma samples from patients and healthy donors were measured in duplicate with an established sGPVI enzyme-linked immunosorbent assay.26
Data analysis
Results are reported as the mean ± standard error of the mean (SEM) or as the median with interquartile range (IQR). All analyses were performed with Prism v6.0 (GraphPad Software, La Jolla, CA) and Microsoft Excel 2017 (Microsoft, Redmond, WA). D’Agostino-Pearson normality tests were used to determine normality. Student t test or 2-way analysis of variance (ANOVA), with Šidák’s multiple comparisons test, was performed for normally distributed data. Correlations were assessed with Pearson correlation coefficients. A 2-tailed Statistical significance was set at P < .05.
Results
We obtained blood samples from severely injured patients (n = 39) with a median injury severity score of 34 (IQR, 18-41; Table 1; supplemental Table 1) and from healthy volunteers (n = 15). Samples were obtained from patients at a median of 106 minutes after injury. Fluids administered before sampling included small volumes of crystalloid (median, 0 mL; IQR, 0-450 mL) and packed red blood cells (median, 1 unit; IQR, 0-4 units), but no patients received platelet transfusion or factor concentrates before blood sampling. Platelet counts were preserved above critical levels (median, 201 × 106/mL; IQR, 156-241); however, we observed that platelets from trauma patients exhibited a profoundly reduced adherence to collagen under conditions of arterial shear (Figure 1Ai-iii). Platelet aggregation in response to collagen under stirring conditions was also reduced compared with that in healthy volunteers, even at maximal concentrations (Figure 1B). Mirroring the platelet responses to collagen, we found that platelets from trauma patients had a profoundly impaired response to the GPVI-specific agonist CRP-XL (Figure 1C). However, aggregation in response to ADP remained largely intact (Figure 1D).

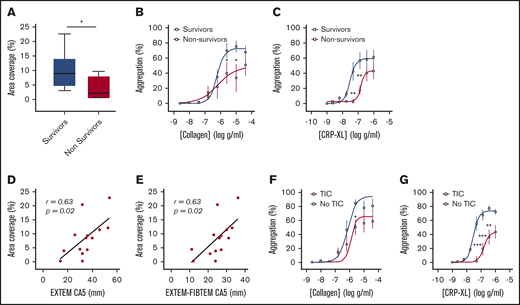
Platelet responsiveness to collagen and GPVI stimulation is reduced in severely injured patients. (A) Platelets from healthy volunteers (n = 10) or trauma patients (n = 14) were labeled with mepacrine (10 μM, green) in whole blood and perfused over collagen at arterial shear rates (1000/s). (i) Representative images after a 5-minute perfusion. Bars represent 25 μm. (ii) Area coverage over time in trauma patients and healthy volunteers. (iii) Area coverage at 5 minutes. (B-D) Platelet aggregometry in PRP from healthy volunteers (blue; n = 10) and trauma patients (red; n = 20) after a 5-minute stimulation with collagen (B), CRP-XL (C) or ADP (D) at 1200 rpm. (E) Platelet P-selectin expression (% positive) measured by flow cytometry after stimulation with CRP-XL (0.1 μg/mL) or ADP (10 μM) for 5 minutes. Two-way ANOVA with Šidák’s multiple-comparisons test (Aii,B-D) or Student t test (Aiii). *P < .05; **P < .01; ***P < .001. n.s., not significant.
Platelet responsiveness to collagen and GPVI stimulation is reduced in severely injured patients. (A) Platelets from healthy volunteers (n = 10) or trauma patients (n = 14) were labeled with mepacrine (10 μM, green) in whole blood and perfused over collagen at arterial shear rates (1000/s). (i) Representative images after a 5-minute perfusion. Bars represent 25 μm. (ii) Area coverage over time in trauma patients and healthy volunteers. (iii) Area coverage at 5 minutes. (B-D) Platelet aggregometry in PRP from healthy volunteers (blue; n = 10) and trauma patients (red; n = 20) after a 5-minute stimulation with collagen (B), CRP-XL (C) or ADP (D) at 1200 rpm. (E) Platelet P-selectin expression (% positive) measured by flow cytometry after stimulation with CRP-XL (0.1 μg/mL) or ADP (10 μM) for 5 minutes. Two-way ANOVA with Šidák’s multiple-comparisons test (Aii,B-D) or Student t test (Aiii). *P < .05; **P < .01; ***P < .001. n.s., not significant.
To evaluate this differential response to agonists further, we measured the ability of platelets to expose P-selectin in response to CRP-XL and ADP. Whereas platelets from trauma patients had higher basal levels of surface P-selectin, it was the response to CRP-XL, not to ADP, that was significantly blunted (Figure 1E). Collectively, these data demonstrate a profound impairment in the platelet response to collagen that can be accounted for by a reduction in the ability to respond via GPVI.
Consistent with previous studies,1-5 we found that patients who died of their injuries had greater impairment of platelet function in response to collagen and CRP-XL than did survivors (Figure 2A-C). In addition, area coverage on collagen was lower in patients with TIC (6.4% ± 1.9% vs 13.7% ± 3.1%; P = .06) and correlated closely with both overall clot strength on ROTEM (EXTEM CA5: r = 0.63; P = .02; n = 14; Figure 2D) and with the platelet-specific contribution to clot strength (EXTEM-FIBTEM CA5: r = 0.61; P = .02; n = 14; Figure 2E). Similarly, platelet aggregation was blunted to a greater degree in patients with TIC than in those without TIC, particularly in response to the GPVI ligand CRP-XL (Figure 2F-G).
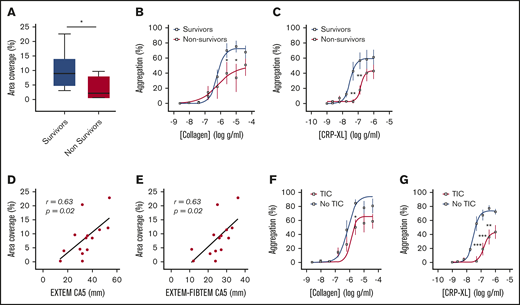
Platelet responses to collagen and CRP-XL in patients stratified by clinical outcome and presence of coagulopathy. (A) Area coverage after adhesion to type 1 collagen under flow conditions in survivors (n = 10) and nonsurvivors (n = 4). (B-C) Aggregation in response to type 1 (Horm) collagen (B) and CRP-XL (C) in survivors (n = 13) and nonsurvivors (n = 7). (D-E) Correlation between adhesion to collagen under flow and CA5 on ROTEM in the presence of tissue factor (EXTEM; D) and after subtraction of cytochalasin D assay to isolate platelet contribution to CA5 (EXTEM-FIBTEM; E). (F-G) Aggregation in response to collagen (F) and CRP-XL (G) in patients with (n = 13) and without (n = 7) TIC. Student t test (A) or 2-way ANOVA with Šidák’s multiple-comparisons test (B-C,F-G). *P < .05; **P < .01; ***P < .001.
Platelet responses to collagen and CRP-XL in patients stratified by clinical outcome and presence of coagulopathy. (A) Area coverage after adhesion to type 1 collagen under flow conditions in survivors (n = 10) and nonsurvivors (n = 4). (B-C) Aggregation in response to type 1 (Horm) collagen (B) and CRP-XL (C) in survivors (n = 13) and nonsurvivors (n = 7). (D-E) Correlation between adhesion to collagen under flow and CA5 on ROTEM in the presence of tissue factor (EXTEM; D) and after subtraction of cytochalasin D assay to isolate platelet contribution to CA5 (EXTEM-FIBTEM; E). (F-G) Aggregation in response to collagen (F) and CRP-XL (G) in patients with (n = 13) and without (n = 7) TIC. Student t test (A) or 2-way ANOVA with Šidák’s multiple-comparisons test (B-C,F-G). *P < .05; **P < .01; ***P < .001.
To investigate a potential mechanism underlying this functional deficit, we next used flow cytometry to assess the surface expression of GPVI and other platelet receptors. We observed significantly lower surface levels of GPVI on circulating platelets from trauma patients compared with healthy volunteers (Figure 3Ai). To confirm that reduced surface GPVI levels may be accounted for by shedding, we measured plasma levels of the shed ectodomain fragment of GPVI. Indeed, trauma patients had significantly higher levels of sGPVI in plasma than did healthy volunteers (Figure 3Aii). The observed reduction in GPVI levels was also mirrored by a reduction in levels of the collagen receptor GPIbα (Figure 3B), but surface expression of GPIIIa was similar to that of healthy volunteers (Figure 3C).
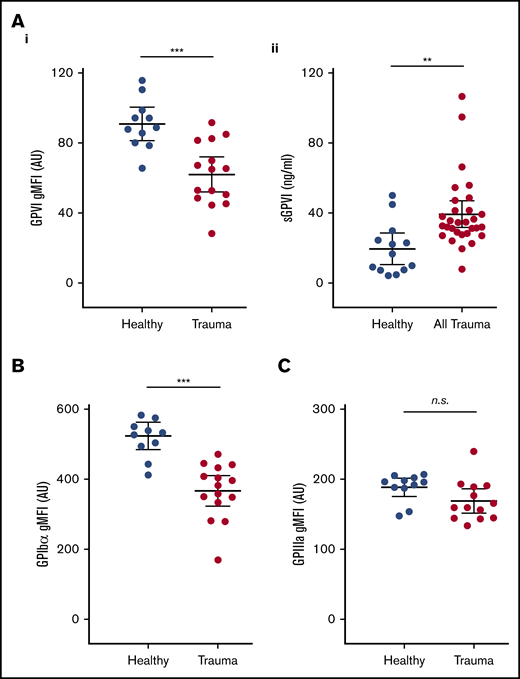
Platelet surface receptor profiles and levels of plasma sGPVI in trauma patients and healthy volunteers. (A) Surface expression of GPVI (i), and levels of sGPVI (ii), in plasma from trauma patients and healthy volunteers. (B-C) Surface expression of GPIb⍺ (B) and GPIIIa (C) on platelets measured by flow cytometry and expressed as geometric mean fluorescence intensity (gMFI). Student t test. **P < .01; ***P < .001.
Platelet surface receptor profiles and levels of plasma sGPVI in trauma patients and healthy volunteers. (A) Surface expression of GPVI (i), and levels of sGPVI (ii), in plasma from trauma patients and healthy volunteers. (B-C) Surface expression of GPIb⍺ (B) and GPIIIa (C) on platelets measured by flow cytometry and expressed as geometric mean fluorescence intensity (gMFI). Student t test. **P < .01; ***P < .001.
Levels of sGPVI were highest in patients with the greatest injury severity (Figure 4A) and were higher in patients who had sustained blunt (n = 25) as opposed to penetrating injuries (n = 5; 41.4 ± 4.3 ng/mL vs 28.5 ± 0.8 ng/mL; P = .007). Patients with TIC also had higher levels of sGPVI (Figure 4B), and we observed an inverse correlation with clot strength on ROTEM (EXTEM CA5: r = −0.419; P = .02; n = 30; Figure 4C). Moreover, as fibrin is known to trigger shedding of GPVI,18 we examined the correlation in patients with measured fibrinogen and sGPVI levels and also found a significant inverse correlation (r = −0.47; P = .04; n = 19; Figure 4D). There was no significant difference in sGPVI levels between survivors (n = 19) and nonsurvivors (n = 11; 35.1 ± 3.1 ng/mL vs 46.6 ± 8.3 ng/mL; P = .13).

sGPVI levels in trauma patients stratified by injury severity and coagulopathy. (A) sGPVI levels in patients with moderate (ISS ≤25; n = 13) and critical (ISS >25; n = 17) injuries. (B) sGPVI levels in patients with (n = 19) and without (n = 11) TIC. (C-D) Correlation between sGPVI levels and CA5 on ROTEM EXTEM (C) and plasma fibrinogen level (D). Bars indicate the mean with 95% CI. Student t test. *P < .05.
sGPVI levels in trauma patients stratified by injury severity and coagulopathy. (A) sGPVI levels in patients with moderate (ISS ≤25; n = 13) and critical (ISS >25; n = 17) injuries. (B) sGPVI levels in patients with (n = 19) and without (n = 11) TIC. (C-D) Correlation between sGPVI levels and CA5 on ROTEM EXTEM (C) and plasma fibrinogen level (D). Bars indicate the mean with 95% CI. Student t test. *P < .05.
Discussion
This study provides new insights into the characteristics of trauma-induced platelet dysfunction and the mechanisms underlying its development. Our observations demonstrate that the ability of platelets to recognize and respond to collagen is profoundly impaired during an active hemorrhage, a phenomenon that is more pronounced in patients with TIC and in those who do not survive their injuries. We have shown that levels of GPVI and GPIbα on the platelet surface are significantly reduced. In the case of GPVI, this response was related to shedding of the receptor ectodomain, translating into reduced responsiveness to stimulation with the GPVI-specific agonist CRP-XL and a blunting of the collagen response.
GPVI is a key mediator of the platelet response to vascular injury,27 by engaging with collagen and fibrin to stabilize a thrombus. Exposure of platelets to GPVI ligands results in proteolytic cleavage of the receptor by platelet metalloproteinases, leading to shedding of the ligand-binding portion of the receptor.28 GPVI signaling occurs in a receptor density–dependent manner and translates into a state of reduced responsiveness that decreases the risk of overexuberant platelet activation and the consequent inappropriate thrombus formation.29,30 However, this agonist-induced desensitization clearly may be detrimental in the setting of active bleeding where there is an ongoing need for platelets to recognize and respond to damaged blood vessels. Major tissue damage and hemorrhage result in widespread exposure of subendothelial collagen and systemic activation of coagulation, both of which are well-described triggers of GPVI shedding.16-18 Our results suggest that exposure of platelets to GPVI ligands during a major hemorrhage leads to receptor shedding and consequently impaired responses to collagen, which is associated with the development of TIC and death caused by bleeding. This conclusion is supported by the existing evidence linking elevated sGPVI levels to blood loss after cardiac surgery31 and to clinical outcomes in thermal injury18 and the finding that soluble factors in plasma from trauma patients can induce impaired responses in healthy platelets.4
GPVI shedding is caused by the proteolytic action of a disintegrin and metalloproteinase (ADAM)-10 and, to a lesser extent, ADAM17.15,17,28 These sheddases can be pharmacologically inhibited,28,32 and thus, targeting GPVI shedding represents a promising therapeutic approach to preserving hemostatic function in bleeding trauma patients. Shedding of GPIbα and GPVI is known to occur during platelet storage,33 which may explain the inability of platelet transfusions to support platelet function during a traumatic hemorrhage.8 Preventing this shedding by ADAM inhibition during storage could represent an innovative approach to providing a product for transfusion, with enhanced capacity to support hemostatic function, and warrants further study.
Given the multitude of alterations in the intravascular milieu after major injury, it is unlikely that a single mediator or pathway is responsible for alterations in platelet behavior in this patient group. TIC involves multiple simultaneous and synergistic mechanisms that combine to produce an overall phenotype of reduced clot strength.2,34 This study identifies acquired GPVI and GPIbα deficiency as one of these mechanisms, but other receptors and pathways are likely to play roles. However, because an effective collagen response is the foundation on which all other aspects of primary platelet–mediated hemostasis are built, GPVI and GPIbα loss may act as “master regulators” of platelet dysfunction in TIC. Furthermore, the negative effects of GPVI deficiency on hemostatic competence are amplified when present in combination with other defects in platelet function or coagulation,35 which reflects the complex coagulopathy present in critically injured patients.
This study is limited by its observational design and, most important, that a cause-and-effect relationship cannot be directly demonstrated. Evaluating circulating platelets in injured patients is confounded by the possibility that they differ in character from those recruited to injured sites in vivo. We also did not examine how our findings relate to clinical (as opposed to laboratory) evidence of coagulopathy. In addition, the sample size in this initial investigation does not permit detailed subgroup comparisons examining the impact of different types of injury. A larger, broader sample of trauma patients is needed to more clearly define the relationship between the phenomena described herein and the development of TIC and adverse clinical outcomes. However, our model is consistent with an extensive body of existing literature illustrating that GPVI depletion can cause a receptor density–dependent reduction in platelet responses to collagen.29,35,36 Finally, because measurement of soluble GPIbα fragments is complicated by wide variation in the circulating levels among healthy individuals, we have not definitively shown that the reduction in surface expression GPIbα is related to shedding.
In summary, we identified that loss of platelet GPVI and GPIbα occurs after trauma, particularly during TIC, and translates into a reduction in the responsiveness of platelets during active hemorrhage, which in turn is associated with reduced hemostatic competence and increased mortality. This study provides new insights into the characteristics of trauma-induced platelet dysfunction and the mechanisms underlying its development. Further study is necessary to determine whether proteolytic shedding of platelet receptors plays a causative role in TIC and whether therapeutic modulation of this process could improve outcomes of traumatic hemorrhage.
Original data are available by e-mail request to the corresponding author.
Acknowledgments
This work was supported by a fellowship grant from the Royal College of Surgeons of the United Kingdom (P.V.), the National Institute for Health Research (United Kingdom), the National Health and Medical Research Council of Australia, and the British Heart Foundation (PG/15/79/31777 [T.D.W. and P.C.A.] and PG/15/47/31591 [T.D.W. and M.V.C.]).
Authorship
Contribution: P.V., T.D.W., K.B., and P.C.A. designed the research; P.V., S.J.M., S.G., M.V.C., and L.A.C. performed the research; P.V., S.J.M., R.K.A., T.D.W., E.E.G., K.B., and P.C.A. analyzed the data; and P.V., S.J.M., E.E.G., and P.C.A. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paul C. Armstrong, Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, 4 Newark St, London E1 2AT, United Kingdom; e-mail: p.c.armstrong@qmul.ac.uk.