

Key Points
Bone marrow–residing tumor-specific CD4+ T cells elicit antitumor responses through interaction with bone marrow–resident macrophages.
The present results demonstrate the potential of resident macrophages as powerful mediators of tumor killing within the bone marrow.

Abstract
CD4+ T cells may induce potent antitumor immune responses through interaction with antigen-presenting cells within the tumor microenvironment. Using a murine model of multiple myeloma, we demonstrated that adoptive transfer of idiotype-specific CD4+ T cells may elicit curative responses against established multifocal myeloma in bone marrow. This finding indicates that the myeloma bone marrow niche contains antigen-presenting cells that may be rendered tumoricidal. Given the complexity of the bone marrow microenvironment, the mechanistic basis of such immunotherapeutic responses is not known. Through a functional characterization of antitumor CD4+ T-cell responses within the bone marrow microenvironment, we found that killing of myeloma cells is orchestrated by a population of bone marrow–resident CD11b+F4/80+MHC-IIHigh macrophages that have taken up and present secreted myeloma protein. The present results demonstrate the potential of resident macrophages as powerful mediators of tumor killing within the bone marrow and provide a basis for novel therapeutic strategies against multiple myeloma and other malignancies that affect the bone marrow.
Introduction
Recent advances in high-throughput methods that enable characterization of the peptide–major histocompatibility complex (MHC) ligandome have made it increasingly apparent that tumor cells express a large number of neoepitopes that form potential targets for immunotherapeutic interventions. MHC-I–restricted neoepitopes have long been the main focus of study, but new studies have revealed that mutated MHC-II epitopes are abundant and may serve as valuable immunogenic targets.1,2 Accordingly, tumor-specific CD4+ T cells have gained increased attention as valuable mediators of immune responses against cancer, and vaccination against MHC-II–restricted neoepitopes has yielded objective responses in human trials.2-4
In hematological malignancies of B-cell origin, immunoglobulin gene rearrangements lead to expression of unique and novel peptide sequences that are not encoded in the germline and thus may serve as neoantigens. Such neoantigens, referred to as idiotypic (Id) peptides, are presented on MHC-II molecules and recognized by Id-specific CD4+ T cells.5 Moreover, B-lymphoma cells spontaneously present Id peptides,6,7 and Id peptides are readily eluted from MHC-II molecules of B-cell lines.8,9 These findings were recently confirmed and extended, and a recent report has demonstrated that Id peptides are commonly presented on MHC-II of human MALT (mucosa-associated lymphoid tissue) lymphomas, whereas other neoepitopes were not clearly identified.10 Hence, the idiotypic immunoglobulin (Id) produced by malignant B cells constitutes an attractive target for tumor-specific immune responses.
Tumor-specific CD4+ T cells have been shown to mediate potent antitumor immune responses through several mechanisms, including licensing of CD8+ T cells,11 cytotoxic killing of MHC-II–expressing tumor cells,12,13 activation of macrophages14 and natural killer (NK) cells,15 and cytokine-mediated effects on tumor vasculature.16 The large number of potential modes of action emphasizes the need for careful analyses, to establish the relative contribution of each candidate mechanism.
Using the bone marrow–homing MOPC315.BM myeloma model,17 we have recently shown that adoptive transfer of Id-specific CD4+ T cells efficiently eliminates advanced-stage myeloma in immunocompetent mice.18 The therapeutic effect was preserved when using MHC-II–deficient MOPC315 myeloma cells, demonstrating that tumor killing occurs in the absence of direct recognition of tumor cells by the tumor-specific CD4+ T cells.18 Hence, we postulate that cytotoxicity is conferred in an indirect manner, involving T-cell–mediated modification of antigen-presenting cells (APCs) within the bone marrow microenvironment. Understanding the mechanistic basis of this process has potential relevance to malignancies that affect bone marrow, notably multiple myeloma, but also advanced stages of other types of cancer.
Previous work using the subcutaneously growing MOPC315 plasmacytoma cell line, which secretes an immunoglobulin A (IgA) Id (M315), has shown that CD4+ T-cell immunoprotection is dependent on M315 secretion by the tumor cells.19,20 In subcutaneously growing tumors, M315 is taken up and presented to T cells by tumor-infiltrating macrophages, resulting in activation of the macrophage upon interaction with Id-specific CD4+ T cells.14
In contrast to subcutaneous solid tumors, the bone marrow microenvironment is highly complex and includes a large number of immature and mature monocyte and leukocyte subsets with potential antigen-presenting function. To identify the effector cells responsible for CD4+ T-cell–mediated killing of myeloma cells within the bone marrow, we evaluated the in vivo role of several candidate APC subsets in CD4+ T-cell responses against MOPC315.BM. Through detailed phenotypic and functional analyses, we identified a subset of bone marrow–resident macrophages as the predominant source of display of secreted Id antigen and the key mediator of cytotoxicity.
Material and methods
Cells and cell lines
The BALB/c-derived MOPC315 plasmacytoma cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA), and the MOPC315.BM variant with a predilection for bone marrow homing was derived by serial in vivo passaging, as previously described.17 MOPC315.BM-Luc2-ZsGreen was generated by lentiviral transduction, using the bicistronic expression vector pHIV-Luc-ZsGreen, encoding firefly luciferase and the green fluorescent protein ZsGreen (generously provided by Bryan Welm, University of Utah, through the Addgene repository, plasmid 39196). Details of the transduction procedure have been published.21 Naive Id-specific CD4+ T cells were isolated by negative selection, using the CD4+ T-Cell Isolation Kit II (Miltenyi Biotech, GmbH) according to the manufacturer’s instructions. Activated Id-specific CD4+ T cells were obtained by in vitro activation and Th1 polarization and expansion, as previously described.22,23 The following magnetic bead–based isolation kits were used according to the manufacturer’s instructions (Miltenyi Biotech, GmbH) on single-cell suspensions from bone marrow, isolated from flushed femurs and using a stainless-steel sieve, to negatively select neutrophils, NK cells, and dendritic cells: a mouse Neutrophil Isolation Kit (cat. no. 130-097-658); an NK Cell Isolation Kit (cat. no. 130-115-818), and a Pan Dendritic Cell Isolation Kit (cat. no. 130-100-875). Similarly, the following magnetic bead–based isolation kits were used according to the manufacturer’s instructions to positively select macrophages and eosinophils: CD11b microbeads (cat. no. 130-049-6019) and anti-Siglec-F microbeads (cat. no. 130-118-513). To obtain Asialo GM1+CD11b+ cells, MOPC315.BM-challenged TCR-Tg BALB/c mice were injected with 50 µg rabbit anti-asialo-GM1 (Poly21460; BioLegend) intraperitoneally (IP). Three hours after antibody treatment, single-cell suspensions from the bone marrow were obtained by flushing femurs of mice with phosphate-buffered saline (PBS) and passing the contents through a stainless-steel sieve. Cells were stained with fluorescein isothiocyanate–conjugated anti-rabbit (to detect Asialo GM1 bound to the surface of cells) and anti-CD11b. Asialo GM1+CD11b+ cells were isolated by fluorescence-activated cell sorting (FACS).
All cells were maintained in RPMI 1640 GlutaMAX (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (Greiner Bio-one GmbH, Frickenhausen, Germany) and penicillin/streptomycin.
In vitro JAM coculture assays
To analyze the cytotoxic potential of different cells within the bone marrow, 104 MOPC315.BM tumor cells were incubated with [3H]-methyl-thymidine in overnight culture. The methyl-thymidine–labeled MOPC315.BM cells were washed carefully with PBS to remove nonincorporated [3H]-methyl-thymidine, before they were cocultured at various effector:target ratios with titrated numbers of Id-specific CD4+ T cells, eosinophils, NK cells, dendritic cells, macrophages or eosinophils. Some experiments were performed by also adding 104 Id-specific CD4+ T cells or synthetic Id antibody (amino acids 88-103; 4 µg/mL), or both, for analysis of cytotoxic potential when cognate interaction with CD4+ T cells is possible. After 6 hours of coculture, the cells were harvested on a TopCount NXT microplate counter (Perkin Elmer).
Mice and in vivo experiments
BALB/c, BALB/c Rag1−/− and Rag2−/−IL2rgc−/− mice (C;129S4-Rag2tm1.1Flv Il2rgtm1.1Flv/J) on a BALB/c background purchased from Taconic (Taconic Farms, Rye, Denmark). GATA1-deficient mice on a BALB/c background (C.129S1(B6)-Gata1tm6Sho/J) were obtained from the Jackson Laboratory (Bar Harbor, ME). Homozygous Id-specific T-cell receptor transgenic (TCR-Tg) BALB/c mice were generated and maintained on a BALB/c or BALB/c Rag1−/− background, as previously described.23 Four- to 8-week-old mice were injected IV with 1 × 106 tumor cells suspended in 100 μL PBS. Tumor growth was observed by bioluminescence imaging with a Xenogen IVIS Spectrum In Vivo Imaging System (Perkin Elmer). For imaging, mice were anesthetized using 2% isoflurane, and injected with d-luciferin substrate (150 mg/kg body weight; BioVision Inc, Milpitas, CA), as previously described.17 The threshold of the bioluminescent signal was automatically determined with Living Image software (Perkin Elmer) and was quantified as photons per second per square centimeter per steradian. Adoptive transfer was performed by IV injection of Id-specific CD4+ cells suspended in 100 µL PBS. Wild-type BALB/c mice were sublethally irradiated (500 cGy) with an RS320 X-ray irradiator (X-strahl Ltd, Camberley, England) 24 hours before T-cell transfer. The sphingosine analogue fingolimod (FTY720, 2-amino-2-propane-1,3-diol hydrochloride) was obtained from SelleckChem (Munich, Germany). Mice were treated with daily IP injections of 2 μg/g bodyweight of FTY720 or vehicle only (0.8% dimethyl sulfoxide; Sigma-Aldrich), starting 15 days before the tumor challenge. Administration of neutralizing antibodies was performed by IP injection with the following dosage regimens: rabbit anti-asialo-GM1 (Poly21460; BioLegend), 50 µg per mouse injected every 3 days beginning on day 3 before the tumor challenge; rat anti-Ly6G/Ly6C (RB6-8C5; BioXCell), 200 µg per mouse injected on days 3 and 1 before the tumor challenge, then every day after the tumor challenge; rat anti-Ly6G (1A8; BioXCell), 200 µg per mouse injected on days 3 and 1 before the tumor challenge, then every day after the challenge; and rat anti-IFN-γ mAb (XMG1.2, BioXcell), 100 µg per mouse injected on days 1 and 4 or isotype-matched control monoclonal antibody (mAb; 187.1) 100 μg per mouse injected on days 1 and 4. The commercially available kit Mouse IFN-γ ELISA MAX Deluxe (BioLegend) was used according to the manufacturer’s instructions to analyze the levels of IFN-γ within the femur of MOPC315.BM-challenged TCR-Tg or wild-type mice.
Experiments were performed according to institutional and governmental guidelines and were approved by the National Committee for Animal Experiments (Oslo, Norway).
Antibodies and flow cytometry
The following commercially available antibodies were used in dye-conjugated formats: F4/80 (BM8), Ly6C (HK1.4), CD64 (FcγRI; X54-5/7.1), mouse IgG1-κ (MOPC-21), rat IgG2a-κ (RTK2758), MHC-II I-A/E (M5.114.15.2), ultra-LEAF purified anti-asialo-GM1 antibody (Poly21460), and rabbit polyclonal IgG (Poly29108; BioLegend); CD80 (1G10) and CD86 (GL1; Southern Biotech); CD3 (17A2), CD4 (GK1.5), CD8a (53-6.7), CD11b (M1/70), CD11c (HL3), rat IgG2b-κ (A95-1), and hamster IgG2-κ (B81-3; BD Biosciences); and CD49b (DX5; eBioscience). The antibodies anti-FcγRII/III (2.4G2; ATCC) and anti-Id–specific TCR clonotype (GB113) were affinity purified and, if necessary, biotinylated. To detect biotinylated mAbs, we used streptavidin conjugated to peridinin chlorophyll protein (BD Biosciences).
The cell-permeable, fluorescent indicator reagent DAF-FM diacetate (DAF-FM; D23844; Thermo Fisher) was used as a stain for nitric oxide measurements, according to the manufacturer’s instructions.
The IEd/L2315-specific TCR mimetic (TCRm) was obtained from a murine single-chain variable fragment phage display library (M.F., manuscript in preparation), produced as a C-terminal 6xHis-tagged protein by periplasmic expression in Escherichia coli, purified by sequential immobilized metal-ion–affinity chromatography and protein l-sepharose affinity chromatography, and biotinylated.
Nonspecific binding was blocked by incubation with heat-inactivated (56°C, 30 minutes) 0.5% normal rat serum in PBS and 100 μg/mL−1 anti-FcγRII/III mAbs (clone 2.4G2). Cells were stained for 15 minutes on ice with specific mAbs in PBS supplemented with 0.5% bovine serum albumin (BioRad). Cells were analyzed on a Fortessa instrument with FACSDiva software (BD Biosciences).
Cytospin
Cytospin preparations of FACS-identified CD11b+CD64+F4/80+MHC-II+ macrophages were fixated by overnight incubation in a 37°C warm room before a 1-minute incubation in methanol and stained by 3 to 4 short immersions in May-Grünwald stain and then a 45-second immersion in Giemsa stain. The cytospin preparations were washed in distilled water and dried. The microscope Nikon Eclipse E 800 and the AnalySIS Soft Imaging System were used to analyze the sections and acquire and process digital images.
Statistics
The Mann-Whitney U test was used for statistical analysis unless stated otherwise. For tumor challenge experiments, differences in survival were analyzed by using the log-rank test. Statistical analysis was performed with Prism 5.0 software (GraphPad Software, La Jolla, CA). Statistical significance was set at P < .05.
Results
Adoptive transfer of Id-specific CD4+ T cells from TCR-Tg (Id+ TCR-Tg) mice induced elimination of established, multifocal MOPC315.BM myeloma in the bone marrow (Figure 1A-B and Haabeth et al18 ). Upon IV challenge, MOPC315.BM growth was observed primarily within the proximal femur and the lumbar spine.18 Similarly, Id+ TCR-Tg mice were protected against IV tumor challenge (Figure 1B).

The bone marrow constitutes a functional secondary lymphoid organ. (A) M315 Id levels in mice challenged IV with 1 × 106 MOPC315.BM cells, as determined by ELISA. Adoptive T-cell therapy (ACT)–treated mice were injected with 2 × 106 Id-spec. TCR-Tg CD4+ T cells on day 18 after the tumor challenge, whereas controls did not receive T cells. Results are shown as means ± standard deviation (SD) (n = 8 mice per treatment group). The dotted line reflects the lower limit of detection (LLD) in ELISA. (B) Survival of mice treated as in panel A, with the addition of a TCR-Tg control group (n = 8 mice per treatment group). (C) Bioluminescence imaging of wild-type BALB/c and TCR-Tg BALB/c mice challenged subcutaneously with 2 × 105 and IV with 1 × 106 MOPC315.BM-Luc cells on the indicated days after tumor cell injection. All mice received a daily IP injection of fingolimod (FTY720) for the duration of the experiment. Red arrows indicate the subcutaneous tumor injection site. (D) Flow cytometry quantitation of Id-specific (GB113+) CD4+ T cells within the femoral bone marrow of nonchallenged mice or IV MOPC315.BM tumor challenged mice receiving daily injections of FTY720 or NaCl. Results are shown as means ± SD (n = 5-8 per treatment group). (E) M315 Id levels of wild-type BALB/c and TCR-Tg BALB/c mice challenged IV with 1 × 106 MOPC315.BM and receiving daily IP injections of FTY720 or NaCl. Results are shown as means ± SD. The dotted line reflects the LLD in the ELISA. (F) Dorsal whole-body bioluminescence quantitation of tumor burden in tumor-challenged mice, as described in panel E. Results are shown as means ± SD.
The bone marrow constitutes a functional secondary lymphoid organ. (A) M315 Id levels in mice challenged IV with 1 × 106 MOPC315.BM cells, as determined by ELISA. Adoptive T-cell therapy (ACT)–treated mice were injected with 2 × 106 Id-spec. TCR-Tg CD4+ T cells on day 18 after the tumor challenge, whereas controls did not receive T cells. Results are shown as means ± standard deviation (SD) (n = 8 mice per treatment group). The dotted line reflects the lower limit of detection (LLD) in ELISA. (B) Survival of mice treated as in panel A, with the addition of a TCR-Tg control group (n = 8 mice per treatment group). (C) Bioluminescence imaging of wild-type BALB/c and TCR-Tg BALB/c mice challenged subcutaneously with 2 × 105 and IV with 1 × 106 MOPC315.BM-Luc cells on the indicated days after tumor cell injection. All mice received a daily IP injection of fingolimod (FTY720) for the duration of the experiment. Red arrows indicate the subcutaneous tumor injection site. (D) Flow cytometry quantitation of Id-specific (GB113+) CD4+ T cells within the femoral bone marrow of nonchallenged mice or IV MOPC315.BM tumor challenged mice receiving daily injections of FTY720 or NaCl. Results are shown as means ± SD (n = 5-8 per treatment group). (E) M315 Id levels of wild-type BALB/c and TCR-Tg BALB/c mice challenged IV with 1 × 106 MOPC315.BM and receiving daily IP injections of FTY720 or NaCl. Results are shown as means ± SD. The dotted line reflects the LLD in the ELISA. (F) Dorsal whole-body bioluminescence quantitation of tumor burden in tumor-challenged mice, as described in panel E. Results are shown as means ± SD.
To further understand the mechanistic basis of antitumor immune responses within the bone marrow, we wanted to test whether a CD4+ T-cell response against bone marrow–homing myeloma cells is dependent on the efflux of T cells from secondary lymphoid organs. To this end, we treated TCR-Tg mice with daily injections of the sphingosine analogue fingolimod (FTY720), which blocks lymphocyte egress from spleen and lymph nodes.24 In line with a previous report,25 fingolimod treatment completely abrogated immunoprotection against subcutaneously injected MOPC315 cells, whereas IV-injected, bone marrow–homing MOPC315.BM cells were still eliminated (Figure 1C).
In line with these findings, accumulation of Id-specific (GB113 clonotype-reactive)26 TCR-Tg cells in affected bone marrow was comparable in FTY720- and mock-treated TCR-Tg mice after IV MOPC315.BM tumor challenge (Figure 1D), despite a dramatic decrease in blood levels of CD4+ and CD8+ T cells after FTY720 treatment (supplemental Figure 1). M315 Id levels remained undetectable in FTY720- and mock-treated TCR-Tg mice challenged with MOPC315 (Figure 1E), and bioluminescence imaging confirmed that immunoprotection against IV-challenged mice was intact in TCR-Tg mice in the presence of FTY720 (Figure 1F).
To further verify the functionality of bone marrow–derived APCs in initiating CD4+ T-cell responses, we isolated CD11b+ and CD11c+ cells from the femur of IV tumor-challenged TCR-Tg and wild-type mice and found that both subsets had the ability to induce in vitro proliferation of naive Id-specific CD4+ T cells when derived from TCR-Tg but not nontransgenic hosts (supplemental Figure 1B).
In summary, the data indicate that the bone marrow microenvironment contains the constituents required for the priming and effector stages of a primary CD4+ T-cell response and thus constitutes a functional secondary lymphoid organ.
IFN-γ has been shown to be an important effector of CD4+ T-cell responses in various subcutaneous tumor models. By enzyme-linked immunosorbent assays (ELISAs), we found a significant increase in IFN-γ levels within the femur of MOPC315.BM-challenged TCR-Tg mice compared with wild-type controls (Figure 2A). To evaluate the importance of IFN-γ in immunoprotection, an IV tumor challenge was performed in mice treated with a neutralizing mAb against IFN-γ. Strikingly, blocking IFN-γ resulted in a complete loss of immunoprotection in MOPC315.BM-challenged TCR-Tg mice (Figure 2B-C).
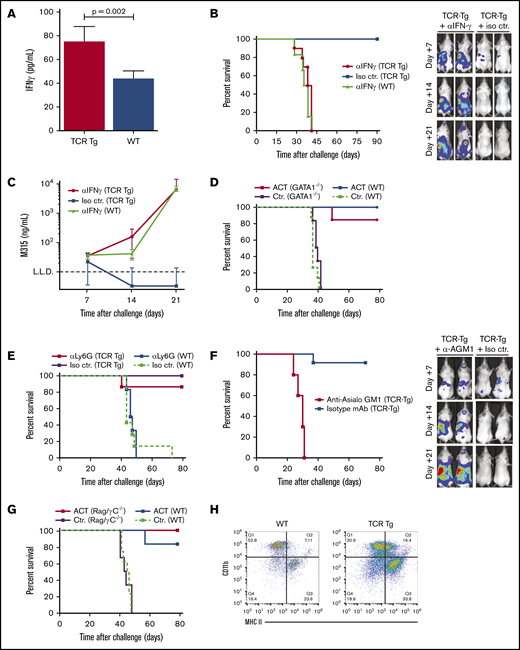
Myeloma killing by CD4+T cells is dependent on IFN-γ and depends on asialo-GM1–expressing effector cells. (A) ELISA quantitation of IFN-γ levels in the cell-free fraction of the tumor-infiltrated femoral bone marrow of TCR-Tg or wild-type BALB/c mice on day 12 after IV challenge with 1 × 106 MOPC315.BM-luc cells (means ± SD; n = 6 mice per treatment group). (B) Survival of wild-type (WT) and TCR-Tg BALB/c mice challenged IV with 1 × 106 MOPC315.BM-Luc cells. Mice were treated every 3 days with IP injection of 200 μg of a blocking mAb against IFN-γ (aIFNg) or isotype control mAb (Iso ctr.) as indicated, starting 1 day before tumor cell injection (n = 8 mice per treatment group). Representative dorsal bioluminescence imaging data for TCR-Tg mice treated with aIFNg or isotype mAb is shown on the right. (C) Serum M315 Id levels from experiment described in panel B. (D) Survival of GATA1−/− BALB/c (GATA1−/−) and wild-type BALB/c (WT) mice challenged IV with 1 × 106 MOPC315.BM cells and treated by adoptive T-cell therapy. Mice were irradiated (500 cGy) on day 18 after tumor challenge and injected IV with 2 × 106 Id-specific CD4+TCR-Tg cells (ACT) or irrelevant, polyclonal CD4+ T cells (Ctr.) the next day (n = 6 mice per treatment group). (E) Survival of TCR-Tg and wild-type BALB/c (WT) IV challenged with 1 × 106 MOPC315.BM cells. Mice received IP injections of 200 μg anti-Ly6C mAb 1A8 (aLy6G) or isotype control mAb (Iso ctr.) every other day, starting 5 days before tumor challenge (n = 6 mice per treatment group). (F) Survival curves and representative bioluminescence imaging data for TCR-Tg BALB/c mice challenged IV with 1 × 106 MOPC315.BM-Luc cells. Mice were treated every 3 days with IP injection of a blocking mAb against asialoGM1 (a-AGM1) or isotype control mAb (Iso ctr.) as indicated, starting 1 day before tumor cell injection (n = 8 mice per treatment group). (G) Survival of IV MOPC315.BM tumor-challenged wild-type BALB/c and BALB/c Rag−/− IL2rgc−/− (Rag/yC−/−) mice treated with 2 × 106 Id-specific CD4+TCR-Tg cells (ACT) or irrelevant, polyclonal CD4+ T cells (Ctr.) on day 18 after tumor cell injection (n = 6 mice per treatment group). (H) Representative flow cytometry staining of asialo-GM1+ cells isolated from femoral bone marrow cells harvested from TCR-Tg or nontransgenic BALB/c (WT) mice on day 5 after IV challenge with 1 × 106 MOPC315.BM cells. Cells were isolated by incubation with anti-asialo GM1 antibody, followed by positive selection by anti-rabbit IgG MicroBeads.
Myeloma killing by CD4+T cells is dependent on IFN-γ and depends on asialo-GM1–expressing effector cells. (A) ELISA quantitation of IFN-γ levels in the cell-free fraction of the tumor-infiltrated femoral bone marrow of TCR-Tg or wild-type BALB/c mice on day 12 after IV challenge with 1 × 106 MOPC315.BM-luc cells (means ± SD; n = 6 mice per treatment group). (B) Survival of wild-type (WT) and TCR-Tg BALB/c mice challenged IV with 1 × 106 MOPC315.BM-Luc cells. Mice were treated every 3 days with IP injection of 200 μg of a blocking mAb against IFN-γ (aIFNg) or isotype control mAb (Iso ctr.) as indicated, starting 1 day before tumor cell injection (n = 8 mice per treatment group). Representative dorsal bioluminescence imaging data for TCR-Tg mice treated with aIFNg or isotype mAb is shown on the right. (C) Serum M315 Id levels from experiment described in panel B. (D) Survival of GATA1−/− BALB/c (GATA1−/−) and wild-type BALB/c (WT) mice challenged IV with 1 × 106 MOPC315.BM cells and treated by adoptive T-cell therapy. Mice were irradiated (500 cGy) on day 18 after tumor challenge and injected IV with 2 × 106 Id-specific CD4+TCR-Tg cells (ACT) or irrelevant, polyclonal CD4+ T cells (Ctr.) the next day (n = 6 mice per treatment group). (E) Survival of TCR-Tg and wild-type BALB/c (WT) IV challenged with 1 × 106 MOPC315.BM cells. Mice received IP injections of 200 μg anti-Ly6C mAb 1A8 (aLy6G) or isotype control mAb (Iso ctr.) every other day, starting 5 days before tumor challenge (n = 6 mice per treatment group). (F) Survival curves and representative bioluminescence imaging data for TCR-Tg BALB/c mice challenged IV with 1 × 106 MOPC315.BM-Luc cells. Mice were treated every 3 days with IP injection of a blocking mAb against asialoGM1 (a-AGM1) or isotype control mAb (Iso ctr.) as indicated, starting 1 day before tumor cell injection (n = 8 mice per treatment group). (G) Survival of IV MOPC315.BM tumor-challenged wild-type BALB/c and BALB/c Rag−/− IL2rgc−/− (Rag/yC−/−) mice treated with 2 × 106 Id-specific CD4+TCR-Tg cells (ACT) or irrelevant, polyclonal CD4+ T cells (Ctr.) on day 18 after tumor cell injection (n = 6 mice per treatment group). (H) Representative flow cytometry staining of asialo-GM1+ cells isolated from femoral bone marrow cells harvested from TCR-Tg or nontransgenic BALB/c (WT) mice on day 5 after IV challenge with 1 × 106 MOPC315.BM cells. Cells were isolated by incubation with anti-asialo GM1 antibody, followed by positive selection by anti-rabbit IgG MicroBeads.
We next proceeded to identify the APC subset responsible for MOPC315.BM tumor killing within the bone marrow. Given the multitude of immature monocyte and leukocyte subsets within this compartment, clear-cut identification of effector subsets involved in CD4+ T-cell–mediated tumor killing is complicated. In vitro tumor-killing assays in ex vivo–sorted candidate APC subsets from bone marrow of TCR-Tg mice revealed that NK cells, eosinophils, neutrophils, macrophages, and dendritic cells showed potent cytotoxic effects that was enhanced in the presence of Id-specific CD4+ T cells (supplemental Figure 2A; data not shown).
To establish whether eosinophils play a role in tumor elimination in vivo, we performed a tumor challenge and adoptive transfer of Id-specific CD4+ T cells in GATA1−/− mice, which are devoid of cells of the eosinophil lineage.27 In line with previous data, preconditioning by sublethal irradiation was required for efficient homeostatic expansion and engraftment of the transferred T cells in immunocompetent hosts.18 GATA1-deficient mice developed tumors with kinetics similar to those in wild-type mice, whereas transfer of Id-specific T cells resulted in complete protection against tumor development (Figure 2D). Similarly, neutrophil depletion with an anti-Ly6G mAb (clone 1A8) did not interfere with immunoprotection in Id+TCR-Tg mice (Figure 2E). In adoptive transfer experiments, mice that had rejected tumors were protected against rechallenge with MOPC315.BM cells via the IV route, indicating the formation of memory T cells (data not shown).
To evaluate the role of bone marrow NK cells, we treated Id+TCR-Tg mice with a polyclonal rabbit anti-asialo GM1 (AGM1) antibody, which is widely recognized as an efficient means of NK cell depletion.28 Upon subsequent tumor challenge, protection against tumor development was completely abrogated, with tumors developing within the bone marrow and spleen with the same kinetics as in untreated wild-type BALB/c controls (Figure 2F). To directly assess the role of the NK cell subset, we performed adoptive transfer of Id-specific CD4+ T cells in tumor-challenged RAG2−/−γc−/− double-mutant mice, which are devoid of T, B, NKT, and NK cells. Surprisingly, double-mutant mice receiving Id-specific CD4+ T cells remained completely protected against tumor development (Figure 2G), suggesting that the effect of the anti-asialo GM1 treatment was mediated by depletion of another cell type. Indeed, reactivity of anti-AGM1 mAbs against other cell types (notably, activated macrophages) is reported in the literature.29 To identify the asialo-GM1–expressing cells involved in tumor killing, we therefore performed a magnetic bead–based isolation of anti-AGM1–reactive cells from the bone marrow of tumor-challenged TCR-Tg mice. Flow cytometry revealed that the resulting cell pool contained a mixture of NK cells (SSCloCD49b+) and CD11b+ cells (SSCInt/HiCD11b+; supplemental Figure 2B). In the CD11b+ fraction, there was an increased fraction of MHC-IIHigh cells in tumor-challenged TCR-Tg mice compared with nontransgenic controls (Figure 2H), reflecting increased activation.
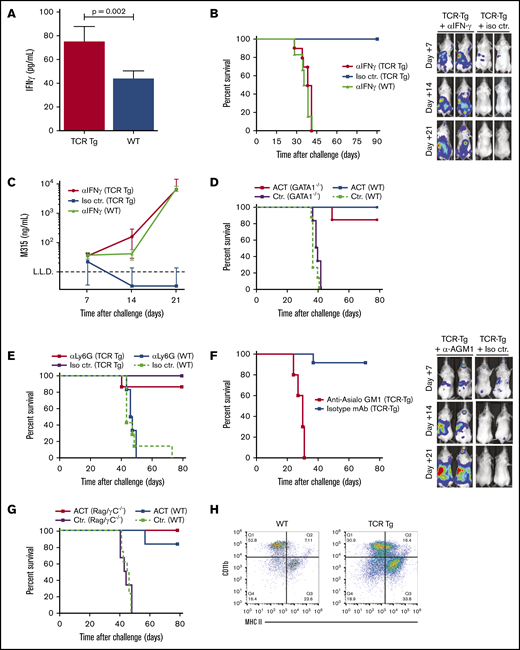
Based on these findings, we turned our focus to bone marrow macrophage subsets. A population of CD11b+CD64+F4/80+MHC-II+ macrophages, constituting ∼15% of CD11b+ cells in the bone marrow of tumor-challenged mice, was found to be significantly reduced by anti-AGM1 treatment (Figure 3A). Cytospin preparation of sorted cells revealed a macrophagelike appearance (supplemental Figure 2C). We next assayed the cytotoxicity of F4/80+ and F4/80− subsets of CD11b+CD64+ cells isolated ex vivo from tumor-challenged TCR-Tg and wild-type mice. CD11b+ cells from TCR-Tg mice showed tumoricidal effects against MOPC315.BM, significantly more pronounced in the F4/80+ fraction. In contrast, both the F4/80+ and F4/80− subsets from wild-type mice showed growth-promoting effects (Figure 3B). As a direct indicator of macrophage activation status, we used the fluorescent nitric oxide indicator DAF-FM, indicative of activated macrophages with an M1-like polarization. Assays of CD11b+ cells from bone marrow of tumor-challenged TCR-Tg and wild-type mice revealed a distinct subset of DAF-FM+ cells that was seen only in TCR-Tg mice (Figure 3C). These cells expressed high levels of MHC-II and the costimulatory receptors CD80 and CD86, as well as AGM1 (Figure 3C).
![CD4+ T-cell–mediated killing of myeloma is mediated by bone marrow macrophages. (A) Flow cytometry quantitation of F4/80+CD64+ macrophages in femoral bone marrow isolated from mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells. Mice were treated with an anti-asialo-GM1 (a-AGM1) or isotype control (Is ctr.) mAb. Gating strategy is shown on the left. The histogram shows means ± SD for 6 mice per treatment group; **P < .01. (B) In vitro coculture of MOPC.BM cells with ex vivo–isolated F4/80+CD64+ macrophages from the experiment described in panel A; effector/target ratio, 10:1. Results are plotted as tumor cell growth of a percentage of tumor cells cultured alone, calculated on the basis of 18-hour [3H]-thymidine release (n = 6-12 per treatment group, mean ± SD; ***P < .001). (C) Flow cytometry, with the nitric oxide–reactive fluorescent dye DAF-FM diacetate. Dot plots show representative results for staining of cells isolated from femoral bone marrow of TCR-Tg and wild-type (WT) BALB/c mice on day 7 after tumor challenge with 1 × 106 MOPC315.BM. Histograms show staining for costimulatory receptors CD80/86, asialo-GM1, and MHC -II on the DAF-FMHigh population found in TCR-Tg mice. (D) Representative flow cytometry staining using a pId315:I-Ed peptide/MHC-II–specific single-chain variable fragment (TCRm). Bone marrow cells were isolated from TCR-Tg and wild-type (WT) mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells, with a rabbit anti-asialo-GM1 antibody, followed by magnetic bead separation. Nonchallenged TCR-Tg mouse bone marrow was used as a control. Dorsal whole-body bioluminescence staining (E) and survival (F) for TCR-Tg and WT BALB/c mice challenged with 1 × 106 MOPC315.BM. Mice received daily injections of 200 μg depleting mAb against Ly6G/C (RB6-8C5) or a relevant isotype control mAb, starting 4 days before tumor challenge (n = 6 to 8 per treatment group). Bioluminescence data are shown as means ± SD of total flux.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/12/10.1182_bloodadvances.2020001434/5/m_advancesadv2020001434f3.png?Expires=1770184080&Signature=vXWm~ugPoiKZ4eMBIfZ1I0aUFHiDpEqnmDhMB5lVLh0mf8hqfrUFL2~rb1d0qpdmQJ9tZw7WHfS8rvVCikAtHZp0bmzf5QXa2efWx945mSzqNftjkqKrhu5AMLLr1fC3zNclPET-3nFBYWK9gshVBY-jscXsgPXt3L-Hnk-s-bjXP6X2JFQFXvrp~5GgYBvB5KoH2nJI219nvw~3v9FVgDloHSa1xlKxlwaD7CvwnKGVN3Zd6fUDNSCKqhungDLTriX6~SDBIsxwsGKPNVT~G8iujd2pJFj-5~eFe7Z41eAijg8fevK1Q4E8deUaqhCEMqJYIc7XNPXb~I-nESFuHQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CD4+ T-cell–mediated killing of myeloma is mediated by bone marrow macrophages. (A) Flow cytometry quantitation of F4/80+CD64+ macrophages in femoral bone marrow isolated from mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells. Mice were treated with an anti-asialo-GM1 (a-AGM1) or isotype control (Is ctr.) mAb. Gating strategy is shown on the left. The histogram shows means ± SD for 6 mice per treatment group; **P < .01. (B) In vitro coculture of MOPC.BM cells with ex vivo–isolated F4/80+CD64+ macrophages from the experiment described in panel A; effector/target ratio, 10:1. Results are plotted as tumor cell growth of a percentage of tumor cells cultured alone, calculated on the basis of 18-hour [3H]-thymidine release (n = 6-12 per treatment group, mean ± SD; ***P < .001). (C) Flow cytometry, with the nitric oxide–reactive fluorescent dye DAF-FM diacetate. Dot plots show representative results for staining of cells isolated from femoral bone marrow of TCR-Tg and wild-type (WT) BALB/c mice on day 7 after tumor challenge with 1 × 106 MOPC315.BM. Histograms show staining for costimulatory receptors CD80/86, asialo-GM1, and MHC -II on the DAF-FMHigh population found in TCR-Tg mice. (D) Representative flow cytometry staining using a pId315:I-Ed peptide/MHC-II–specific single-chain variable fragment (TCRm). Bone marrow cells were isolated from TCR-Tg and wild-type (WT) mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells, with a rabbit anti-asialo-GM1 antibody, followed by magnetic bead separation. Nonchallenged TCR-Tg mouse bone marrow was used as a control. Dorsal whole-body bioluminescence staining (E) and survival (F) for TCR-Tg and WT BALB/c mice challenged with 1 × 106 MOPC315.BM. Mice received daily injections of 200 μg depleting mAb against Ly6G/C (RB6-8C5) or a relevant isotype control mAb, starting 4 days before tumor challenge (n = 6 to 8 per treatment group). Bioluminescence data are shown as means ± SD of total flux.
CD4+ T-cell–mediated killing of myeloma is mediated by bone marrow macrophages. (A) Flow cytometry quantitation of F4/80+CD64+ macrophages in femoral bone marrow isolated from mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells. Mice were treated with an anti-asialo-GM1 (a-AGM1) or isotype control (Is ctr.) mAb. Gating strategy is shown on the left. The histogram shows means ± SD for 6 mice per treatment group; **P < .01. (B) In vitro coculture of MOPC.BM cells with ex vivo–isolated F4/80+CD64+ macrophages from the experiment described in panel A; effector/target ratio, 10:1. Results are plotted as tumor cell growth of a percentage of tumor cells cultured alone, calculated on the basis of 18-hour [3H]-thymidine release (n = 6-12 per treatment group, mean ± SD; ***P < .001). (C) Flow cytometry, with the nitric oxide–reactive fluorescent dye DAF-FM diacetate. Dot plots show representative results for staining of cells isolated from femoral bone marrow of TCR-Tg and wild-type (WT) BALB/c mice on day 7 after tumor challenge with 1 × 106 MOPC315.BM. Histograms show staining for costimulatory receptors CD80/86, asialo-GM1, and MHC -II on the DAF-FMHigh population found in TCR-Tg mice. (D) Representative flow cytometry staining using a pId315:I-Ed peptide/MHC-II–specific single-chain variable fragment (TCRm). Bone marrow cells were isolated from TCR-Tg and wild-type (WT) mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells, with a rabbit anti-asialo-GM1 antibody, followed by magnetic bead separation. Nonchallenged TCR-Tg mouse bone marrow was used as a control. Dorsal whole-body bioluminescence staining (E) and survival (F) for TCR-Tg and WT BALB/c mice challenged with 1 × 106 MOPC315.BM. Mice received daily injections of 200 μg depleting mAb against Ly6G/C (RB6-8C5) or a relevant isotype control mAb, starting 4 days before tumor challenge (n = 6 to 8 per treatment group). Bioluminescence data are shown as means ± SD of total flux.
For definitive identification of APCs displaying Id-derived antigens to TCR-Tg CD4+ cells, we generated a single-chain variable fragment specific for the λ2315/I-Ed peptide/MHC-II (pId315:I-Ed), by using a phage display, effectively yielding a TCRm. Flow cytometry of bead-isolated AGM1+CD11b+ cells from the bone marrow of tumor-challenged mice revealed a distinct population of TCRm+ cells with a high expression of F4/80, most apparent in TCR-Tg mice (Figure 3D). Conversely, the AGM1− fraction of bone marrow from TCR-Tg mice showed only sparse TCRm staining, with an almost complete disappearance of the pId315:I-Ed +F4/80High subset (Figure 3D).
Attempts at specific macrophage depletion using liposomal clodronate were unsuccessful, because of the severe side effects of clodronate therapy in tumor-bearing mice. Based on the high expression of Ly6C in the identified CD64+F4/80+ macrophage subset, we therefore took advantage of the Ly6G/C (Gr-1)–specific neutralizing antibody RB6-8C5, which depletes neutrophils as well as Gr-1+ monocyte–derived cells.30 Treatment of TCR-Tg mice with RB6-8C5 resulted in a significant reduction in CD11b+F4/80+ cells within the bone marrow (data not shown) and loss of immunoprotection in most of the mice, albeit with prolonged survival compared with wild-type mice (Fig. 3E-F). In light of the lack of effect of neutrophil depletion using 1A8 (Figure 2E), these findings provide further support to the conclusion that macrophages play a dominant role as effectors of CD4+ T-cell immunoprotection within the bone marrow.
Discussion
In addition to its role as a primary hematopoietic organ, it is increasingly clear that the bone marrow functions as a secondary lymphoid tissue and contains a variety of differentiated and functional immune cells. In addition to primary hematological malignancies such as myeloma, the bone marrow is a predilection site for metastatic disease in many solid malignancies. Gaining an understanding of the mechanisms governing immune responses within the bone marrow is therefore of general interest within the field of immunotherapy. Nonetheless, current knowledge of primary antitumor T-cell responses within the bone marrow is limited.
Infiltration by immune cells is a hallmark of most forms of malignancy. In the context of solid tumors, tumor-associated macrophages represent key regulators of the complex interplay between the immune system and cancer. A recent publication by Lossos et al31 highlights the potential antitumor activity of bone marrow macrophages. The authors showed, in a xenograph model, that VEGFA-stimulated bone marrow macrophages could phagocytose antibody-resistant primary double-hit lymphoma cells.
Previous work on the roles of tumor-specific CD4+ in the MOPC315 myeloma model has provided detailed insight into a specialized form of tumor cell recognition and elimination that involves indirect detection of a secreted tumor antigen by T cells occurring via uptake and presentation in stromal macrophages.18,32-35 This process triggers activation and repolarization of the macrophages, resulting in spatially constrained, nitric-oxide–mediated killing of neighboring tumor cells.32 These results stem predominantly from studies of subcutaneously injected tumor cells, which form a stroma through recruitment of cells from surrounding tissue and circulation. Hence, it is not clear whether similar processes of indirect antitumor immune responses would be operational in physiological sites of myeloma cell growth, such as the bone marrow. Moreover, the nature and effects of APCs operating in such an environment has not been determined. For instance, macrophages may have different effects on tumor growth and survival, depending on their localization and growth condition.36 In this study, tumor-specific CD4+ T cells elicited protective immune responses within the bone marrow through interaction with a population of bone marrow–resident macrophages. Our data indicate that, at least in the context of a TCR-Tg model, a primary T-cell response including priming, expansion, and effector phases may occur within an isolated bone marrow environment. TCR-Tg mice harbor supraphysiological levels of antigen-specific T cells within the naive T-cell repertoire. The relevance of such localized primary antitumor responses in the setting of established cancer in a healthy individual is thus questionable, although a role of such processes in cancer immunosurveillance cannot be excluded. Regardless, in patients with established cancer that has undergone immunoediting, eliciting a therapeutic T-cell response is likely to require the provision of an expanded population of tumor-specific T cells (eg, adoptive transfer, as demonstrated in the present and previous work18 ) or possibly invigoration of preexisting T-cell responses (eg, checkpoint inhibitor therapy), as substantiated by the present and previous findings in non-TCR-Tg mice.18
CD4+ T cells may elicit tumor killing through several direct or indirect mechanisms, as previously reviewed.37 Although the existence and impact of cytotoxic CD4+ T-cell effector populations have been well documented, the lack of MHC-II expression in MOPC315.BM myeloma cells preclude direct tumor cell killing in our model. This enables direct assessment of the roles of other putative effector subsets. Despite the abundance of several other potential effector leukocyte populations, including neutrophils, eosinophils, and NK cells, we find that none of these cell types is necessary for tumor killing in vivo. In evaluating the role of NK cells, we discovered that the commonly used approach of depleting these cells by using a polyclonal anti-asialo-GM1 mAb also resulted in the depletion of a substantial population of AGM1-expressing CD11b+ cells, including a large population of macrophages. These results call for caution in interpreting the results of previous reports claiming a role of NK cells based on anti-AGM1-based depletion.
Although the diverse network of monocyte-derived cells within the bone marrow has been extensively studied, less is known about the functional characteristics of resident cells with macrophage characteristics. Previous reports have identified F4/80 staining on nonadherent precursor cells from bone marrow, these cells were found to be committed to the macrophage lineage.38 Bone marrow resident macrophages have been reported to play a role in the development of B lineage cells by providing prosurvival factors.39 Macrophages and the broader class of monocyte-derived suppressor cells are considered important prosurvival factors in the myeloma niche (reviewed in Berardi et al40 ). Indeed, CD11b+F4/80+ cells isolated ex vivo from bone marrow of tumor challenged wild-type mice significantly promoted in vitro tumor cell growth, contrasting the pronounced cytotoxicity of these cells when isolated from mice harboring tumor-specific CD4+ T cells. Within this population of bone marrow macrophages, a subset showing high levels of nitric oxide production could be specifically identified in bone marrow explants from tumor-bearing TCR-Tg mice, consistent with an activated, M1-like macrophage phenotype.
Using a TCRm specific for the pId315:I-Ed complex, we found that detectable presentation of the Id-derived epitope underlying the Id-specific CD4+ T-cell response was restricted to a subset of bone marrow macrophages. We have previously shown that CD4+ T-cell immunosurveillance against MOPC315 cells is critically dependent on secretion of the tumor-specific antigen.19 Hence, it seems highly likely that the macrophages take up Id antigen secreted by myeloma cells in their vicinity.
Attempts at specific macrophage depletion using liposomal clodronate were unsuccessful, because of adverse side effects of clodronate therapy in tumor-bearing mice. Nonetheless, through a detailed assessment of the role of the various APC subsets present within the immunological environment of the bone marrow, our present data clearly implicate macrophages as the key source of antigen presentation to bone marrow-resident CD4+ T cells based on the following observations: (1) macrophages isolated ex vivo from bone marrow of tumor-challenged TCR-Tg mice were highly cytotoxic toward myeloma cells in vitro; (2) in the presence of Id-specific T cells, a population of macrophages was activated in an M1-like fashion, including increased MHC-II expression and nitric oxide synthesis; (3) pId315:I-Ed TCRm staining revealed selective presentation of the tumor-specific antigen in an AGM1-expressing subset of bone marrow macrophages; and (4) depletion of AGM1+ cells abrogated immunoprotection. Based on these findings, we therefore propose that CD4+ T cells act through a process of macrophage re-education within the bone marrow tumor microenvironment, thus inducing a shift from a tumor-supportive to a cytotoxic, M1-like phenotype.41
The MOPC315.BM model is derived from a mineral-oil–induced peritoneal plasmacytoma,42 and a subclone with bone marrow–homing properties is promoted by serial passaging in syngeneic mice.17 Although this model recapitulates several key aspects of the clinical presentation of myeloma, the characteristics of its interaction with other bone marrow constituents, and the composition of its surrounding stroma may be different from that of a spontaneously occurring myeloma. Compared with the vast literature on the impact of macrophages in solid tumors, their role in the microenvironment in hematological malignancies is less well characterized. Nonetheless, myeloma bone marrow biopsies have been found to contain abundant CD68+ macrophages, compared with only scattered appearance in healthy bone marrow specimens,43 and macrophages have been shown to promote the growth, survival, and resistance to therapy of primary myeloma cells in an in vitro model system.43,44 High levels of macrophage infiltration, in particular cases with a dominance of macrophages with M2-like phenotypic traits have been reported to be negative prognostic factors for myeloma patient survival.45-47
In line with previous reports,14 we found that intact IFN-γ signaling was necessary for macrophage-mediated tumor rejection. Although we have previously shown that intratumoral delivery of IFN-γ may significantly delay tumor outgrowth in a T-cell–independent manner, presumably through direct effects on tumor-associated macrophages,14 IFN-γ in itself is insufficient to induce a tumoricidal phenotype in tumor-associated macrophages ex vivo,22 suggesting that this cytokine is required, but not sufficient for tumor rejection. We have previously shown that treatment with an agonistic anti-CD40 mAb induces an M1-like, tumoricidal phenotype in tumor-infiltrating macrophages in subcutaneously growing plasmacytomas.48 The present results indicate that therapeutic interventions targeted against such bone marrow–resident macrophages may serve as an effective means of eliciting cytotoxicity against myeloma cells.
Original data are available by e-mail request to the corresponding author.
The full-text version of this article contains a data supplement.
Acknowledgment
This work was supported by Norwegian Cancer Society grant 420042.
Authorship
Contribution: O.A.W.H., K.H., and A.T. conceptualized the work and strategy; O.A.W.H., K.H., M.F., and A.T. planned and analyzed the experiments; G.Å.L. provided reagents and technical advice on the development of the pId315:I-Ed TCRm; G.Å.L., M.F., B.B., and A.T. developed the IEd/L2315-specific TCRm; and O.A.H. and A.T., supported by K.H., M.F., B.B., and G.Å.L., wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ole Audun W. Haabeth, Division of Oncology, Department of Medicine, Stanford Cancer Institute, Stanford University, 269 Campus Dr, CCSR Room 1110, Stanford, CA 94305; e-mail: haabeth@stanford.edu.



![CD4+ T-cell–mediated killing of myeloma is mediated by bone marrow macrophages. (A) Flow cytometry quantitation of F4/80+CD64+ macrophages in femoral bone marrow isolated from mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells. Mice were treated with an anti-asialo-GM1 (a-AGM1) or isotype control (Is ctr.) mAb. Gating strategy is shown on the left. The histogram shows means ± SD for 6 mice per treatment group; **P < .01. (B) In vitro coculture of MOPC.BM cells with ex vivo–isolated F4/80+CD64+ macrophages from the experiment described in panel A; effector/target ratio, 10:1. Results are plotted as tumor cell growth of a percentage of tumor cells cultured alone, calculated on the basis of 18-hour [3H]-thymidine release (n = 6-12 per treatment group, mean ± SD; ***P < .001). (C) Flow cytometry, with the nitric oxide–reactive fluorescent dye DAF-FM diacetate. Dot plots show representative results for staining of cells isolated from femoral bone marrow of TCR-Tg and wild-type (WT) BALB/c mice on day 7 after tumor challenge with 1 × 106 MOPC315.BM. Histograms show staining for costimulatory receptors CD80/86, asialo-GM1, and MHC -II on the DAF-FMHigh population found in TCR-Tg mice. (D) Representative flow cytometry staining using a pId315:I-Ed peptide/MHC-II–specific single-chain variable fragment (TCRm). Bone marrow cells were isolated from TCR-Tg and wild-type (WT) mice on day 5 after IV challenge with 1 × 106 MOPC.BM cells, with a rabbit anti-asialo-GM1 antibody, followed by magnetic bead separation. Nonchallenged TCR-Tg mouse bone marrow was used as a control. Dorsal whole-body bioluminescence staining (E) and survival (F) for TCR-Tg and WT BALB/c mice challenged with 1 × 106 MOPC315.BM. Mice received daily injections of 200 μg depleting mAb against Ly6G/C (RB6-8C5) or a relevant isotype control mAb, starting 4 days before tumor challenge (n = 6 to 8 per treatment group). Bioluminescence data are shown as means ± SD of total flux.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/12/10.1182_bloodadvances.2020001434/5/m_advancesadv2020001434f3.png?Expires=1770184081&Signature=ndnv-F2YZXujQhXvXDB3mfWtZvGTO8uPpZ5HvZESlJGrWZKBaGn9uUoiH9nnaAFW~4NjqCBPGBxCVr4oilbN6QRCRZIv0VqW4Jyj5yhLbhSZ3jqFufj1xiDGEDNqulnViHOCGDAPGi4aT9xdaKyV~ib~zbfmn85PK1P7~a543W6ir4rLis3~PmyPj8jbajIJtaHIEK0D1VyovhLvoMxGHYWeRO36cPaUyBSkRpG5g2JCVacD16QfIVsL9cnTbl21oSHfVJkZqrV80pQLo3-OUb6crzmqNFVxr042-DG~MA5RGkZzOFZAJgR57KgG7x2hohfTw2uGp2Op2X8nwwFvEw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)

