

Key Points
Selective activation of macrophages with agonistic anti-CD40 Ab induces the full clinical spectrum of hemophagocytic syndrome in mice.

Abstract
Hemophagocytic syndromes comprise a cluster of hyperinflammatory disorders, including hemophagocytic lymphohistiocytosis and macrophage activation syndrome. Overwhelming macrophage activation has long been considered a final common pathway in the pathophysiology of hemophagocytic syndromes leading to the characteristic cytokine storm, laboratory abnormalities, and organ injuries that define the clinical spectrum of the disease. So far, it is unknown whether primary macrophage activation alone can induce the disease phenotype. In this study, we established a novel mouse model of a hemophagocytic syndrome by treating mice with an agonistic anti-CD40 antibody (Ab). The response in wild-type mice is characterized by a cytokine storm, associated with hyperferritinemia, high soluble CD25, erythrophagocytosis, secondary endothelial activation with multiple organ vaso-occlusion, necrotizing hepatitis, and variable cytopenias. The disease is dependent on a tumor necrosis factor-α–interferon-γ–driven amplification loop. After macrophage depletion with clodronate liposomes or in mice with a macrophage-selective deletion of the CD40 gene (CD40flox/flox/LysMCre), the disease was abolished. These data provide a new preclinical model of a hemophagocytic syndrome and reinforce the key pathophysiological role of macrophages.
Introduction
Hemophagocytic syndromes are a group of rare diseases that are characterized by uncontrolled immune activation and hyperinflammation leading to a variable disease spectrum characterized by fever, cytopenias, hepatosplenomegaly, lymphadenopathy, central nervous system dysfunction, and coagulopathy.1,2 A growing number of mutations in immune-associated genes cause primary hemophagocytic syndromes or hemophagocytic lymphohistiocytosis (HLH), whereas secondary variants of the disease are related to infections, malignancies, drugs, or primary inflammatory diseases.3-5 The term macrophage activation syndrome (MAS) has been coined for secondary hemophagocytic syndromes that occur in the context of rheumatic disorders, most frequently in patients with adult-onset Still disease (AOSD) and systemic-onset juvenile idiopathic arthritis (JIA).6-8 Although underlying genetic mutations have rarely been detected in rheumatic disease–associated MAS, gain-of-function mutations of the inflammasome have been identified in some patients.9
As the terminology implies, macrophages are considered key effector cells in the disease process leading to hemophagocytic syndromes.10-13 Early reports of hemophagocytic syndromes defined phagocytosis of blood cells and their precursors by well-differentiated macrophages in the bone marrow, spleen, or liver as a histopathological hallmark feature of the diagnosis coining the term hemophagocytic syndrome.14-16 Since then, overwhelming activation of macrophages has been hypothesized as the final common pathway of all hemophagocytic syndromes leading to cytopenias, hyperinflammation with a cytokine storm as well as fever, hyperferritinemia, coagulopathy, and organ failure.17,18 However, since the discovery of loss-of-function mutations in the perforin gene and in other genes of the granule exocytosis pathway as molecular causes of primary HLH, the focus of research in the field has moved toward dysfunctional T lymphocytes and natural killer (NK) cells as inciting drivers of hemophagocytic syndromes.19-22 So far, the extent to which activation of macrophages contributes to the multiple manifestations of the clinical disease remains experimentally undefined.
The costimulatory receptor CD40 is an archetypical activation-receptor expressed on antigen-presenting cells (APCs) such as dendritic cells (DCs), macrophages, and B cells. In vivo administration of agonistic anti-CD40 antibodies mimics the effects of CD40 ligand (CD40L) on CD40-expressing cells, inducing a strong activation of innate and adaptive immunity. Agonistic anti-CD40 antibodies have been explored in cancer immunotherapy.23 In preclinical studies and clinical trials, administration of the agonistic anti-CD40 antibody was linked to acute cytokine release syndrome, with concomitant liver damage resembling secondary HLH/MAS in some patients.24,25 Based on these data, we generated the hypothesis that a new macrophage-centric mouse model of a hemophagocytic syndrome could be designed by antibody-activated CD40 signaling and that this model could be used to explore whether direct macrophage activation alone, in the absence of less cell-line–restricted Toll-like receptor (TLR) agonists or antigen persistence, could trigger the pathophysiological cascade leading to the disease-defining spectrum of manifestations.
Inflammation is induced through mutually interactive pathway crosstalk, nonlinear feed-forward mechanisms, and redundant signaling. Defining the contribution of a specific immune cell type, such as the macrophage, to a complex systemic inflammatory disease phenotype is therefore challenging. In this study, we found that treating mice with agonistic anti-CD40 antibodies induces a tumor necrosis factor-α (TNF-α)– and interferon-γ (IFN-γ)–promoted pathology, driving the typical clinical, pathological, and laboratory features of hemophagocytic syndromes. To define the role of macrophages in this disease phenotype, we generated a macrophage-specific conditional CD40-knockout mouse (LysMCre, CD40flox/flox) and found that all features of this hemophagocytic syndrome were completely abolished in the absence of CD40-expressing macrophages. Therefore, our data suggest that inflammatory macrophage activation is sufficient to trigger the full disease phenotype, reinforcing the key effector role of the phagocyte in the pathophysiology of hemophagocytic syndromes.
Methods
Animal models and experiments
Mice 8 to 10 weeks of age were used. C57BL/6J mice were obtained from Charles River (Wilmington, MA). B6.FVB-Tg(Cdh5-cre)7Mlia/J (VE-cadCre) mice were obtained from The Jackson Laboratory. B6;129S7-IFNgtm1 Ts/J (INF-γ KO), B6.129S-Tnfrsf1atm1ImxTnfrsf1btm1Imx/J (TNFR1/2 KO), B6N.129P2(B6)-Lyz2tm1(cre)Ifo/J (LysMCre), B6-Tg(CD11c-DTR-eGFP)Littm (inducible depletion of DCs), B6;129S7-Rag1tm1 Mom/J (RAG1 KO) mice were obtained from the Swiss Immunological Mouse repository (SwImMR).
The CD40 mouse strain used for this research project was created from embryonic stem cell clone EPD0901_3_A02, obtained from the Knockout Mouse Project (KOMP) Repository, and generated by the Wellcome Trust Sanger Institute (WTSI). Targeting vectors used were generated by the WTSI and the Children’s Hospital Oakland Research Institute as part of the Knockout Mouse Project (3U01HG004080). We crossed the gene-targeted CD40flox/flox mice with FLP recombinase mice that recognize the FLP recombinase target site to remove the LacZ reporter and neomycin selection cassette. The spliced exons, which are flanked by loxP sites before CD40 exon 2 and after CD40 exon 3, were removed by crossing to the LysMCre or VE-cadCre mouse, resulting in macrophage-specific or endothelial-specific CD40-knockout animals. Littermate CD40flox/flox mice were used as controls.26
Ethical approval
All experimental protocols were reviewed and approved by the Veterinary Office of the canton of Zürich. ZH202-1830747. All animals were maintained at the animal facility of the University of Zurich (LSC) and were treated in accordance with guidelines of the Swiss federal Veterinary Office.
Mouse treatments
Mice were treated intraperitoneally with 20 mg/kg agonistic anti-CD40 antibody (clone FGK4.5/FGK45; InVivoPlus Bio Cell) or saline solution as control. Blood was collected in heparin tubes by terminal heart puncture and tissues were harvested for further analysis, either 12, 24, 30, or 48 hours post–anti-CD40 administration.
Mice were treated with dexamethasone (Mephameson, 3 mg/kg intraperitoneally; Mepha Pharma) 12 and 2 hours before anti-CD40. To neutralize IFN-γ, anti-mouse IFN-γ antibody (clone XMG1.2, 0.5 mg intraperitoneally; InVivoPlus Bio Cell) was injected 48 and 2 hours before anti-CD40.
Liposomal clodronate injections
Macrophage depletion was performed with a protocol modified from Elmore et al.27 To selectively remove the macrophages, mice were given 2 intraperitoneally doses of 150 to 200 µL of clodronate liposomes (Clophosome-A-clodronate liposomes [anionic]; FormuMax), on days 1 and 3. Mice were injected with anti-CD40 48 hours after the last clodronate injection and euthanized 30 hours later.
DC depletion
To selectively remove the DCs, diphtheria toxin (DT; 4 ng/g body weight, Sigma Aldrich) was administered intraperitoneally in CD11c-DTR/GFP-transgenic mice. After 24 hours from DT injection, mice were treated with anti-CD40 and euthanized 30 hours later.
Blood analyses
Plasma transaminases.
Alanine aminotransferase (ALT) levels (Reflotron; Roche) were measured from mice plasma after anti-CD40 antibody treatment.
Blood count and LDH.
Blood analysis (leukocytes, monocytes, lymphocytes, and platelets count) and plasma lactate dehydrogenase (LDH) measurements were performed by the Veterinary Laboratory of the University of Zurich.
Plasma ferritin, CD25/IL-2R, sVCAM-1.
Blood-released ferritin (mouse ferritin enzyme-linked immunosorbent assay [ELISA] kit; ALPCO), CD25/interleukin 2 receptor α (IL-2R α) (mouse CD25/IL-2 R α DuoSet ELISA; R&D Systems), and soluble vascular cell adhesion protein 1 (sVCAM-1) (mouse sVCAM-1/CD106 ELISA Kit; R&D Biosystems) were quantified in the plasma after anti-CD40 treatment in mice.
Plasma cytokines.
The concentration of plasma cytokines and plasma intracellular adhesion molecule 1 (ICAM-1) were measured using the Bio-Plex Pro Mouse Cytokine 23-plex assay (Bio-Rad) and the Bio-Plex Pro Mouse Cytokine ICAM-1 Set, High Dilution Reagent Kit and mouse Th17 cytokine standards (Bio-Rad) according to the manufacturer’s instructions. The Bio-Plex 200 system (Bio-Rad) and Bio-Plex Data Pro software (Bio-Rad) were used for the detection and the analysis of the data.
Histology
An Olympus IX71 microscope was used for macroscopic photographs of fresh livers, spleens, and lungs. The organs were fixed in 10% formalin for 24 hours at room temperature, before embedding in paraffin. Microtome sections, 5-μm thick, were stained with hematoxylin and eosin. F4/80 immunohistochemistry staining was performed by using rat anti-mouse F4/80 (1:100; Bio-Rad) as primary antibody and biotinylated anti-rat immunoglobulin G (IgG) (1:1000; Vector Laboratories) as secondary antibody. A 3,3-diaminobenzidine substrate kit (DAB; Abcam) was used for antigen detection. The stained sections were imaged using a Zeiss Apotome.2 microscope.
Spleen single-cell suspensions
Mouse spleen single-cell suspensions were prepared by mechanical disaggregation and filtration through a 70-μm cell strainer (BD Biosciences). Red blood cells were lysed with an erythrocyte lysis buffer (155 mM NH4Cl, 100 mM KHCO3, 0.1 mM Na2 EDTA).
Liver cell isolation
Liver digestion was performed with a protocol modified from Cabral et al.28 The abdominal cavity of a living, deeply anesthetized mouse was opened, and the portal vein was catheterized for in situ liver perfusion and digestion with 50 mL of 0.06 U/mL collagenase B buffered solution (Roche). The liver was removed and the mouse was euthanized. The digested liver was mechanically disaggregated in a petri dish on ice and filtered through a 100-μm cell strainer. The cell suspension was centrifuged twice at 60g for 2 minutes at 4°C, and the pellets of hepatocytes were discarded. The supernatant was then centrifuged at 300g for 5 minutes at 4°C to obtain a pellet of nonparenchymal liver cells containing liver macrophages and endothelial cells.
Dynabeads-based F4/80+ cell enrichment
To select for F4/80+ macrophages from liver cell suspensions, Dynabeads Sheep anti-Rat IgG (Invitrogen), in combination with purified rat anti-mouse F4/80 antibody (0.5 mg/mL; BD Pharmingen) and avDynaMag-2 Magnet (Thermo Fisher Scientific) were used according to the manufacturer’s protocol (Invitrogen). The purity of the separated cells was tested by flow cytometry.
Fluorescence-activated cell sorting–based endothelial cell isolation
To isolate endothelial cells from liver cell suspensions, CD45− CD31/CD102high cells were sorted using a BD FACSAria flow cytometer.
Statistical analysis
Data plotting and statistical analysis were performed with Prism 7 (GraphPad) or JMP 13 (SAS). For comparison of multiple groups, we used unpaired Student t test analysis or analysis of variance with Tukey post hoc testing.
Results
Mice treated with an agonistic anti-CD40 antibody develop typical clinical and biochemical features of a hemophagocytic syndrome
A hemophagocytic syndrome is characterized by overwhelming systemic inflammation often leading to multiorgan injury.10,11 We treated wild-type mice with a single bolus injection of an agonistic anti-CD40 antibody. Thirty hours after administration, we observed large spots of necrotic tissue on the surface of the liver (Figure 1A). Histopathology revealed thrombotic vaso-occlusion adjacent to large areas of necrotic liver parenchyma, suggesting acute ischemic liver disease (Figure 1B). Mice exhibited splenomegaly with expansion of the white pulp reflecting CD40-induced B-cell activation and proliferation (Figures 1C and 2A). In plasma chemistry and peripheral blood counts, we found the classical changes that have been reported in patients with hemophagocytic syndromes, including elevated transaminases (ALT), LDH, soluble IL-2R (sCD25), and hyperferritinemia (Figure 1D). In peripheral blood, all mice developed cytopenias characterized by a leukopenia, accounted for by a reduction in lymphocytes and monocyte numbers, as well as low platelet counts (Figure 1D). A massive elevation of inflammatory cytokines reflecting a cytokine release syndrome was observed in plasma 12 hours after antibody administration (Figure 1E; supplemental Figure 1A).
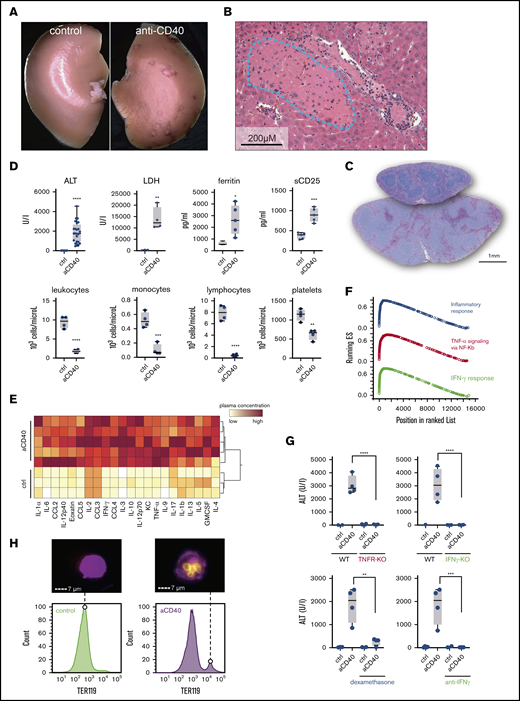
Anti-CD40–treated mice develop clinical and biochemical features of hemophagocytic syndrome. (A) Macroscopic picture of representative liver lobes of a control and anti-CD40–treated mouse at 30 hours after antibody treatment (original magnification ×1.25). Ischemic infarcts were detected in the anti-CD40 antibody-treated wild-type mouse (white spots on the liver lobe). (B) Representative photographs of hematoxylin and eosin (H&E)–stained liver tissue sections from a mouse 30 hours after antibody injection. Ischemic infarcts were detected in the area within the blue dashed line. (C) Representative photographs of H&E-stained spleen tissue sections from saline (upper section of the right panel) or anti-CD40–treated mice (bottom section of the right panel). Mice were euthanized 48 hours after antibody treatment. (D) Plasma concentrations of ALT, LDH, ferritin, sCD25 (top panels), and blood count analysis for total leukocytes, monocytes, lymphocytes, and platelet count (bottom panels) in saline (control) and anti-CD40–treated mice (n ≥ 4). (E) Hierarchical clustering analysis of cytokines and chemokines in saline or anti-CD40–treated mice, 12 hours postinjection. Plasma protein concentrations were measured by Bioplex and are color-coded. Red indicates high concentration; yellow low concentration. Each line identifies an individual saline or anti-CD40–treated animal. (F) Gene-set enrichment analysis enrichment score plots for the top 3 most positively enriched hallmark gene sets for the differentially expressed genes in F4/80+ liver macrophages of anti-CD40 antibody-treated animals vs saline-treated animals. (G) Top panels, Plasma ALT concentrations in saline (n = 2-3) or anti-CD40 antibody-treated wild-type mice, TNFR1/2 knockout mice, and IFN-γ knockout mice (n = 4). Bottom panels, Plasma ALT concentrations in wild-type mice treated with dexamethasone or neutralizing anti-IFN-γ, with or without anti-CD40 antibody (n = 2-4). (H) Left panels, Representative histograms of liver cell suspensions stained for F4/80 (macrophages) and intracellular TER119 (red blood cells) in saline or anti-CD40-treated wild-type mice. The displayed cells were gated from live CD45high leukocytes. Data are representative of 3 independent experiments. Right panels, Corresponding pictures generated by Image Stream X flow cytometer showing intracellular TER119high erythrocytes (yellow) in F4/80high macrophages (magenta). Individual symbols represent 1 mouse; ****P < .0001; ***P < .001; **P < .01; *P < .05 for all panels.
Anti-CD40–treated mice develop clinical and biochemical features of hemophagocytic syndrome. (A) Macroscopic picture of representative liver lobes of a control and anti-CD40–treated mouse at 30 hours after antibody treatment (original magnification ×1.25). Ischemic infarcts were detected in the anti-CD40 antibody-treated wild-type mouse (white spots on the liver lobe). (B) Representative photographs of hematoxylin and eosin (H&E)–stained liver tissue sections from a mouse 30 hours after antibody injection. Ischemic infarcts were detected in the area within the blue dashed line. (C) Representative photographs of H&E-stained spleen tissue sections from saline (upper section of the right panel) or anti-CD40–treated mice (bottom section of the right panel). Mice were euthanized 48 hours after antibody treatment. (D) Plasma concentrations of ALT, LDH, ferritin, sCD25 (top panels), and blood count analysis for total leukocytes, monocytes, lymphocytes, and platelet count (bottom panels) in saline (control) and anti-CD40–treated mice (n ≥ 4). (E) Hierarchical clustering analysis of cytokines and chemokines in saline or anti-CD40–treated mice, 12 hours postinjection. Plasma protein concentrations were measured by Bioplex and are color-coded. Red indicates high concentration; yellow low concentration. Each line identifies an individual saline or anti-CD40–treated animal. (F) Gene-set enrichment analysis enrichment score plots for the top 3 most positively enriched hallmark gene sets for the differentially expressed genes in F4/80+ liver macrophages of anti-CD40 antibody-treated animals vs saline-treated animals. (G) Top panels, Plasma ALT concentrations in saline (n = 2-3) or anti-CD40 antibody-treated wild-type mice, TNFR1/2 knockout mice, and IFN-γ knockout mice (n = 4). Bottom panels, Plasma ALT concentrations in wild-type mice treated with dexamethasone or neutralizing anti-IFN-γ, with or without anti-CD40 antibody (n = 2-4). (H) Left panels, Representative histograms of liver cell suspensions stained for F4/80 (macrophages) and intracellular TER119 (red blood cells) in saline or anti-CD40-treated wild-type mice. The displayed cells were gated from live CD45high leukocytes. Data are representative of 3 independent experiments. Right panels, Corresponding pictures generated by Image Stream X flow cytometer showing intracellular TER119high erythrocytes (yellow) in F4/80high macrophages (magenta). Individual symbols represent 1 mouse; ****P < .0001; ***P < .001; **P < .01; *P < .05 for all panels.

DCs and lymphocytes aggravate the disease expression. (A) Representative flow cytometric histograms of major histocompatibility complex class II (MHCII), CD80, and CD69 expression of the indicated splenic cell subsets in saline (filled blue histogram) and anti-CD40–treated (filled red histogram) wild-type mice. DCs (CD45high, CD11chigh), B cells (B220high, CD19high), T cells (CD3high, CD8high), NK cells (B220low, CD3low, NK1.1high). (B) Plasma ALT concentrations in saline (n = 2-3) or anti-CD40–treated wild-type (WT) (n = 5-8), CD11cDTg mice (n = 4-5), and RAG1KO mice (n = 8-11). (C) Schematic representation of the anti-CD40–induced hemophagocytic syndrome. Individual symbols represent 1 mouse; ****P < .0001; **P < .01 for all panels.
DCs and lymphocytes aggravate the disease expression. (A) Representative flow cytometric histograms of major histocompatibility complex class II (MHCII), CD80, and CD69 expression of the indicated splenic cell subsets in saline (filled blue histogram) and anti-CD40–treated (filled red histogram) wild-type mice. DCs (CD45high, CD11chigh), B cells (B220high, CD19high), T cells (CD3high, CD8high), NK cells (B220low, CD3low, NK1.1high). (B) Plasma ALT concentrations in saline (n = 2-3) or anti-CD40–treated wild-type (WT) (n = 5-8), CD11cDTg mice (n = 4-5), and RAG1KO mice (n = 8-11). (C) Schematic representation of the anti-CD40–induced hemophagocytic syndrome. Individual symbols represent 1 mouse; ****P < .0001; **P < .01 for all panels.
To characterize the CD40 ligation-induced macrophage response, we performed a bulk RNA-sequencing analysis of isolated F4/80+ liver macrophages that were isolated with magnetic beads from 3 anti-CD40–treated and 3 saline-treated wild-type mice. A gene-set enrichment analysis of the macrophage response to anti-CD40 treatment revealed the strongest enrichment for hallmark gene sets defining an inflammatory response, TNF signaling, and the IFN-γ response (Figure 1F; supplemental Figure 2A). To determine the role of these 2 cytokines in our model, we explored the response to anti-CD40 antibody treatment in Tnf receptor knockout (TnfR−/−) and Ifn-γ knockout (Ifnγ−/−) animals. In both knockout mice, the anti-CD40–induced disease was abolished as highlighted by the absence of liver enzyme and ferritin elevation in plasma (Figure 1G; supplemental Figure 1B,D) along with the absence of systemic inflammation (supplemental Figure 1C,E). Likewise, treatment with neutralizing anti–IFN-γ antibodies or with the broad anti-inflammatory drug dexamethasone protects mice against anti-CD40–driven pathology (Figure 1G).
Hemophagocytic macrophages in bone marrow aspirates, liver, spleen, or lymph node biopsy samples are a hallmark of activated macrophages supporting the diagnosis of a hemophagocytic syndrome.11 With imaging flow cytometry of liver cell suspensions, we identified a distinct population of F4/80+ macrophages that were also positive for ingested TER119+ erythrocytes, suggesting erythrophagocytosis (Figure 1H). However, as in most patients with a hemophagocytic syndrome, the percentages of hemophagocytes in the liver appeared to be too low to fully explain the severe cytopenias in peripheral blood. This suggests that other processes in addition to erythrophagocytosis are contributing to the hematological abnormalities such as cytokine-driven cell recruitment into tissues, acute splenic sequestration, and bone marrow suppression.
Anti-CD40 treatment results in macrophage-driven endothelial cell activation and systemic vaso-occlusive disease
Patients with hemophagocytic syndromes may also develop disseminated intravascular coagulopathy (DIC).1 Because the fibrinogen measurement for diagnosis of DIC has a low sensitivity, we performed a quantitative liquid chromatography tandem mass spectrometry analysis of plasma samples from mice treated with and without anti-CD40 antibody. Proteins identified by at least 2 unique peptides were retained for further analysis. Supplemental Figure 3A shows a volcano plot of log-transformed and mean normalized ion-intensity ratios of plasma proteins that were significantly regulated by anti-CD40 antibody treatment compared with saline-treated animals. Among the proteins with higher levels in the diseased animals, we identified serum amyloid A and several liver enzymes reflecting the massive anti-CD40–induced necroinflammation of the liver as described in Figure 1D. Among the top 20 proteins with a reduced abundance in the diseased animals, we identified coagulation factors XIII (A and B), prothrombin, and plasminogen. These results suggest that concomitant coagulation factor consumption, enhanced fibrin generation, and activation of fibrinolysis occur during anti-CD40-triggered hemophagocytic syndrome. In the context of widespread thromboses and low platelet counts these findings are consistent with DIC.
Endothelial cell activation plays a pivotal role in the initiation of coagulation and thrombotic vaso-occlusion. In our model, this process was reflected by the enhanced transcriptional expression of Vcam-1 in sorted CD45− CD31/CD102high liver endothelial cells, as well as increased VCAM-1 and ICAM1 in plasma of anti-CD40–treated mice (Figure 3A-B). Histological analysis revealed microthrombi in multiple tissue sections across the liver, spleen, and the lung, indicating the presence of widespread coagulopathy (Figure 3C-E). We used the release of liver enzymes as a surrogate marker for the multistep pathophysiology in our disease model, reflecting a sequence starting with anti-CD40–induced macrophage stimulation, endothelial cell activation leading to DIC, thrombosis formation, tissue ischemia, and necrosis. Low-level CD40 expression has also been demonstrated on endothelial cells.29 To exclude a direct anti-CD40 antibody-mediated activation of endothelial cells in our model, we generated endothelial cell–specific conditional VE-cadCre, CD40flox/flox knockout mice. The disease phenotype was not affected by the absence of CD40 expressing endothelial cells, suggesting that endothelial cell activation is a secondary response driven by activated macrophages (Figure 3F).
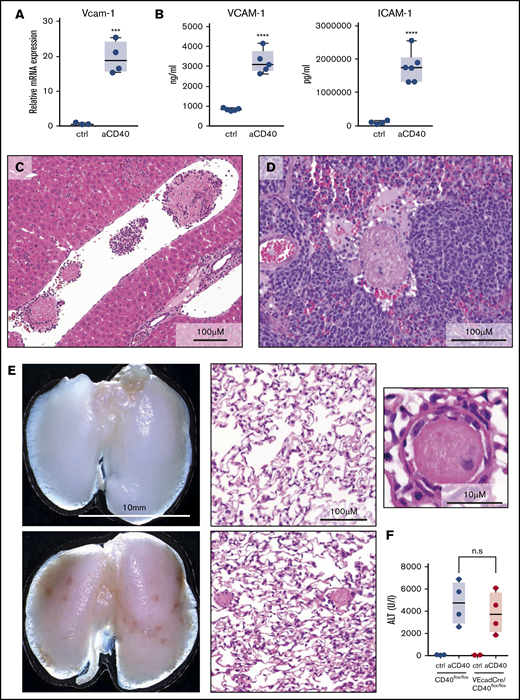
Endothelial cell activation and vaso-occlusion after anti-CD40 injection. Mice were treated with anti-CD40 antibody and euthanized 30 hours postinjection. (A) messenger RNA (mRNA) expression of Vcam-1 measured by real-time polymerase chain reaction (RT-PCR) in fluorescence-activated cell sorting (FACS)-sorted CD45low CD31/CD102high liver endothelial cells of saline (control) (n = 3) and anti-CD40–treated (n = 4) wild-type mice. (B) Left panel, VCAM1 quantification in plasma of saline (n = 5) and anti-CD40–treated (n = 5) mice. Right panel, ICAM1 quantification in plasma of saline (n = 4) and anti-CD40–treated (n = 6) mice. (C) Representative photograph of a H&E-stained liver tissue section from an anti-CD40–treated mouse. (D) Representative photograph of a H&E-stained splenic tissue section from an anti-CD40–treated mouse. (E) Macroscopic image of representative lungs of saline (top left panel) and anti-CD40–treated mice (bottom left panel). Ischemic infarcts can be detected in the anti-CD40–treated mouse lungs (dark spots on the lungs). Representative photographs of H&E-stained lung tissue sections from control (top middle panel) and anti-CD40–treated mice (bottom middle panel). A higher magnification of an intravascular thrombus from the lung section of the anti-CD40-treated mouse is appreciable in the top right panel. (F) Plasma ALT concentrations in CD40flox/flox mice (wild-type) (black dots) or VE-cadCre CD40flox/flox mice (red dots) after saline (n = 2-3) or anti-CD40 antibody treatment (n = 4). Individual symbols represent 1 mouse; ****P < .0001, *** P < .001, not significant (n.s.) P > .05, for all panels.
Endothelial cell activation and vaso-occlusion after anti-CD40 injection. Mice were treated with anti-CD40 antibody and euthanized 30 hours postinjection. (A) messenger RNA (mRNA) expression of Vcam-1 measured by real-time polymerase chain reaction (RT-PCR) in fluorescence-activated cell sorting (FACS)-sorted CD45low CD31/CD102high liver endothelial cells of saline (control) (n = 3) and anti-CD40–treated (n = 4) wild-type mice. (B) Left panel, VCAM1 quantification in plasma of saline (n = 5) and anti-CD40–treated (n = 5) mice. Right panel, ICAM1 quantification in plasma of saline (n = 4) and anti-CD40–treated (n = 6) mice. (C) Representative photograph of a H&E-stained liver tissue section from an anti-CD40–treated mouse. (D) Representative photograph of a H&E-stained splenic tissue section from an anti-CD40–treated mouse. (E) Macroscopic image of representative lungs of saline (top left panel) and anti-CD40–treated mice (bottom left panel). Ischemic infarcts can be detected in the anti-CD40–treated mouse lungs (dark spots on the lungs). Representative photographs of H&E-stained lung tissue sections from control (top middle panel) and anti-CD40–treated mice (bottom middle panel). A higher magnification of an intravascular thrombus from the lung section of the anti-CD40-treated mouse is appreciable in the top right panel. (F) Plasma ALT concentrations in CD40flox/flox mice (wild-type) (black dots) or VE-cadCre CD40flox/flox mice (red dots) after saline (n = 2-3) or anti-CD40 antibody treatment (n = 4). Individual symbols represent 1 mouse; ****P < .0001, *** P < .001, not significant (n.s.) P > .05, for all panels.
Primary activation of macrophages by CD40 ligation is sufficient to initiate a hemophagocytic syndrome
In the next step, we aimed to prove that activation of CD40 on macrophages, and not other CD40-expressing cells, drives the hemophagocytic syndrome in our model. Macrophage depletion with liposomal clodronate led to a highly efficient macrophage depletion as evidenced by the absence of F4/80+ cells in immunohistochemistry of liver and spleen (supplemental Figure 4A). This macrophage depletion completely prevented anti-CD40 antibody-induced pathology. Hierarchical clustering analysis of the expression of various inflammatory genes in whole liver tissue RNA shows a clear segregation of the gene signatures of the depleted vs the nondepleted animals, which were all treated with anti-CD40 antibody (Figure 4A). Accordingly, liver enzymes and inflammatory cytokines in plasma remained almost at baseline in the macrophage-depleted animals after anti-CD40 antibody treatment (Figure 4B-C). Moreover, in the absence of macrophages, the anti-CD40–induced direct B-cell activation is maintained (supplemental Figure 4B).
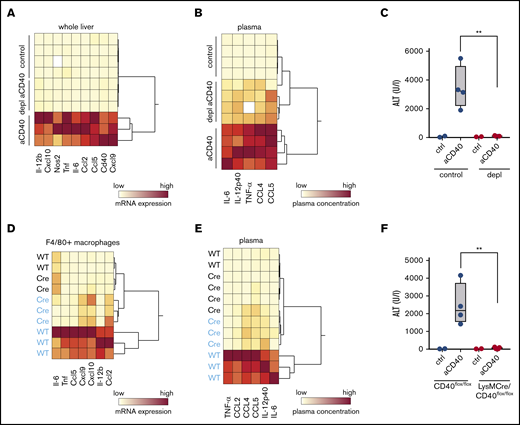
Macrophages as driver of a hemophagocytic syndrome. Mice were treated with anti-CD40 antibody and euthanized 30 hours postinjection. (A) Hierarchical clustering analysis of mRNA expression levels of proinflammatory genes in the whole liver RNA of saline and anti-CD40–treated mice, subjected or not to macrophage depletion. mRNA expression was measured by RT-PCR. Each line identifies an individual saline (control) or anti-CD40–treated animal (yellow, low concentration; red, high concentration). (B) Hierarchical clustering analysis of plasma concentrations of cytokines and chemokines in saline and anti-CD40–ttreated mice, subjected or not to macrophage depletion (yellow, low concentration; red, high concentration). (C) Plasma ALT concentrations in saline (n = 2) or anti-CD40 antibody (n = 4) injected mice, subjected (red dots) or not (black dots) to macrophage depletion. (D) Hierarchical clustering analysis of mRNA expression levels of cytokines and chemokines in F4/80+ isolated liver macrophages from saline (black) and anti-CD40 (blue)-treated CD40flox/flox (wild-type) and LysMCre CD40flox/floxmice. mRNA expression was measured by RT-PCR (yellow, low concentration; red, high concentration). (E) Hierarchical clustering analysis of plasma concentrations of cytokines and chemokines in saline (black) and anti-CD40 (blue)–treated CD40flox/flox (wild-type) and LysMCre CD40flox/flox mice (yellow, low concentration; red, high concentration). (F) Plasma ALT concentrations in saline (n = 2) or anti-CD40–treated CD40flox/flox mice (wild-type, black dots) or LysMCre CD40flox/flox mice (red dots) (n = 4). Individual symbols represent 1 mouse; **P < .01 for all panels.
Macrophages as driver of a hemophagocytic syndrome. Mice were treated with anti-CD40 antibody and euthanized 30 hours postinjection. (A) Hierarchical clustering analysis of mRNA expression levels of proinflammatory genes in the whole liver RNA of saline and anti-CD40–treated mice, subjected or not to macrophage depletion. mRNA expression was measured by RT-PCR. Each line identifies an individual saline (control) or anti-CD40–treated animal (yellow, low concentration; red, high concentration). (B) Hierarchical clustering analysis of plasma concentrations of cytokines and chemokines in saline and anti-CD40–ttreated mice, subjected or not to macrophage depletion (yellow, low concentration; red, high concentration). (C) Plasma ALT concentrations in saline (n = 2) or anti-CD40 antibody (n = 4) injected mice, subjected (red dots) or not (black dots) to macrophage depletion. (D) Hierarchical clustering analysis of mRNA expression levels of cytokines and chemokines in F4/80+ isolated liver macrophages from saline (black) and anti-CD40 (blue)-treated CD40flox/flox (wild-type) and LysMCre CD40flox/floxmice. mRNA expression was measured by RT-PCR (yellow, low concentration; red, high concentration). (E) Hierarchical clustering analysis of plasma concentrations of cytokines and chemokines in saline (black) and anti-CD40 (blue)–treated CD40flox/flox (wild-type) and LysMCre CD40flox/flox mice (yellow, low concentration; red, high concentration). (F) Plasma ALT concentrations in saline (n = 2) or anti-CD40–treated CD40flox/flox mice (wild-type, black dots) or LysMCre CD40flox/flox mice (red dots) (n = 4). Individual symbols represent 1 mouse; **P < .01 for all panels.
To ultimately validate the macrophage specificity of our model, we generated a macrophage-specific CD40 knockout mouse (LysMCre, CD40flox/flox) and confirmed the absence of Cd40 messenger RNA (mRNA) in purified F4/80+ liver macrophages by RT-PCR (supplemental Figure 4C). The size of the F4/80+ Kupffer cell population was not significantly affected by the depletion of CD40 in macrophages (supplemental Figure 4D). However, based on plasma cytokines and liver enzymes, we found no evidence of disease in LysMCre CD40flox/flox animals after treatment with anti-CD40 antibody (Figure 4E-F) and the transcriptional inflammatory response in purified liver macrophages remained suppressed in the knockout compared with wild-type liver macrophages (Figure 4D).
A DC–lymphocyte amplification loop aggravates disease expression
In the last section, we have established the essential role of macrophages in general and, specifically, of macrophage-CD40 expression as the initial driver of the anti-CD40–induced disease manifestations in mice. We assumed that other subsets of leukocytes might function as amplifiers of the disease process. Anti-CD40 antibody-treated mice demonstrated an activated phenotype of DCs, NK cells, T cells, and B cells. We revealed cell activation by enhanced expression of MHCII and costimulatory receptor CD80 on DCs and B cells, and by enhanced CD69 on T lymphocytes and NK cells in the spleen, respectively (Figure 2A). To determine to what extent DC and lymphocyte activation augmented disease expression, we used the CD11c-DTR mouse, which transiently depletes CD11c+ DCs after DT administration, and recombination activation gene-deficient mice (RAG1−/−) lacking T and B cells. In both models, the disease expression was significantly attenuated (Figure 2B) but not completely abolished. Figure 2C shows a synoptic framework of our model.
Discussion
This study provides the first description of line-selective activation of macrophages promoting key clinical and biochemical features of a hemophagocytic syndrome in mice, reinforcing the pivotal role of macrophages in this cluster of hyperinflammatory disorders. The HLH-2004 study defined fever, splenomegaly, bicytopenia, hypertriglyceridemia or hypofibrinogenemia, hemophagocytosis, hyperferritinemia, and elevation of sCD25 in plasma as the most specific features of the disease.4 Additionally, most patients display liver abnormalities including elevations of liver enzymes, coagulopathies, and elevated LDH.30 In our mouse model, we specifically searched for all of these disease manifestations and found them to be impressively present during the acute phase of the disease, except for fever and hypertriglyceridemia, which we did not measure.
In the past, understanding the role of adaptive immunity and persistent antigen stimulation became the focus of many hemophagocytic syndrome mechanistic studies.20,31 More recently, the innate immune system has come to the forefront and evidence is now emerging that hyperactivity of innate immunity may be sufficient to induce and propagate the disease. Behrens et al found that repetitive administration of CpG, a TLR9 agonist, mainly expressed by DCs, mimics the clinical picture of hemophagocytic syndrome in mice.18 Macrophages were also proposed as an important driver of hyperinflammation in a hemophagocytic model induced by TLR4 agonists in wild-type and IL-6 transgenic mice.32,33 However, in these models, the explicit function of phagocytes in the initiation and propagation of disease pathology was not specifically studied.
In contrast to other approaches of mimicking a hemophagocytic syndrome in an animal, our strategy was to use a cell line–selective exogenous trigger. The combination of exploring the effect of agonistic anti-CD40 antibody in combination with a macrophage-specific CD40 knockout mouse allowed us to prove that an initial inflammatory activation of LysM-expressing macrophages was sufficient to induce the disease. Similarly, macrophage depletion with clodronate liposomes completely prevented CD40-induced hemophagocytic syndrome. Our findings further suggest that macrophages, beyond their role in propagating hyperinflammation, are the key initiators of the pathophysiological cascade of endothelial priming, DIC, and vaso-occlusion, finally resulting in multiple organ ischemia and tissue damage.
When DCs or lymphocytes were absent in our model, the hemophagocytic syndrome was attenuated, suggesting a secondary amplification loop, which propagates hyperinflammation and organ damage downstream of the initial macrophage activation. Within this amplification loop, IFN-γ and TNF-α appear to be the central cytokine mediators. Knockout of either of the 2 signaling pathways conferred tolerance against the anti-CD40 antibody–triggered disease. This finding reinforces the nonredundant role of IFN-γ in the pathophysiology of hemophagocytic syndromes, which has been demonstrated across different disease models triggered by persistent antigen-presentation20,34 or by primary stimulation of innate immunity pathways.18,35 The critical role of interferon signaling is also reflected by the efficacy of the humanized anti–IFN-γ antibody emapalumab, which has been approved for the treatment of adult and pediatric patients with primary HLH.36,37 The role of TNF-α appears to be more controversial. In some mouse models of virus-induced hemophagocytic syndrome in perforin-deficient mice, TNF-α signaling was a critical contributor to liver damage and death,34,38 whereas other studies reported no significant role of this cytokine.20
In clinical trials, the administration of agonistic anti-CD40 antibodies to cancer patients has occasionally induced cytokine release syndrome with liver toxicity, thromboembolic complications, anemia, and thrombocytopenia.25,39-44 However, the pathophysiology of this severe complication, which ultimately limits the clinical use of anti-CD40–based immunotherapies, remained unclear. In light of our results, it is likely that these adverse effects are primarily mediated by inflammatory stimulation of CD40-expressing macrophages mirroring the clinical and biochemical features of the hemophagocytic syndrome in our mouse model. This idea is also supported by the observation that specific macrophage depletion by clodronate liposomes, macrophage deactivation by anti–colony stimulating factor 1 receptor (CSF 1R) antibody, or inhibition of TNF signaling attenuates the systemic inflammatory response and multiorgan damage induced by anti-CD40 or combined anti-CD40/IL-2 immunotherapy.39,45,46 The largest pool of macrophages resides in the liver.47 This may explain why the primary organ toxicity of CD40 ligation in our model, and in patients with hemophagocytic syndromes, occurs in the liver. Collectively, our study underlines the central role of macrophages as an essential driver of hemophagocytic syndromes.
For original data, please contact the corresponding author, Florence Vallelian, at florence.vallelian@usz.ch.
Acknowledgments
This work was supported by Innosuisse 19300.1 PFLS-L (D.J.S.), the Promedica Foundation (F.V.), the Olga Mayenfisch Stiftung (F.V.), and the Novartis Foundation (F.V.).
Authorship
Contribution: G.I. performed experiments, analyzed data, and wrote the paper; A.Y., K.V., K.H., and N.S. performed experiments; M.P. and I.L.D. performed experiments and analyzed data; C.A.S. analyzed data; R.H. designed the study, performed experiments, and designed graphical abstract; and D.J.S. and F.V. designed the study, performed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Florence Vallelian, Division of Internal Medicine, University Hospital, CH-8091 Zurich, Switzerland; e-mail: florence.vallelian@usz.ch.

