

Key Points
A definite genetic diagnosis was made in 10.4% of 1892 patients with suspected HLH by a panel approach including 15 HLH-associated genes.
HLH next-generation sequencing panels were ∼400 test orders per year; single-gene tests related to HLH have drastically decreased.

Abstract
This article explores the distribution and mutation spectrum of potential disease-causing genetic variants in hemophagocytic lymphohistiocytosis (HLH)–associated genes observed in a large tertiary clinical referral laboratory. Samples from 1892 patients submitted for HLH genetic analysis were studied between September 2013 and June 2018 using a targeted next-generation sequencing panel approach. Patients ranged in age from 1 day to 78 years. Analysis included 15 genes associated with HLH. A potentially causal genetic finding was observed in 227 (12.0%) samples in this cohort. A total of 197 patients (10.4%) had a definite genetic diagnosis. Patients with pathogenic variants in familial HLH genes tended to be diagnosed significantly younger compared with other genes. Pathogenic or likely pathogenic variants in the PRF1 gene were the most frequent. However, mutations in genes associated with degranulation defects (STXBP2, UNC13D, RAB27A, LYST, and STX11) were more common than previously appreciated and collectively represented >50% of cases. X-linked conditions (XIAP, SH2D1A, and MAGT1) accounted for 17.8% of the 197 cases. Pathogenic variants in the SLC7A7 gene were the least encountered. These results describe the largest cohort of genetic variation associated with suspected HLH in North America. Merely 10.4% of patients were identified with a clearly genetic cause by this diagnostic approach; other possible etiologies of HLH should be investigated. These results suggest that careful thought should be given regarding whether patients have a clinical phenotype most consistent with HLH vs other clinical and disease phenotypes. The gene panel identified known pathogenic and novel variants in 10 HLH-associated genes.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive and life-threatening syndrome of hyperinflammation characterized by pathologic activation and proliferation of T cells and macrophages. Although HLH frequently affects infants, it is also observed in children and adults of all ages. HLH can occur as a typical or principal manifestation of several genetically heterogeneous disorders. A group of diseases known as familial HLH types 2-5 are caused by pathogenic variants in PRF1, UNC13D, STX11, and STXBP2, respectively, which are all critical for normal cytotoxic lymphocyte granule-mediated cytotoxicity.1-5 In addition, loss-of-function mutations in the LYST, RAB27A, and AP3B1 genes cause problems in the formation of the cytotoxic granules or transport of the granules through the cytoplasm6-8 and can also lead to HLH as well as other problems such as pigmentary dilution in the disorders known as Chediak-Higashi syndrome, Griscelli syndrome type 2, and Hermansky-Pudlak syndrome type 2, respectively. Other genetic disorders with more complex mechanisms of diseases that are associated with a high risk of HLH include X-linked lymphoproliferative syndrome type 1 and 2 (XLP1 and XLP2) caused by mutations in the SH2D1A9 and XIAP10 genes, respectively, X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection and neoplasia caused by defects in the MAGT1 gene,11 CD27 deficiency from loss of function in CD27,12 and interleukin-2 inducible T-cell kinase (ITK) deficiency from ITK dysfunction.13 Some metabolic disorders can also be complicated by the development of HLH, notably including lysinuric protein intolerance caused by mutations in the SLC7A7 gene.14
Defects in these genes are sometimes indistinguishable from each other clinically. Historically, genetic investigations started with Sanger sequencing of the most commonly defective gene, PRF1. Sequential examination of other HLH-related genes was then pursued if PRF1 testing was negative. This process not only prolonged the diagnosis in many cases but was also expensive. Next-generation sequencing (NGS) technology has allowed the creation of targeted gene panels in which several genes can be interrogated at once in a time and cost-efficient manner. At Cincinnati Children’s Hospital Medical Center (CCHMC), an NGS-based HLH sequencing panel including 15 HLH-associated genes was launched in September 2013. In this study, we aimed to examine the impact of NGS HLH panel on genetic testing ordering patterns and examine the distribution and details of genetic variants observed in 1892 patient samples submitted for NGS HLH panel sequencing.
Materials and methods
Patient samples and clinical information
The present study was approved by the institutional review board at Cincinnati Children’s Hospital, Cincinnati, OH. A total of 1892 patient samples and submitted clinical information were analyzed and reviewed after HLH sequencing panel testing was submitted to the Molecular Diagnostic Laboratory at CCHMC between September 2013 and June 2018. Of these, 33 orders clearly stated that the reason for testing was for mutation carrier status evaluation and these were excluded from further analysis for molecular diagnosis. Of the remaining 1859 samples, 1632 had only variants classified as benign, likely benign, or variants of uncertain significance. At least 1 pathogenic or likely pathogenic variant was detected in 227 samples. Among them, 197 samples were identified with either homozygous or compound heterozygous pathologic variants in an autosomal recessive condition, or a hemizygous pathologic variant in an X-linked disorder. In addition, 30 patient samples were identified carrying only 1 heterozygous pathogenic or likely pathogenic variant in a recessive condition (supplemental Figure 1). Patients were referred by physicians from 300 institutions across North America and the included patients were either referred with a clinical diagnosis of HLH or suspected related conditions. Patient age at time of referral ranged from 1 day to 78 years. Forty-seven percent (895/1892) were female and 53% (997/1892) were male. The reported cohort of 197 patients ranged from 1 day to 57.8 years, including 44% (86/197) female and 56% (111/197) male. Fifty-eight percent (114/197) were younger than 2 years old, and 30% (59/197) were between 2 and 18 years old (Table 1). Clinical information was collected using a standardized clinical checklist completed by the ordering physician that captured information such as age of onset, and general clinical history such as fever, liver and spleen abnormalities, infections, skin abnormalities, laboratory findings, neurological abnormalities, family history, and results of previous testing. Our laboratory did not systematically confirm clinical characteristics or prior laboratory investigations of patients reported by referring clinicians.
Pathogenic or likely pathogenic variants identified in 197 HLH patients with a definite genetic diagnosis
| Patient no. | Sex | Age at testing, y (unless indicated otherwise) | Ethnicity | Gene | Variant | Zygosity | Population frequency (gnomAD*), % | Symptoms/immunology testing/family history† |
|---|---|---|---|---|---|---|---|---|
| 1 | Female | 53 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 2 | Male | 63 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 3 | Male | 0.4 | Middle Eastern | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 4 | Female | 27 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression |
| 5 | Female | 32 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression |
| 6 | Male | 4 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression; sibling died of HLH |
| 7 | Female | 0.4 | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 8 | Male | 6 d | African American + European-white | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression |
| 9 | Male | 59 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 10 | Male | 13 d | African | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 11 | Female | 7 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent NK cell function |
| PRF1 | c.266C>T(p.Pro89Leu) | Heterozygous | ND | |||||
| 12 | Female | 0.3 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.350_356delinsATGC (p.Val117_Arg119delinsAspAla) | Heterozygous | ND | |||||
| 13 | Male | 0.4 | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | |||||
| 14 | Male | 3.3 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression; brother with HLH |
| PRF1 | c.527G>A(p.Cys176Tyr) | Heterozygous | ND | |||||
| 15 | Female | 0.5 | Latino-Hispanic | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression |
| PRF1 | c.659G>A(p.Gly220Asp) | Heterozygous | 0.0008 | |||||
| 16 | Female | 0.2 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent NK cell function |
| PRF1 | c.853_855del(p.Lys285del) | Heterozygous | 0.0056 | |||||
| 17 | Male | 66 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.895C>T(p.Arg299Cys) | Heterozygous | 0.0012 | |||||
| 18 | Male | 20.6 | Latino-Hispanic | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.902C>T(p.Ser301Leu) | Heterozygous | ND | |||||
| 19 | Male | 54 d | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Symptoms of HLH |
| PRF1 | c.916G>T(p.Gly306Cys) | Heterozygous | ND | |||||
| 20 | Female | 45 d | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression |
| PRF1 | c.916G>T(p.Gly306Cys) | Heterozygous | ND | |||||
| 21 | Male | 32 d | Latino-Hispanic | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Symptoms of HLH |
| PRF1 | c.985dup(p.Val329fs) | Heterozygous | ND | |||||
| 22 | Male | 0.3 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression |
| PRF1 | c.1385C>A(p.Ser462*) | Heterozygous | ND | |||||
| 23 | Male | 36.1 | Unknown | PRF1 | c.116C>A(p.Pro39His) | Heterozygous | 0.00081 | NA |
| PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | |||||
| 24 | Female | 1.2 | Asian-American | PRF1 | c.133G>A(p.Gly45Arg) | Homozygous | 0.0012 | Absent NK cell function |
| 25 | Female | 37 d | Non-Hispanic white | PRF1 | c.150del(p.Thr51fs) | Heterozygous | 0.0004 | Absent perforin expression |
| PRF1 | c.227G>A(p.Cys76Tyr) | Heterozygous | 0.00071 | |||||
| 26 | Male | 69 d | Latino-Hispanic | PRF1 | c.218G>C(p.Cys73Ser) | Homozygous | 0.0004 | Symptoms of HLH |
| 27 | Female | 20 | European-American | PRF1 | c.227G>A(p.Cys76Tyr) | Heterozygous | 0.00071 | Absent perforin expression |
| PRF1 | c.626A>C(p.Gln209Pro) | Heterozygous | 0.0012 | |||||
| 28 | Female | 21.8 | Unknown | PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | Absent NK cell function, decreased perforin expression |
| PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | |||||
| 29 | Male | 17 | European-American | PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | Absent perforin expression |
| PRF1 | c.635A>C(p.Tyr212Ser) | Heterozygous | ND | |||||
| 30 | Female | 41.2 | European-American | PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | Absent NK cell function, decreased perforin expression |
| PRF1 | c.666C>A(p.His222Gln) | Heterozygous | 0.0039 | |||||
| 31 | Female | 8.3 | European-American | PRF1 | c.443C>G(p.Ala148Gly) | Heterozygous | 0.0004 | NA |
| PRF1 | c.666C>A(p.His222Gln) | Heterozygous | 0.0039 | |||||
| 32 | Male | 2.6 | Unknown | PRF1 | c.445G>A(p.Gly149Ser) | Homozygous | 0.014 | Absent NK cell function |
| 33 | Male | 0.8 | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Homozygous | 0.014 | Symptoms of HLH |
| 34 | Female | 6 | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Homozygous | 0.014 | NA |
| 35 | Female | 0.3 | European-American | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | Family history of HLH |
| PRF1 | c.614A>G(p.Asn205Ser) | Heterozygous | 0.0043 | |||||
| 36 | Male | 42 d | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | NA |
| PRF1 | c.938A>T(p.Asp313Val) | Heterozygous | 0.0012 | |||||
| 37 | Male | 4.6 | Unknown | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | Absent perforin expression |
| PRF1 | c.1081A>T(p.Arg361Trp) | Heterozygous | 0.0011 | |||||
| 38 | Female | 32 d | Middle Eastern | PRF1 | c.501C>G(p.Tyr167*) | Homozygous | ND | Symptoms of HLH |
| 39 | Female | 0.3 | Unknown | PRF1 | c.512C>A(p.Thr171Asn) | Homozygous | 0.0028 | Absent perforin expression |
| 40 | Male | 9.5 | European-American | PRF1 | c.786_801del(p.Gln263fs) | Heterozygous | ND | Absent NK cell function |
| PRF1 | c.886T>C(p.Tyr296His) | Heterozygous | 0.0012 | |||||
| 41 | Male | 59 d | Unknown | PRF1 | c.853_855del(p.Lys285del) | Heterozygous | 0.0057 | NA |
| PRF1 | c.921del(p.His308fs) | Heterozygous | 0.002 | |||||
| 42 | Female | 0.7 | Middle Eastern | PRF1 | c.880del(p.Gln294fs) | Homozygous | ND | Symptoms of HLH |
| 43 | Female | 2 | Middle Eastern | PRF1 | c.895C>T(p.Arg299Cys) | Homozygous | 0.0012 | Symptoms of HLH |
| 44 | Female | 0.2 | Latino-Hispanic | PRF1 | c.904G>T(p.Glu302*) | Homozygous | ND | Absent perforin expression |
| 45 | Female | 1.8 | Unknown | PRF1 | c.949G>A(p.Gly317Arg) | Homozygous | 0.0008 | Symptoms of HLH |
| 46 | Female | 10.3 | European-American | PRF1 | c.973T>C(p.Tyr325His) | Heterozygous | ND | Absent perforin expression |
| PRF1 | c.1326_1328del(p.Phe443del) | Heterozygous | ND | |||||
| 47 | Male | 1.1 | Middle Eastern | PRF1 | c.1070G>C(p.Arg357Pro) | Homozygous | ND | Symptoms of HLH |
| 48 | Male | 12.5 | Middle Eastern | PRF1 | c.1081A>T(p.Arg361Trp) | Homozygous | 0.0011 | Abnormal brain lesions and seizures |
| 49 | Female | 2.6 | Unknown | PRF1 | c.1229_1230delinsCC (p.Arg410Pro) | Homozygous | ND | NA |
| 50 | Female | 0.2 | African American | PRF1 | c.1304C>T(p.Thr435Met) | Heterozygous | 0.0028 | Absent perforin expression |
| PRF1 | c.1314T>A(p.Tyr438*) | Heterozygous | 0.0032 | |||||
| 51 | Female | 2.6 | Latino-Hispanic | PRF1 | c.1337A>C(p.Gln446Pro) | Homozygous | 0.0016 | NA |
| 52 | Female | 2.6 | Unknown | PRF1 | c.1337A>C(p.Gln446Pro) | Homozygous | 0.0016 | Symptoms of HLH |
| 53 | Female | 0.4 | Middle Eastern | STXBP2 | c.37+2T>C | Heterozygous | ND | Absent NK cell function |
| STXBP2 | c.1430C>T(p.Pro477Leu) | Heterozygous | 0.00074 | |||||
| 54 | Male | 0.6 | Unknown | STXBP2 | c.37+5G>A | Heterozygous | ND | NA |
| STXBP2 | c.1057T>C (p.Cys353Arg) | Heterozygous | 0.0004 | |||||
| 55 | Female | 63 d | Asian-American | STXBP2 | c.193C>T(p.Arg65Trp) | Homozygous | 0.00071 | Absent NK cell function |
| 56 | Female | 5.5 | Unknown | STXBP2 | c.194G>A(p.Arg65Gln) | Heterozygous | 0.0028 | Absent NK cell function |
| STXBP2 | c.560C>T (p.Pro187Leu) | Heterozygous | 0.00064 | |||||
| 57 | Male | 4.1 | European-American | STXBP2 | c.194G>A(p.Arg65Gln) | Heterozygous | 0.0028 | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 58 | Female | 4.2 | European-American | STXBP2 | c.326-30_326-23del | Heterozygous | 0.0068 | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 59 | Male | 0.6 | Latino-Hispanic | STXBP2 | c.389T>C(p.Leu130Ser) | Homozygous | 0.0032 | Symptoms of HLH |
| 60 | Male | 45 d | African American | STXBP2 | c.389T>C(p.Leu130Ser) | Heterozygous | 0.0032 | Symptoms of HLH; family history of HLH |
| STXBP2 | exon 14-19 deletion | Heterozygous | ND | |||||
| 61 | Male | 0.7 | Middle Eastern | STXBP2 | c.481del(p.Arg161fs) | Homozygous | ND | Symptoms of HLH |
| 62 | Male | 0.4 | Unknown | STXBP2 | c.481del(p.Arg161fs) | Homozygous | ND | Symptoms of HLH |
| 63 | Female | 11 | European-American | STXBP2 | c.539_540delinsAA(p.Cys180*) | Heterozygous | ND | Symptoms of HLH |
| STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | |||||
| 64 | Male | 0.3 | Latino-Hispanic | STXBP2 | c.703C>G(p.Arg235Gly) | Homozygous | 0.00071 | Absent NK cell function |
| 65 | Female | 52.7 | European-American | STXBP2 | c.752C>T(p.Ala251Val) | Heterozygous | ND | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 66 | Male | 0.9 | Unknown | STXBP2 | c.902+5G>A | Heterozygous | 0.0036 | NA |
| STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | |||||
| 67 | Male | 3.1 | Unknown | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Symptoms of HLH |
| 68 | Female | 22.7 | Latino-Spanish | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Decreased NK cell function |
| 69 | Male | 26.4 | European-American | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Symptoms of HLH |
| 70 | Male | 25.6 | European-American | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Symptoms of HLH |
| 71 | Female | 29.7 | European-American | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | NA |
| 72 | Male | 4 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Absent NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 73 | Female | 15.8 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Absent NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 74 | Female | 19 | Unknown | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Decreased NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 75 | Female | 26.9 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 76 | Female | 57.8 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Absent NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 77 | Female | 0.2 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 78 | Female | 0.3 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | NA |
| 79 | Female | 0.3 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 80 | Female | 0.6 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 81 | Male | 0.2 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | NA |
| 82 | Male | 0.7 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 83 | Female | 0.8 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 84 | Male | 0.8 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 85 | Male | 0.6 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Family history of HLH |
| 86 | Male | 1.6 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 87 | Male | 0.3 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 88 | Female | 63 d | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Family history of HLH |
| 89 | Male | 0.5 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 90 | Male | 0.2 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 91 | Male | 11.1 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Absent NK cell function |
| 92 | Female | 10.1 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Heterozygous | 0.00074 | Symptoms of HLH |
| STXBP2 | c.1696+5G>T | Heterozygous | ND | |||||
| 93 | Male | 0.5 | Middle Eastern | STXBP2 | c.1452+1G>A | Homozygous | ND | Symptoms of HLH |
| 94 | Male | 1 | Middle Eastern | STXBP2 | c.1452+1G>A | Homozygous | ND | Abnormal NK cell function |
| 95 | Female | 49 d | European-American | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Dysmorphic facies, decreased NK cell function |
| UNC13D | c.2258_2267delinsTACCTTGTTCGA (p.Gly753fs) | Heterozygous | ND | |||||
| 96 | Male | 0.7 | European-American | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Decreased NK cell function |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 97 | Male | 0.2 | European-American + Latino-Spanish | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Decreased NK cell function |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 98 | Female | 1.3 | Non-Hispanic white | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Symptoms of HLH, seizures, normal NK cell function |
| UNC13D | c.2867C>T(p.Pro956Leu) | Heterozygous | ND | |||||
| 99 | Female | 1 | Non-Hispanic white | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Absent NK cell function |
| UNC13D | c.3193C>T(p.Arg1065*) | Heterozygous | 0.0011 | |||||
| 100 | Female | 3.2 | European-American | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Absent NK cell function |
| UNC13D | 253Kb inversion | Heterozygous | ND | |||||
| 101 | Male | 2.6 | Unknown | UNC13D | 253Kb inversion | Heterozygous | ND | Decreased NK cell function |
| UNC13D | c.154-1G>C | Heterozygous | ND | |||||
| 102 | Male | 0.2 | Unknown | UNC13D | 253Kb inversion | Heterozygous | ND | Symptoms of HLH |
| UNC13D | c.551G>A(p.Trp184*) | Heterozygous | 0.0011 | |||||
| 103 | Female | 0.2 | European-American | UNC13D | 253Kb inversion | Heterozygous | ND | NA |
| UNC13D | c.1389+1G>A | Heterozygous | 0.0071 | |||||
| 104 | Female | 0.2 | European-American | UNC13D | 253Kb inversion | Heterozygous | ND | Absent NK cell function |
| UNC13D | c.2447+1G>A | Heterozygous | 0.00051 | |||||
| 105 | Female | 0.6 | Non-Hispanic white | UNC13D | 253Kb inversion | Heterozygous | ND | Decreased NK cell function |
| UNC13D | c.2695C>T(p.Arg899*) | Heterozygous | 0.0018 | |||||
| 106 | Male | 11.4 | Hispanic white | UNC13D | c.182A>G(p.Tyr61Cys) | Heterozygous | ND | Symptoms of HLH |
| UNC13D | c.778T>C(p.Trp260Arg) | Heterozygous | ND | |||||
| 107 | Male | 0.4 | European-American | UNC13D | c.262-1G>A | Heterozygous | ND | Symptoms of HLH, abnormal NK cell function, family history of HLH |
| UNC13D | c.766C>T(p.Arg256*) | Heterozygous | 0.0025 | |||||
| 108 | Male | 0.4 | Unknown | UNC13D | c.321+1_321+2del | Heterozygous | ND | Decreased NK cell function |
| UNC13D | c.753+1G>T | Heterozygous | 0.0044 | |||||
| 109 | Male | 0.3 | Unknown | UNC13D | c.322-2A>T | Heterozygous | 0.0024 | Symptoms of HLH, decreased NK cell function |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 110 | Male | 0.3 | Unknown | UNC13D | c.419T>C(p.Ile140Thr) | Heterozygous | 0.0004 | NA |
| UNC13D | c.460C>T(p.Arg154Trp) | Heterozygous | 0.011 | |||||
| 111 | Female | 5.4 | Middle Eastern | UNC13D | c.424dup(p.Gln142fs) | Homozygous | ND | Symptoms of HLH |
| 112 | Male | 7.7 | Latino-Hispanic | UNC13D | c.518C>T(p.Thr173Met) | Heterozygous | 0.0028 | NA |
| UNC13D | c.1803_1819dup(p.Arg607fs) | Heterozygous | ND | |||||
| 113 | Female | 39 d | European-American | UNC13D | c.551G>A(p.Trp184*) | Heterozygous | 0.0011 | Abnormal NK cell function |
| UNC13D | c.766C>T(p.Arg256*) | Heterozygous | 0.0025 | |||||
| 114 | Male | 4.9 | European-American | UNC13D | c.570-2A>T | Heterozygous | ND | Absent NK cell function |
| UNC13D | c.3049G>A(p.Glu1017Lys) | Heterozygous | 0.00044 | |||||
| 115 | Female | 0.2 | Middle Eastern | UNC13D | c.753+1G>T | Homozygous | 0.0044 | Symptoms of HLH |
| 116 | Female | 0.6 | European-American | UNC13D | c.766C>T(p.Arg256*) | Heterozygous | 0.0025 | Symptoms of HLH, abnormal NK cell function |
| UNC13D | c.2447+1G>A | Heterozygous | 0.00051 | |||||
| 117 | Female | 1.2 | Latino-Spanish | UNC13D | c.859del(p.Arg287fs) | Homozygous | ND | Decreased NK cell function |
| 118 | Female | 1 | Non-Hispanic white | UNC13D | c.1055+1G>T | Heterozygous | ND | Decreased NK cell function, family history of HLH |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 119 | Male | 0.7 | European-American | UNC13D | c.1229_1230dup(p.Arg411fs) | Heterozygous | 0.00 | Symptoms of HLH, hypertelorism |
| UNC13D | c.2298+1G>T | Heterozygous | ND | |||||
| 120 | Female | 0.2 | European-American | UNC13D | c.1259_1260del(p.Ser420fs) | Heterozygous | ND | symptoms of HLH, decreased NK cell function |
| UNC13D | c.1848+1G>C | Heterozygous | ND | |||||
| 121 | Male | 18.3 | European-American | UNC13D | c.1387C>T(p.Gln463*) | Heterozygous | ND | symptoms of HLH, decreased NK cell function |
| UNC13D | c.1820G>C(p.Arg607Pro) | Heterozygous | 0.011 | |||||
| 122 | Female | 58 d | European-American + African American | UNC13D | c.1389+1G>A | Heterozygous | 0.0071 | Symptoms of HLH |
| UNC13D | c.1848+1G>C | Heterozygous | ND | |||||
| 123 | Female | 0.2 | Middle Eastern | UNC13D | c.1423C>T(p.Gln475*) | Homozygous | ND | NA |
| 124 | Male | 10 d | Pacific Islander | UNC13D | c.2296C>T(p.Gln766*) | Homozygous | ND | Decreased NK cell function |
| 125 | Female | 13.2 | Unknown | UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | NA |
| UNC13D | c.2588G>A(p.Gly863Asp) | Heterozygous | 0.029 | |||||
| 126 | Female | 2 | Unknown | UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | NA |
| UNC13D | c.3065T>C(p.Leu1022Pro) | Heterozygous | ND | |||||
| 127 | Female | 0.2 | Middle Eastern | UNC13D | c.2553+1G>T | Homozygous | ND | Symptoms of HLH |
| 128 | Female | 0.3 | Middle Eastern | UNC13D | c.2553+1G>T | Homozygous | ND | Symptoms of HLH |
| 129 | Female | 1 | Asian | UNC13D | c.2588G>A(p.Gly863Asp) | Homozygous | 0.029 | Decreased NK cell function |
| 130 | Female | 0.4 | African American | UNC13D | c.2695C>T(p.Arg899*) | Homozygous | 0.0018 | Symptoms of HLH, absent NK cell function, dysmorphic facies |
| 131 | Male | 0.7 | Non-Hispanic white | UNC13D | c.2819del(p.Leu940fs) | Homozygous | ND | symptoms of HLH |
| 132 | Female | 0.6 | Middle Eastern | UNC13D | c.3048dup(p.Glu1017fs) | Homozygous | ND | NA |
| 133 | Female | 0.7 | Unknown | UNC13D | c.3053C>A(p.Ala1018Asp) | Homozygous | 0.00088 | 2 affected siblings |
| 134 | Male | 13 | European-American | RAB27A | c.121A>G(p.Thr41Ala) | Heterozygous | ND | NA |
| RAB27A | c.352C>T(p.Gln118*) | Heterozygous | ND | |||||
| 135 | Male | 9.6 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | NA |
| 136 | Male | 9.9 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | NA |
| 137 | Female | 2 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Failure to thrive, bone marrow failure |
| 138 | Female | 9.4 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Symptoms of HLH |
| 139 | Male | 10.6 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Symptoms of HLH |
| 140 | Male | 19.5 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Symptoms of HLH |
| 141 | Female | 0.3 | Latino-Hispanic | RAB27A | c.335del(p.Asn112fs) | Homozygous | 0.0044 | Symptoms of HLH |
| 142 | Female | 0.4 | Middle Eastern | RAB27A | c.400A>C(p.Lys134Gln) | Homozygous | ND | NA |
| 143 | Female | 53.9 | Pacific Islander | RAB27A | c.476A>G(p.Tyr159Cys) | Homozygous | ND | Symptoms of HLH, absent NK cell function |
| 144 | Male | 1.2 | Middle Eastern | RAB27A | c.598C>T(p.Arg200*) | Homozygous | 0.0004 | NA |
| 145 | Female | 0.4 | European-American | RAB27A | c.638_642del(p.Glu213fs) | Homozygous | 0.0008 | Symptoms of HLH |
| 146 | Female | 0.7 | African American | LYST | c.925C>T(p.Arg309*) | Heterozygous | ND | Oculocutaneous albinism, neutropenia |
| LYST | c.2015dup(p.Tyr672*) | Heterozygous | ND | |||||
| 147 | Male | 3.4 | Unknown | LYST | c.3194del(p.Leu1065*) | Homozygous | 0.0004 | NA |
| 148 | Female | 0.3 | Middle Eastern | LYST | c.4159dup(p.Thr1387fs) | Homozygous | ND | Premature gray hair, anemia |
| 149 | Male | 0.9 | European-American | LYST | c.5715del(p.Asn1905fs) | Heterozygous | ND | Oculocutaneous albinism, neutropenia, absent NK cell function |
| LYST | c.8802-2A>G | Heterozygous | ND | |||||
| 150 | Female | 6.9 | Unknown | LYST | c.5784+1G>T | Homozygous | ND | Oculocutaneous albinism, dysmorphic facies, neutropenia |
| 151 | Male | 3.2 | Non-Hispanic white | LYST | c.6159_6160del(p.Met2053fs) | Homozygous | ND | NA |
| 152 | Male | 1.8 | Middle Eastern | LYST | c.7291del(p.Leu2431fs) | Homozygous | ND | Hypopigmentation, anemia |
| 153 | Male | 1.2 | African American | LYST | c.8770C>T(p.Gln2924*) | Heterozygous | ND | Silver hair, hypopigmented skin lesions, pancytopenia |
| LYST | c.9844_9845del(p.Ser3282fs) | Heterozygous | ND | |||||
| 154 | Female | 7.1 | Middle Eastern | LYST | c.10776C>G(p.Tyr3592*) | Homozygous | ND | Abnormal pigmentation, neutropenia |
| 155 | Female | 1.2 | Unknown | STX11 | c.73G>T(p.Glu25*) | Heterozygous | 0.0004 | Decreased NK cell function |
| STX11 | c.748C>T(p.Gln250*) | Heterozygous | 0.00081 | |||||
| 156 | Female | 5.6 | Middle Eastern | STX11 | c.173T>C(p.Leu58Pro) | Homozygous | 0.0008 | Symptoms of HLH, grayish hair |
| 157 | Female | 11.6 | Middle Eastern | STX11 | c.173T>C(p.Leu58Pro) | Homozygous | 0.0008 | NA |
| 158 | Male | 2.9 | European-American + Latino-Hispanic | STX11 | c.462_463delinsA(p.Asp155fs) | Heterozygous | ND | Decreased NK cell function |
| STX11 | c.784C>T(p.Gln262*) | Heterozygous | ND | |||||
| 159 | Male | 1.4 | Asian-Indian | STX11 | c.687dup(p.Gln230fs) | Homozygous | ND | NA |
| 160 | Female | 12 d | Middle Eastern | SLC7A7 | c.1429+1G>C | Homozygous | ND | Family history of HLH |
| 161 | Male | 1.2 | African American | SLC7A7 | c.701del(p.Ser234fs) | Heterozygous | 0.0016 | NA |
| SLC7A7 | c.895-1G>A | Heterozygous | ND | |||||
| 162 | Female | 13.7 | European-American | SLC7A7 | c.360_361delinsAA (p.Trp121Arg) | Homozygous | ND | NA |
| 163 | Male | 5.4 | African American | XIAP | c.145C>T(p.Arg49*) | Hemizygous | ND | Markedly decreased XIAP expression |
| 164 | Male | 18.7 | European-American | XIAP | c.345C>G(p.Tyr115*) | Hemizygous | ND | Symptoms of HLH |
| 165 | Male | 30.3 | European-American | XIAP | c.608G>T(p.Cys203Phe) | Hemizygous | ND | NA |
| 166 | Male | 2 | European-American | XIAP | c.664C>T(p.Arg222*) | Hemizygous | ND | Symptoms of HLH |
| 167 | Male | 8.9 | European-American | XIAP | c.738del(p.Asp247fs) | Hemizygous | ND | NA |
| 168 | Male | 16 | European-American | XIAP | c.738del(p.Asp247fs) | Hemizygous | ND | Absent XIAP expression |
| 169 | Male | 3.6 | African American | XIAP | c.889A>T(p.Lys297*) | Hemizygous | ND | NA |
| 170 | Male | 0.4 | Unknown | XIAP | c.894_898del(p.Lys299fs) | Hemizygous | ND | NA |
| 171 | Male | 17.2 | European-American | XIAP | c.894_898del(p.Lys299fs) | Hemizygous | ND | Symptoms of HLH |
| 172 | Male | 19.6 | African American | XIAP | c.926_929del(p.Asp309fs) | Hemizygous | ND | Symptoms of HLH |
| 173 | Male | 17.2 | European-American | XIAP | c.969G>A(p.Trp323*) | Hemizygous | ND | Decreased XIAP expression |
| 174 | Male | 22.6 | Unknown | XIAP | c.1021_1022del(p.Asn341fs) | Hemizygous | ND | NA |
| 175 | Male | 4.5 | Unknown | XIAP | c.1056+1G>A | Hemizygous | ND | NA |
| 176 | Male | 2.7 | Latino-Hispanic | XIAP | c.1141C>T(p.Arg381*) | Hemizygous | ND | NA |
| 177 | Male | 11.8 | Unknown | XIAP | c.1141C>T(p.Arg381*) | Hemizygous | ND | Absent XIAP expression |
| 178 | Male | 1 d | Pacific-Islander | XIAP | c.1239_1242dup(p.Val415fs) | Hemizygous | ND | Markedly decreased XIAP expression |
| 179 | Male | 1.9 | Unknown | XIAP | c.1239_1242dup(p.Val415fs) | Hemizygous | ND | NA |
| 180 | Male | 1.1 | Unknown | XIAP | c.1301-1G>A | Hemizygous | ND | Symptoms of HLH |
| 181 | Male | 1.5 | Unknown | XIAP | c.1445C>G(p.Pro482Arg) | Hemizygous | ND | NA |
| 182 | Male | 35 | European-American | XIAP | c.1456dup(p.Thr486fs) | Hemizygous | ND | Symptoms of HLH |
| 183 | Male | 4.5 | European-American | SH2D1A | c.20A>G(p.Tyr7Cys) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 184 | Male | 1.8 | Unknown | SH2D1A | c.117C>T(p.Gly39Gly) | Hemizygous | ND | NA |
| 185 | Male | 3.5 | Unknown | SH2D1A | c.130T>C(p.Cys44Arg) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 186 | Male | 27 | European-American | SH2D1A | c.163C>T(p.Arg55*) | Hemizygous | ND | History of pneumonia |
| 187 | Male | 8 d | Unknown | SH2D1A | c.172C>T(p.Gln58*) | Hemizygous | ND | NA |
| 188 | Male | 6.8 | African American | SH2D1A | c.172C>T(p.Gln58*) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 189 | Male | 4.7 | Latino-Hispanic | SH2D1A | c.199_201+19del(p.Glu67del) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 190 | Male | 1.3 | African American | SH2D1A | c.201G>A(p.Glu67Glu) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 191 | Male | 7 d | European-American | SH2D1A | c.245del(p.Asn82fs) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 192 | Male | 34 d | European-American + Pacific-Islander | SH2D1A | c.295C>T(p.Gln99*) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 193 | Male | 5.4 | Middle-Eastern | MAGT1 | c.154_161delinsC(p.Ile52fs) | Hemizygous | ND | Symptoms of HLH, bone pain in low extremities |
| 194 | Male | 10.4 | European-American | MAGT1 | c.223C>T(p.Gln75*) | Hemizygous | ND | NA |
| 195 | Male | 18.4 | African American | MAGT1 | c.407G>A(p.Trp136*) | Hemizygous | ND | NA |
| 196 | Male | 27.6 | European-American | MAGT1 | c.443_444del(p.Phe148fs) | Hemizygous | ND | Symptoms of HLH |
| 197 | Male | 17.3 | European-American | MAGT1 | c.774del(p.Phe258fs) | Hemizygous | ND | NA |
| Patient no. | Sex | Age at testing, y (unless indicated otherwise) | Ethnicity | Gene | Variant | Zygosity | Population frequency (gnomAD*), % | Symptoms/immunology testing/family history† |
|---|---|---|---|---|---|---|---|---|
| 1 | Female | 53 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 2 | Male | 63 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 3 | Male | 0.4 | Middle Eastern | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 4 | Female | 27 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression |
| 5 | Female | 32 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression |
| 6 | Male | 4 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression; sibling died of HLH |
| 7 | Female | 0.4 | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 8 | Male | 6 d | African American + European-white | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Absent perforin expression |
| 9 | Male | 59 d | African American | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 10 | Male | 13 d | African | PRF1 | c.50del(p.Leu17fs) | Homozygous | 0.033 | Symptoms of HLH |
| 11 | Female | 7 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent NK cell function |
| PRF1 | c.266C>T(p.Pro89Leu) | Heterozygous | ND | |||||
| 12 | Female | 0.3 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.350_356delinsATGC (p.Val117_Arg119delinsAspAla) | Heterozygous | ND | |||||
| 13 | Male | 0.4 | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | |||||
| 14 | Male | 3.3 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression; brother with HLH |
| PRF1 | c.527G>A(p.Cys176Tyr) | Heterozygous | ND | |||||
| 15 | Female | 0.5 | Latino-Hispanic | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression |
| PRF1 | c.659G>A(p.Gly220Asp) | Heterozygous | 0.0008 | |||||
| 16 | Female | 0.2 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent NK cell function |
| PRF1 | c.853_855del(p.Lys285del) | Heterozygous | 0.0056 | |||||
| 17 | Male | 66 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.895C>T(p.Arg299Cys) | Heterozygous | 0.0012 | |||||
| 18 | Male | 20.6 | Latino-Hispanic | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | NA |
| PRF1 | c.902C>T(p.Ser301Leu) | Heterozygous | ND | |||||
| 19 | Male | 54 d | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Symptoms of HLH |
| PRF1 | c.916G>T(p.Gly306Cys) | Heterozygous | ND | |||||
| 20 | Female | 45 d | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression |
| PRF1 | c.916G>T(p.Gly306Cys) | Heterozygous | ND | |||||
| 21 | Male | 32 d | Latino-Hispanic | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Symptoms of HLH |
| PRF1 | c.985dup(p.Val329fs) | Heterozygous | ND | |||||
| 22 | Male | 0.3 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent perforin expression |
| PRF1 | c.1385C>A(p.Ser462*) | Heterozygous | ND | |||||
| 23 | Male | 36.1 | Unknown | PRF1 | c.116C>A(p.Pro39His) | Heterozygous | 0.00081 | NA |
| PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | |||||
| 24 | Female | 1.2 | Asian-American | PRF1 | c.133G>A(p.Gly45Arg) | Homozygous | 0.0012 | Absent NK cell function |
| 25 | Female | 37 d | Non-Hispanic white | PRF1 | c.150del(p.Thr51fs) | Heterozygous | 0.0004 | Absent perforin expression |
| PRF1 | c.227G>A(p.Cys76Tyr) | Heterozygous | 0.00071 | |||||
| 26 | Male | 69 d | Latino-Hispanic | PRF1 | c.218G>C(p.Cys73Ser) | Homozygous | 0.0004 | Symptoms of HLH |
| 27 | Female | 20 | European-American | PRF1 | c.227G>A(p.Cys76Tyr) | Heterozygous | 0.00071 | Absent perforin expression |
| PRF1 | c.626A>C(p.Gln209Pro) | Heterozygous | 0.0012 | |||||
| 28 | Female | 21.8 | Unknown | PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | Absent NK cell function, decreased perforin expression |
| PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | |||||
| 29 | Male | 17 | European-American | PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | Absent perforin expression |
| PRF1 | c.635A>C(p.Tyr212Ser) | Heterozygous | ND | |||||
| 30 | Female | 41.2 | European-American | PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | Absent NK cell function, decreased perforin expression |
| PRF1 | c.666C>A(p.His222Gln) | Heterozygous | 0.0039 | |||||
| 31 | Female | 8.3 | European-American | PRF1 | c.443C>G(p.Ala148Gly) | Heterozygous | 0.0004 | NA |
| PRF1 | c.666C>A(p.His222Gln) | Heterozygous | 0.0039 | |||||
| 32 | Male | 2.6 | Unknown | PRF1 | c.445G>A(p.Gly149Ser) | Homozygous | 0.014 | Absent NK cell function |
| 33 | Male | 0.8 | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Homozygous | 0.014 | Symptoms of HLH |
| 34 | Female | 6 | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Homozygous | 0.014 | NA |
| 35 | Female | 0.3 | European-American | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | Family history of HLH |
| PRF1 | c.614A>G(p.Asn205Ser) | Heterozygous | 0.0043 | |||||
| 36 | Male | 42 d | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | NA |
| PRF1 | c.938A>T(p.Asp313Val) | Heterozygous | 0.0012 | |||||
| 37 | Male | 4.6 | Unknown | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | Absent perforin expression |
| PRF1 | c.1081A>T(p.Arg361Trp) | Heterozygous | 0.0011 | |||||
| 38 | Female | 32 d | Middle Eastern | PRF1 | c.501C>G(p.Tyr167*) | Homozygous | ND | Symptoms of HLH |
| 39 | Female | 0.3 | Unknown | PRF1 | c.512C>A(p.Thr171Asn) | Homozygous | 0.0028 | Absent perforin expression |
| 40 | Male | 9.5 | European-American | PRF1 | c.786_801del(p.Gln263fs) | Heterozygous | ND | Absent NK cell function |
| PRF1 | c.886T>C(p.Tyr296His) | Heterozygous | 0.0012 | |||||
| 41 | Male | 59 d | Unknown | PRF1 | c.853_855del(p.Lys285del) | Heterozygous | 0.0057 | NA |
| PRF1 | c.921del(p.His308fs) | Heterozygous | 0.002 | |||||
| 42 | Female | 0.7 | Middle Eastern | PRF1 | c.880del(p.Gln294fs) | Homozygous | ND | Symptoms of HLH |
| 43 | Female | 2 | Middle Eastern | PRF1 | c.895C>T(p.Arg299Cys) | Homozygous | 0.0012 | Symptoms of HLH |
| 44 | Female | 0.2 | Latino-Hispanic | PRF1 | c.904G>T(p.Glu302*) | Homozygous | ND | Absent perforin expression |
| 45 | Female | 1.8 | Unknown | PRF1 | c.949G>A(p.Gly317Arg) | Homozygous | 0.0008 | Symptoms of HLH |
| 46 | Female | 10.3 | European-American | PRF1 | c.973T>C(p.Tyr325His) | Heterozygous | ND | Absent perforin expression |
| PRF1 | c.1326_1328del(p.Phe443del) | Heterozygous | ND | |||||
| 47 | Male | 1.1 | Middle Eastern | PRF1 | c.1070G>C(p.Arg357Pro) | Homozygous | ND | Symptoms of HLH |
| 48 | Male | 12.5 | Middle Eastern | PRF1 | c.1081A>T(p.Arg361Trp) | Homozygous | 0.0011 | Abnormal brain lesions and seizures |
| 49 | Female | 2.6 | Unknown | PRF1 | c.1229_1230delinsCC (p.Arg410Pro) | Homozygous | ND | NA |
| 50 | Female | 0.2 | African American | PRF1 | c.1304C>T(p.Thr435Met) | Heterozygous | 0.0028 | Absent perforin expression |
| PRF1 | c.1314T>A(p.Tyr438*) | Heterozygous | 0.0032 | |||||
| 51 | Female | 2.6 | Latino-Hispanic | PRF1 | c.1337A>C(p.Gln446Pro) | Homozygous | 0.0016 | NA |
| 52 | Female | 2.6 | Unknown | PRF1 | c.1337A>C(p.Gln446Pro) | Homozygous | 0.0016 | Symptoms of HLH |
| 53 | Female | 0.4 | Middle Eastern | STXBP2 | c.37+2T>C | Heterozygous | ND | Absent NK cell function |
| STXBP2 | c.1430C>T(p.Pro477Leu) | Heterozygous | 0.00074 | |||||
| 54 | Male | 0.6 | Unknown | STXBP2 | c.37+5G>A | Heterozygous | ND | NA |
| STXBP2 | c.1057T>C (p.Cys353Arg) | Heterozygous | 0.0004 | |||||
| 55 | Female | 63 d | Asian-American | STXBP2 | c.193C>T(p.Arg65Trp) | Homozygous | 0.00071 | Absent NK cell function |
| 56 | Female | 5.5 | Unknown | STXBP2 | c.194G>A(p.Arg65Gln) | Heterozygous | 0.0028 | Absent NK cell function |
| STXBP2 | c.560C>T (p.Pro187Leu) | Heterozygous | 0.00064 | |||||
| 57 | Male | 4.1 | European-American | STXBP2 | c.194G>A(p.Arg65Gln) | Heterozygous | 0.0028 | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 58 | Female | 4.2 | European-American | STXBP2 | c.326-30_326-23del | Heterozygous | 0.0068 | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 59 | Male | 0.6 | Latino-Hispanic | STXBP2 | c.389T>C(p.Leu130Ser) | Homozygous | 0.0032 | Symptoms of HLH |
| 60 | Male | 45 d | African American | STXBP2 | c.389T>C(p.Leu130Ser) | Heterozygous | 0.0032 | Symptoms of HLH; family history of HLH |
| STXBP2 | exon 14-19 deletion | Heterozygous | ND | |||||
| 61 | Male | 0.7 | Middle Eastern | STXBP2 | c.481del(p.Arg161fs) | Homozygous | ND | Symptoms of HLH |
| 62 | Male | 0.4 | Unknown | STXBP2 | c.481del(p.Arg161fs) | Homozygous | ND | Symptoms of HLH |
| 63 | Female | 11 | European-American | STXBP2 | c.539_540delinsAA(p.Cys180*) | Heterozygous | ND | Symptoms of HLH |
| STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | |||||
| 64 | Male | 0.3 | Latino-Hispanic | STXBP2 | c.703C>G(p.Arg235Gly) | Homozygous | 0.00071 | Absent NK cell function |
| 65 | Female | 52.7 | European-American | STXBP2 | c.752C>T(p.Ala251Val) | Heterozygous | ND | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 66 | Male | 0.9 | Unknown | STXBP2 | c.902+5G>A | Heterozygous | 0.0036 | NA |
| STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | |||||
| 67 | Male | 3.1 | Unknown | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Symptoms of HLH |
| 68 | Female | 22.7 | Latino-Spanish | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Decreased NK cell function |
| 69 | Male | 26.4 | European-American | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Symptoms of HLH |
| 70 | Male | 25.6 | European-American | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | Symptoms of HLH |
| 71 | Female | 29.7 | European-American | STXBP2 | c.1247-1G>C | Homozygous | 0.02 | NA |
| 72 | Male | 4 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Absent NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 73 | Female | 15.8 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Absent NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 74 | Female | 19 | Unknown | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Decreased NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 75 | Female | 26.9 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Symptoms of HLH |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 76 | Female | 57.8 | European-American | STXBP2 | c.1247-1G>C | Heterozygous | 0.02 | Absent NK cell function |
| STXBP2 | c.1621G>A(p.Gly541Ser) | Heterozygous | 0.023 | |||||
| 77 | Female | 0.2 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 78 | Female | 0.3 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | NA |
| 79 | Female | 0.3 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 80 | Female | 0.6 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 81 | Male | 0.2 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | NA |
| 82 | Male | 0.7 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 83 | Female | 0.8 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 84 | Male | 0.8 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 85 | Male | 0.6 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Family history of HLH |
| 86 | Male | 1.6 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 87 | Male | 0.3 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 88 | Female | 63 d | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Family history of HLH |
| 89 | Male | 0.5 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 90 | Male | 0.2 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Symptoms of HLH |
| 91 | Male | 11.1 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Homozygous | 0.00074 | Absent NK cell function |
| 92 | Female | 10.1 | Unknown | STXBP2 | c.1430C>T(p.Pro477Leu) | Heterozygous | 0.00074 | Symptoms of HLH |
| STXBP2 | c.1696+5G>T | Heterozygous | ND | |||||
| 93 | Male | 0.5 | Middle Eastern | STXBP2 | c.1452+1G>A | Homozygous | ND | Symptoms of HLH |
| 94 | Male | 1 | Middle Eastern | STXBP2 | c.1452+1G>A | Homozygous | ND | Abnormal NK cell function |
| 95 | Female | 49 d | European-American | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Dysmorphic facies, decreased NK cell function |
| UNC13D | c.2258_2267delinsTACCTTGTTCGA (p.Gly753fs) | Heterozygous | ND | |||||
| 96 | Male | 0.7 | European-American | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Decreased NK cell function |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 97 | Male | 0.2 | European-American + Latino-Spanish | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Decreased NK cell function |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 98 | Female | 1.3 | Non-Hispanic white | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Symptoms of HLH, seizures, normal NK cell function |
| UNC13D | c.2867C>T(p.Pro956Leu) | Heterozygous | ND | |||||
| 99 | Female | 1 | Non-Hispanic white | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Absent NK cell function |
| UNC13D | c.3193C>T(p.Arg1065*) | Heterozygous | 0.0011 | |||||
| 100 | Female | 3.2 | European-American | UNC13D | c.118-308C>T | Heterozygous | 0.019 | Absent NK cell function |
| UNC13D | 253Kb inversion | Heterozygous | ND | |||||
| 101 | Male | 2.6 | Unknown | UNC13D | 253Kb inversion | Heterozygous | ND | Decreased NK cell function |
| UNC13D | c.154-1G>C | Heterozygous | ND | |||||
| 102 | Male | 0.2 | Unknown | UNC13D | 253Kb inversion | Heterozygous | ND | Symptoms of HLH |
| UNC13D | c.551G>A(p.Trp184*) | Heterozygous | 0.0011 | |||||
| 103 | Female | 0.2 | European-American | UNC13D | 253Kb inversion | Heterozygous | ND | NA |
| UNC13D | c.1389+1G>A | Heterozygous | 0.0071 | |||||
| 104 | Female | 0.2 | European-American | UNC13D | 253Kb inversion | Heterozygous | ND | Absent NK cell function |
| UNC13D | c.2447+1G>A | Heterozygous | 0.00051 | |||||
| 105 | Female | 0.6 | Non-Hispanic white | UNC13D | 253Kb inversion | Heterozygous | ND | Decreased NK cell function |
| UNC13D | c.2695C>T(p.Arg899*) | Heterozygous | 0.0018 | |||||
| 106 | Male | 11.4 | Hispanic white | UNC13D | c.182A>G(p.Tyr61Cys) | Heterozygous | ND | Symptoms of HLH |
| UNC13D | c.778T>C(p.Trp260Arg) | Heterozygous | ND | |||||
| 107 | Male | 0.4 | European-American | UNC13D | c.262-1G>A | Heterozygous | ND | Symptoms of HLH, abnormal NK cell function, family history of HLH |
| UNC13D | c.766C>T(p.Arg256*) | Heterozygous | 0.0025 | |||||
| 108 | Male | 0.4 | Unknown | UNC13D | c.321+1_321+2del | Heterozygous | ND | Decreased NK cell function |
| UNC13D | c.753+1G>T | Heterozygous | 0.0044 | |||||
| 109 | Male | 0.3 | Unknown | UNC13D | c.322-2A>T | Heterozygous | 0.0024 | Symptoms of HLH, decreased NK cell function |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 110 | Male | 0.3 | Unknown | UNC13D | c.419T>C(p.Ile140Thr) | Heterozygous | 0.0004 | NA |
| UNC13D | c.460C>T(p.Arg154Trp) | Heterozygous | 0.011 | |||||
| 111 | Female | 5.4 | Middle Eastern | UNC13D | c.424dup(p.Gln142fs) | Homozygous | ND | Symptoms of HLH |
| 112 | Male | 7.7 | Latino-Hispanic | UNC13D | c.518C>T(p.Thr173Met) | Heterozygous | 0.0028 | NA |
| UNC13D | c.1803_1819dup(p.Arg607fs) | Heterozygous | ND | |||||
| 113 | Female | 39 d | European-American | UNC13D | c.551G>A(p.Trp184*) | Heterozygous | 0.0011 | Abnormal NK cell function |
| UNC13D | c.766C>T(p.Arg256*) | Heterozygous | 0.0025 | |||||
| 114 | Male | 4.9 | European-American | UNC13D | c.570-2A>T | Heterozygous | ND | Absent NK cell function |
| UNC13D | c.3049G>A(p.Glu1017Lys) | Heterozygous | 0.00044 | |||||
| 115 | Female | 0.2 | Middle Eastern | UNC13D | c.753+1G>T | Homozygous | 0.0044 | Symptoms of HLH |
| 116 | Female | 0.6 | European-American | UNC13D | c.766C>T(p.Arg256*) | Heterozygous | 0.0025 | Symptoms of HLH, abnormal NK cell function |
| UNC13D | c.2447+1G>A | Heterozygous | 0.00051 | |||||
| 117 | Female | 1.2 | Latino-Spanish | UNC13D | c.859del(p.Arg287fs) | Homozygous | ND | Decreased NK cell function |
| 118 | Female | 1 | Non-Hispanic white | UNC13D | c.1055+1G>T | Heterozygous | ND | Decreased NK cell function, family history of HLH |
| UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | |||||
| 119 | Male | 0.7 | European-American | UNC13D | c.1229_1230dup(p.Arg411fs) | Heterozygous | 0.00 | Symptoms of HLH, hypertelorism |
| UNC13D | c.2298+1G>T | Heterozygous | ND | |||||
| 120 | Female | 0.2 | European-American | UNC13D | c.1259_1260del(p.Ser420fs) | Heterozygous | ND | symptoms of HLH, decreased NK cell function |
| UNC13D | c.1848+1G>C | Heterozygous | ND | |||||
| 121 | Male | 18.3 | European-American | UNC13D | c.1387C>T(p.Gln463*) | Heterozygous | ND | symptoms of HLH, decreased NK cell function |
| UNC13D | c.1820G>C(p.Arg607Pro) | Heterozygous | 0.011 | |||||
| 122 | Female | 58 d | European-American + African American | UNC13D | c.1389+1G>A | Heterozygous | 0.0071 | Symptoms of HLH |
| UNC13D | c.1848+1G>C | Heterozygous | ND | |||||
| 123 | Female | 0.2 | Middle Eastern | UNC13D | c.1423C>T(p.Gln475*) | Homozygous | ND | NA |
| 124 | Male | 10 d | Pacific Islander | UNC13D | c.2296C>T(p.Gln766*) | Homozygous | ND | Decreased NK cell function |
| 125 | Female | 13.2 | Unknown | UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | NA |
| UNC13D | c.2588G>A(p.Gly863Asp) | Heterozygous | 0.029 | |||||
| 126 | Female | 2 | Unknown | UNC13D | c.2346_2349del(p.Arg782fs) | Heterozygous | 0.01 | NA |
| UNC13D | c.3065T>C(p.Leu1022Pro) | Heterozygous | ND | |||||
| 127 | Female | 0.2 | Middle Eastern | UNC13D | c.2553+1G>T | Homozygous | ND | Symptoms of HLH |
| 128 | Female | 0.3 | Middle Eastern | UNC13D | c.2553+1G>T | Homozygous | ND | Symptoms of HLH |
| 129 | Female | 1 | Asian | UNC13D | c.2588G>A(p.Gly863Asp) | Homozygous | 0.029 | Decreased NK cell function |
| 130 | Female | 0.4 | African American | UNC13D | c.2695C>T(p.Arg899*) | Homozygous | 0.0018 | Symptoms of HLH, absent NK cell function, dysmorphic facies |
| 131 | Male | 0.7 | Non-Hispanic white | UNC13D | c.2819del(p.Leu940fs) | Homozygous | ND | symptoms of HLH |
| 132 | Female | 0.6 | Middle Eastern | UNC13D | c.3048dup(p.Glu1017fs) | Homozygous | ND | NA |
| 133 | Female | 0.7 | Unknown | UNC13D | c.3053C>A(p.Ala1018Asp) | Homozygous | 0.00088 | 2 affected siblings |
| 134 | Male | 13 | European-American | RAB27A | c.121A>G(p.Thr41Ala) | Heterozygous | ND | NA |
| RAB27A | c.352C>T(p.Gln118*) | Heterozygous | ND | |||||
| 135 | Male | 9.6 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | NA |
| 136 | Male | 9.9 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | NA |
| 137 | Female | 2 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Failure to thrive, bone marrow failure |
| 138 | Female | 9.4 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Symptoms of HLH |
| 139 | Male | 10.6 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Symptoms of HLH |
| 140 | Male | 19.5 | Middle Eastern | RAB27A | c.244C>T(p.Arg82Cys) | Homozygous | 0.0016 | Symptoms of HLH |
| 141 | Female | 0.3 | Latino-Hispanic | RAB27A | c.335del(p.Asn112fs) | Homozygous | 0.0044 | Symptoms of HLH |
| 142 | Female | 0.4 | Middle Eastern | RAB27A | c.400A>C(p.Lys134Gln) | Homozygous | ND | NA |
| 143 | Female | 53.9 | Pacific Islander | RAB27A | c.476A>G(p.Tyr159Cys) | Homozygous | ND | Symptoms of HLH, absent NK cell function |
| 144 | Male | 1.2 | Middle Eastern | RAB27A | c.598C>T(p.Arg200*) | Homozygous | 0.0004 | NA |
| 145 | Female | 0.4 | European-American | RAB27A | c.638_642del(p.Glu213fs) | Homozygous | 0.0008 | Symptoms of HLH |
| 146 | Female | 0.7 | African American | LYST | c.925C>T(p.Arg309*) | Heterozygous | ND | Oculocutaneous albinism, neutropenia |
| LYST | c.2015dup(p.Tyr672*) | Heterozygous | ND | |||||
| 147 | Male | 3.4 | Unknown | LYST | c.3194del(p.Leu1065*) | Homozygous | 0.0004 | NA |
| 148 | Female | 0.3 | Middle Eastern | LYST | c.4159dup(p.Thr1387fs) | Homozygous | ND | Premature gray hair, anemia |
| 149 | Male | 0.9 | European-American | LYST | c.5715del(p.Asn1905fs) | Heterozygous | ND | Oculocutaneous albinism, neutropenia, absent NK cell function |
| LYST | c.8802-2A>G | Heterozygous | ND | |||||
| 150 | Female | 6.9 | Unknown | LYST | c.5784+1G>T | Homozygous | ND | Oculocutaneous albinism, dysmorphic facies, neutropenia |
| 151 | Male | 3.2 | Non-Hispanic white | LYST | c.6159_6160del(p.Met2053fs) | Homozygous | ND | NA |
| 152 | Male | 1.8 | Middle Eastern | LYST | c.7291del(p.Leu2431fs) | Homozygous | ND | Hypopigmentation, anemia |
| 153 | Male | 1.2 | African American | LYST | c.8770C>T(p.Gln2924*) | Heterozygous | ND | Silver hair, hypopigmented skin lesions, pancytopenia |
| LYST | c.9844_9845del(p.Ser3282fs) | Heterozygous | ND | |||||
| 154 | Female | 7.1 | Middle Eastern | LYST | c.10776C>G(p.Tyr3592*) | Homozygous | ND | Abnormal pigmentation, neutropenia |
| 155 | Female | 1.2 | Unknown | STX11 | c.73G>T(p.Glu25*) | Heterozygous | 0.0004 | Decreased NK cell function |
| STX11 | c.748C>T(p.Gln250*) | Heterozygous | 0.00081 | |||||
| 156 | Female | 5.6 | Middle Eastern | STX11 | c.173T>C(p.Leu58Pro) | Homozygous | 0.0008 | Symptoms of HLH, grayish hair |
| 157 | Female | 11.6 | Middle Eastern | STX11 | c.173T>C(p.Leu58Pro) | Homozygous | 0.0008 | NA |
| 158 | Male | 2.9 | European-American + Latino-Hispanic | STX11 | c.462_463delinsA(p.Asp155fs) | Heterozygous | ND | Decreased NK cell function |
| STX11 | c.784C>T(p.Gln262*) | Heterozygous | ND | |||||
| 159 | Male | 1.4 | Asian-Indian | STX11 | c.687dup(p.Gln230fs) | Homozygous | ND | NA |
| 160 | Female | 12 d | Middle Eastern | SLC7A7 | c.1429+1G>C | Homozygous | ND | Family history of HLH |
| 161 | Male | 1.2 | African American | SLC7A7 | c.701del(p.Ser234fs) | Heterozygous | 0.0016 | NA |
| SLC7A7 | c.895-1G>A | Heterozygous | ND | |||||
| 162 | Female | 13.7 | European-American | SLC7A7 | c.360_361delinsAA (p.Trp121Arg) | Homozygous | ND | NA |
| 163 | Male | 5.4 | African American | XIAP | c.145C>T(p.Arg49*) | Hemizygous | ND | Markedly decreased XIAP expression |
| 164 | Male | 18.7 | European-American | XIAP | c.345C>G(p.Tyr115*) | Hemizygous | ND | Symptoms of HLH |
| 165 | Male | 30.3 | European-American | XIAP | c.608G>T(p.Cys203Phe) | Hemizygous | ND | NA |
| 166 | Male | 2 | European-American | XIAP | c.664C>T(p.Arg222*) | Hemizygous | ND | Symptoms of HLH |
| 167 | Male | 8.9 | European-American | XIAP | c.738del(p.Asp247fs) | Hemizygous | ND | NA |
| 168 | Male | 16 | European-American | XIAP | c.738del(p.Asp247fs) | Hemizygous | ND | Absent XIAP expression |
| 169 | Male | 3.6 | African American | XIAP | c.889A>T(p.Lys297*) | Hemizygous | ND | NA |
| 170 | Male | 0.4 | Unknown | XIAP | c.894_898del(p.Lys299fs) | Hemizygous | ND | NA |
| 171 | Male | 17.2 | European-American | XIAP | c.894_898del(p.Lys299fs) | Hemizygous | ND | Symptoms of HLH |
| 172 | Male | 19.6 | African American | XIAP | c.926_929del(p.Asp309fs) | Hemizygous | ND | Symptoms of HLH |
| 173 | Male | 17.2 | European-American | XIAP | c.969G>A(p.Trp323*) | Hemizygous | ND | Decreased XIAP expression |
| 174 | Male | 22.6 | Unknown | XIAP | c.1021_1022del(p.Asn341fs) | Hemizygous | ND | NA |
| 175 | Male | 4.5 | Unknown | XIAP | c.1056+1G>A | Hemizygous | ND | NA |
| 176 | Male | 2.7 | Latino-Hispanic | XIAP | c.1141C>T(p.Arg381*) | Hemizygous | ND | NA |
| 177 | Male | 11.8 | Unknown | XIAP | c.1141C>T(p.Arg381*) | Hemizygous | ND | Absent XIAP expression |
| 178 | Male | 1 d | Pacific-Islander | XIAP | c.1239_1242dup(p.Val415fs) | Hemizygous | ND | Markedly decreased XIAP expression |
| 179 | Male | 1.9 | Unknown | XIAP | c.1239_1242dup(p.Val415fs) | Hemizygous | ND | NA |
| 180 | Male | 1.1 | Unknown | XIAP | c.1301-1G>A | Hemizygous | ND | Symptoms of HLH |
| 181 | Male | 1.5 | Unknown | XIAP | c.1445C>G(p.Pro482Arg) | Hemizygous | ND | NA |
| 182 | Male | 35 | European-American | XIAP | c.1456dup(p.Thr486fs) | Hemizygous | ND | Symptoms of HLH |
| 183 | Male | 4.5 | European-American | SH2D1A | c.20A>G(p.Tyr7Cys) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 184 | Male | 1.8 | Unknown | SH2D1A | c.117C>T(p.Gly39Gly) | Hemizygous | ND | NA |
| 185 | Male | 3.5 | Unknown | SH2D1A | c.130T>C(p.Cys44Arg) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 186 | Male | 27 | European-American | SH2D1A | c.163C>T(p.Arg55*) | Hemizygous | ND | History of pneumonia |
| 187 | Male | 8 d | Unknown | SH2D1A | c.172C>T(p.Gln58*) | Hemizygous | ND | NA |
| 188 | Male | 6.8 | African American | SH2D1A | c.172C>T(p.Gln58*) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 189 | Male | 4.7 | Latino-Hispanic | SH2D1A | c.199_201+19del(p.Glu67del) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 190 | Male | 1.3 | African American | SH2D1A | c.201G>A(p.Glu67Glu) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 191 | Male | 7 d | European-American | SH2D1A | c.245del(p.Asn82fs) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 192 | Male | 34 d | European-American + Pacific-Islander | SH2D1A | c.295C>T(p.Gln99*) | Hemizygous | ND | Absent SAP in CD8+ T cells |
| 193 | Male | 5.4 | Middle-Eastern | MAGT1 | c.154_161delinsC(p.Ile52fs) | Hemizygous | ND | Symptoms of HLH, bone pain in low extremities |
| 194 | Male | 10.4 | European-American | MAGT1 | c.223C>T(p.Gln75*) | Hemizygous | ND | NA |
| 195 | Male | 18.4 | African American | MAGT1 | c.407G>A(p.Trp136*) | Hemizygous | ND | NA |
| 196 | Male | 27.6 | European-American | MAGT1 | c.443_444del(p.Phe148fs) | Hemizygous | ND | Symptoms of HLH |
| 197 | Male | 17.3 | European-American | MAGT1 | c.774del(p.Phe258fs) | Hemizygous | ND | NA |
NA, no data; ND, no data; NK, natural killer.
gnomAD v2.1.1 total population frequency.
HLH hemophagocytic lymphohistiocytosis, symptoms of HLH reported included any or all of the following “fever, hepatosplenomegaly, anemia/cytopenias, neutropenia/leukopenia, elevated ferritin/triglycerides, and/or decreased fibrinogen.”
According to the ACMG guideline, c.272C>T(p.Ala91Val) in PRF1 was classified as a variant of unknown significance.
HLH NGS panel
Fifteen genes that have been associated with HLH or HLH-like conditions were included in our HLH NGS panel: PRF1, UNC13D, STX11, STXBP2, ITK, CD27, SH2D1A, XIAP, MAGT1, LYST, RAB27A, AP3B1, BLOC1S6, SLC7A7, and GATA2. Their associated OMIM diseases, transcripts, and inheritance pattern are listed in supplemental Table 1. A typical turnaround time for this clinical testing is 4 weeks. Expedited turnaround time is available upon request.
NGS, data analysis, and Sanger confirmation
NGS was performed on the genomic DNA isolated from the patient samples using microdroplet polymerase chain reaction technology (RainDance Technologies Inc.) and sequenced on an Illumina HiSeq2500 instrument (Illumina Inc.). All exons, flanking intronic (±20 base pairs) and 5′ and 3′ untranslated regions of the 15 genes in the HLH panel (supplemental Table 1) were captured. Data for each sample were assessed for quality and confirmed they had at least 20× read depth at every target base. Sanger sequencing was performed to rescue all low coverage (<20× read depth) regions. Variants within those regions were identified and evaluated using a validated, custom bioinformatic pipeline. The American College of Medical Genetics and Genomics (ACMG) guidelines for sequence variant classification were used to categorize variants. All reported variants were confirmed by Sanger sequencing. In addition, allele-specific analysis for the 253-kb inversion as well as targeted analysis of the c.118-308 and c.118-307 regions in the UNC13D gene were performed for each sample because these variants have been reported to disrupt UNC13D transcription in lymphocytes and abolish Munc13-4 expression.15
We reviewed the results of the 1892 patient samples, excluded potential carriers based on clinical information provided, and reported the number of samples that were abnormal with pathogenic or likely pathogenic variants associated with HLH. Samples were classified according to the genes affected, types of mutations, and predicted impact on protein sequencing or structure. Pathogenic or likely pathogenic variants were identified in 10 genes: PRF1, STXBP2, UNC13D, LYST, RAB27A, STX11, SLC7A7, XIAP, SH2D1A, and MAGT1.
Results
At CCHMC, a 15-gene NGS panel for the molecular diagnosis of HLH disorders was offered from September 2013. Since then, the number of orders for traditionally sequential single-gene tests related to HLH disorders drastically decreased. As shown in Figure 1, from 2013 to 2018, the orders for single-gene Sanger sequencing such as PRF1, UNC13D, STXBP2, RAB27A, XIAP, and SH2D1A decreased from 308, 302, 277, 249, 132, and 104 in 2013 to 21, 3, 4, 1, 9, and 10 in 2018, respectively. On the other hand, the orders of HLH NGS panel jumped and maintained ∼400 test orders per year from 2014 to 2018.

Volume of HLH-related single gene and HLH panel testing in Cincinnati Children’s Hospital Medical Center from 2013 to 2018.
Volume of HLH-related single gene and HLH panel testing in Cincinnati Children’s Hospital Medical Center from 2013 to 2018.
A total of 1892 HLH panel testing results were analyzed, and clearly pathogenic and likely pathogenic variants were identified in 227 patients. Of these, 197 samples had a definite molecular genetic diagnosis: 87 samples with homozygotes and 75 with compound heterozygotes observed in a recessive condition, respectively, and 35 samples with hemizygotes observed in an X-linked disorder. This resulted in a positive molecular diagnostic rate of 10.4% (supplemental Figure 1). Table 1 lists the genetic variants identified in these 197 patients. Pathogenic or likely causal variants in the PRF1 gene were the most frequent and were identified in 26.4% (52/197) of patients (Figure 2A). Mutations in the genes associated with degranulation defects were more common than previously appreciated: 21.3% (42/197) of the patients had pathogenic or likely causal variants in STXBP2, 19.8% (39/197) in UNC13D, 6.1% (12/197) in RAB27A, 4.6% (9/197) in LYST, and 2.5% (5/197) in STX11. Pathogenic variants in the lysinuric protein intolerance gene SLC7A7 were identified in 1.5% (3/197) of the patients, the least frequent group of patients in our cohort. X-linked conditions accounted for 17.8% (35/197) of the patients: 20 (10.2%) patients had pathogenic or suspected diagnostic variants in XIAP, 10 (5.1%) in SH2D1A, and 5 (2.5%) in MAGT1 (Figure 2A). In addition, 30 of 227 patients with clinically suspected HLH were identified to carry only 1 pathogenic or likely pathogenic variant in a recessive condition by this panel approach. Among them, 50% (15/30) had a PRF1 pathogenic variant, 3 had a STXBP2 pathogenic variant, 4 had a UNC13D pathogenic variant, 5 had a RAB27 pathogenic variant, 2 had a LYST pathogenic variant, and 1 patient had a STX11 pathogenic variant. Of these 30 patients, 3 (cases S18, S23, and S30) also carried another common PRF1 variant c.272C>T (p.Ala91Val) in the heterozygous state (Table 2).

Characteristics of genetic findings and age ranges for 197 HLH patients. (A) Distribution of genetic findings in 197 HLH patients with a definite genetic diagnosis. (B) Whisker-box plot of the age ranges at referral for 197 HLH patients.
Characteristics of genetic findings and age ranges for 197 HLH patients. (A) Distribution of genetic findings in 197 HLH patients with a definite genetic diagnosis. (B) Whisker-box plot of the age ranges at referral for 197 HLH patients.
List of 30 patients in whom only 1 heterozygous pathogenic or likely pathogenic variant was identified
| Patient no. | Sex | Age at testing, y (unless indicated otherwise) | Ethnicity | Gene | Variant | Zygosity | Population frequency (gnomAD*), % | Symptoms/immunology testing/family history† |
|---|---|---|---|---|---|---|---|---|
| S01 | Female | 28.3 | Unknown | PRF1 | c.35_46del(p.Leu12_Leu15del) | Heterozygous | ND | NA |
| S02 | Male | 20 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Decreased NK cell function and perforin expression |
| S03 | Female | 0.4 | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Symptoms of HLH |
| S04 | Male | 7.8 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Decreased perforin expression |
| S05 | Female | 11.2 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent NK cell function |
| S06 | Male | 52 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | HLH, lymphoma |
| S07 | Male | 13.3 | Native American | PRF1 | c.112G>A(p.Val38Met) | Heterozygous | 0.0073 | Symptoms of HLH |
| S08 | Male | 16.6 | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | Symptoms of HLH |
| S09 | Female | 9.9 | Unknown | PRF1 | c.563C>T(p.Pro188Leu) | Heterozygous | 0.013 | Symptoms of HLH |
| S10 | Male | 17.8 | Unknown | PRF1 | c.853_855del(p.Lys285del) | Heterozygous | 0.0057 | Thrombocytopenia, absent NK cell function |
| S11 | Female | 3.1 | European-American | PRF1 | c.1066C>T(p.Arg356Trp) | Heterozygous | 0.0014 | NA |
| S12 | Female | 3 | Unknown | PRF1 | c.1117C>T(p.Arg373Cys) | Heterozygous | 0.0051 | Symptoms of HLH; normal NK cell function |
| S13 | Female | 16.7 | Unknown | PRF1 | c.1117C>T(p.Arg373Cys) | Heterozygous | 0.0051 | Absent NK cell function |
| S14 | Female | 12 d | Middle Eastern | PRF1 | c.1122G>A(p.Trp374*) | Heterozygous | 0.0016 | NA |
| S15 | Female | 9.7 | Malaysian-Chinese | PRF1 | c.1349C>T(p.Thr450Met) | Heterozygous | 0.0028 | History of HLH |
| S16 | Female | 0.5 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Heterozygous | 0.00074 | Symptoms of HLH |
| S17 | Female | 0.5 | Latino-Hispanic | STXBP2 | c.1717C>T(p.Pro573Ser) | Heterozygous | ND | NA |
| S18 | Male | 37 d | European-American | STXBP2 | c.1717C>T(p.Pro573Ser) | Heterozygous | ND | Absent NK cell function |
| PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | |||||
| S19 | Male | 14.9 | Asian-American | UNC13D | c.118-307G>A | Heterozygous | ND | Absent NK cell function |
| S20 | Male | 20.3 | Unknown | UNC13D | c.247C>T(p.Arg83*) | Heterozygous | 0.0004 | NA |
| S21 | Female | 62.9 | European-American | UNC13D | c.1759C>T(p.Arg587Cys) | Heterozygous | 0.019 | Symptoms of HLH |
| S22 | Female | 13.1 | European-American | UNC13D | c.2037_2038insG(p.Arg680fs) | Heterozygous | ND | Symptoms of HLH, one sibling deceased due to HLH |
| S23 | Male | 0.8 | Middle Eastern | RAB27A | c.148_149delinsC(p.Arg50fs) | Heterozygous | ND | Gray hair, suspected for GS, consanguinity |
| PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | |||||
| S24 | Male | 3 | European-American | RAB27A | c.240-47_240delins20 | Heterozygous | ND | Rash, neutropenia |
| S25 | Male | 11.1 | Indian | RAB27A | c.244C>T(p.Arg82Cys) | Heterozygous | 0.0016 | Symptoms of HLH |
| S26 | Female | 24.9 | Latino-Hispanic | RAB27A | c.335del(p.Asn112fs) | Heterozygous | 0.0044 | Symptoms of HLH |
| S27 | Male | 2.1 | Unknown | RAB27A | c.400_401del(p.Lys134fs) | Heterozygous | 0.0004 | Abnormal brain MRI, decreased NK cell function |
| S28 | Male | 29 | European-American | LYST | c.465_466del(p.Asp157fs) | Heterozygous | ND | Pancytopenia, increased ferritin level |
| S29 | Male | 6.4 | Middle Eastern | LYST | c.4159dup(p.Thr1387fs) | Heterozygous | ND | Gray hair |
| S30 | Female | 23.2 | Unknown | STX11 | c.650T>A(p.Leu217Gln) | Heterozygous | 0.0004 | NA |
| PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 |
| Patient no. | Sex | Age at testing, y (unless indicated otherwise) | Ethnicity | Gene | Variant | Zygosity | Population frequency (gnomAD*), % | Symptoms/immunology testing/family history† |
|---|---|---|---|---|---|---|---|---|
| S01 | Female | 28.3 | Unknown | PRF1 | c.35_46del(p.Leu12_Leu15del) | Heterozygous | ND | NA |
| S02 | Male | 20 d | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Decreased NK cell function and perforin expression |
| S03 | Female | 0.4 | Unknown | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Symptoms of HLH |
| S04 | Male | 7.8 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Decreased perforin expression |
| S05 | Female | 11.2 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | Absent NK cell function |
| S06 | Male | 52 | African American | PRF1 | c.50del(p.Leu17fs) | Heterozygous | 0.033 | HLH, lymphoma |
| S07 | Male | 13.3 | Native American | PRF1 | c.112G>A(p.Val38Met) | Heterozygous | 0.0073 | Symptoms of HLH |
| S08 | Male | 16.6 | Latino-Hispanic | PRF1 | c.445G>A(p.Gly149Ser) | Heterozygous | 0.014 | Symptoms of HLH |
| S09 | Female | 9.9 | Unknown | PRF1 | c.563C>T(p.Pro188Leu) | Heterozygous | 0.013 | Symptoms of HLH |
| S10 | Male | 17.8 | Unknown | PRF1 | c.853_855del(p.Lys285del) | Heterozygous | 0.0057 | Thrombocytopenia, absent NK cell function |
| S11 | Female | 3.1 | European-American | PRF1 | c.1066C>T(p.Arg356Trp) | Heterozygous | 0.0014 | NA |
| S12 | Female | 3 | Unknown | PRF1 | c.1117C>T(p.Arg373Cys) | Heterozygous | 0.0051 | Symptoms of HLH; normal NK cell function |
| S13 | Female | 16.7 | Unknown | PRF1 | c.1117C>T(p.Arg373Cys) | Heterozygous | 0.0051 | Absent NK cell function |
| S14 | Female | 12 d | Middle Eastern | PRF1 | c.1122G>A(p.Trp374*) | Heterozygous | 0.0016 | NA |
| S15 | Female | 9.7 | Malaysian-Chinese | PRF1 | c.1349C>T(p.Thr450Met) | Heterozygous | 0.0028 | History of HLH |
| S16 | Female | 0.5 | Middle Eastern | STXBP2 | c.1430C>T(p.Pro477Leu) | Heterozygous | 0.00074 | Symptoms of HLH |
| S17 | Female | 0.5 | Latino-Hispanic | STXBP2 | c.1717C>T(p.Pro573Ser) | Heterozygous | ND | NA |
| S18 | Male | 37 d | European-American | STXBP2 | c.1717C>T(p.Pro573Ser) | Heterozygous | ND | Absent NK cell function |
| PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | |||||
| S19 | Male | 14.9 | Asian-American | UNC13D | c.118-307G>A | Heterozygous | ND | Absent NK cell function |
| S20 | Male | 20.3 | Unknown | UNC13D | c.247C>T(p.Arg83*) | Heterozygous | 0.0004 | NA |
| S21 | Female | 62.9 | European-American | UNC13D | c.1759C>T(p.Arg587Cys) | Heterozygous | 0.019 | Symptoms of HLH |
| S22 | Female | 13.1 | European-American | UNC13D | c.2037_2038insG(p.Arg680fs) | Heterozygous | ND | Symptoms of HLH, one sibling deceased due to HLH |
| S23 | Male | 0.8 | Middle Eastern | RAB27A | c.148_149delinsC(p.Arg50fs) | Heterozygous | ND | Gray hair, suspected for GS, consanguinity |
| PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 | |||||
| S24 | Male | 3 | European-American | RAB27A | c.240-47_240delins20 | Heterozygous | ND | Rash, neutropenia |
| S25 | Male | 11.1 | Indian | RAB27A | c.244C>T(p.Arg82Cys) | Heterozygous | 0.0016 | Symptoms of HLH |
| S26 | Female | 24.9 | Latino-Hispanic | RAB27A | c.335del(p.Asn112fs) | Heterozygous | 0.0044 | Symptoms of HLH |
| S27 | Male | 2.1 | Unknown | RAB27A | c.400_401del(p.Lys134fs) | Heterozygous | 0.0004 | Abnormal brain MRI, decreased NK cell function |
| S28 | Male | 29 | European-American | LYST | c.465_466del(p.Asp157fs) | Heterozygous | ND | Pancytopenia, increased ferritin level |
| S29 | Male | 6.4 | Middle Eastern | LYST | c.4159dup(p.Thr1387fs) | Heterozygous | ND | Gray hair |
| S30 | Female | 23.2 | Unknown | STX11 | c.650T>A(p.Leu217Gln) | Heterozygous | 0.0004 | NA |
| PRF1 | c.272C>T(p.Ala91Val)‡ | Heterozygous | 2.92 |
gnomAD v2.1.1 total population frequency.
HLH hemophagocytic lymphohistiocytosis, symptoms of HLH reported included any or all of the following “fever, hepatosplenomegaly, anemia/cytopenias, neutropenia/leukopenia, elevated ferritin/triglycerides, and/or decreased fibrinogen.”
According to the ACMG guideline, c.272C>T(p.Ala91Val) in PRF1 was classified as a variant of unknown significance.
When the patients were divided based on age ranges, the diagnostic rates in patients aged 0 to 12 months, 1 to 5 years, 5 to 12 years, 12 to 18 years, and older than 18 years are 28.6% (95/332), 11.3% (43/380), 6.7% (25/371), 3.3% (10/304), and 4.8% (24/505), respectively. Moreover, patients with a molecular diagnosis in familial HLH type 2-5 genes (PRF1, UNC13D, STX11, and STXBP2; supplemental Table 1) tended to be referred and diagnosed at an earlier age compared with other genes (median age, 0.7 years [4 days-57.8 years] vs 4.5 years [1 day-53.9 years]; P = .009). Patients with X-linked conditions (XIAP, SH2D1A, and MAGT1) were referred and diagnosed at relatively older ages (median age, 5.4 years [1 day-35 years]) (Figure 2B).
Ten of 15 genes in the HLH sequencing panel were identified with pathogenic or likely pathogenic variants in our patient cohort, with the highest allele number and unique variants in the PRF1 gene (115 alleles; 45 unique variants). The majority of the identified PRF1 variants (66.7%; 30/45) were missense changes that were distributed along the exons without a particular hot spot. The most frequent pathogenic variant identified in our cohort was c.50del (p.Leu17fs), which appeared in 37 alleles in 27 patients including 10 homozygotes, 12 compound heterozygotes, and 5 heterozygotes. Of the 27 patients carrying this variant, 16 were African American, 3 were Latino-Spanish, 1 was Middle Eastern, and 7 were unknown. The other frequently detected variant was c.445G>A (p.Gly149Ser), which was identified in 19.2% (10/52) of the patients including 3 homozygotes, 6 compound heterozygotes, and 1 heterozygote. Among these 10 patients, 4 were Latino-Spanish, 1 was European-American, and 5 were unknown. (Tables 1 and 2; Figures 3 and 4A).

Distribution of unique pathogenic or likely pathogenic variants identified in 10 HLH-associated genes in our patient cohort.
Distribution of unique pathogenic or likely pathogenic variants identified in 10 HLH-associated genes in our patient cohort.
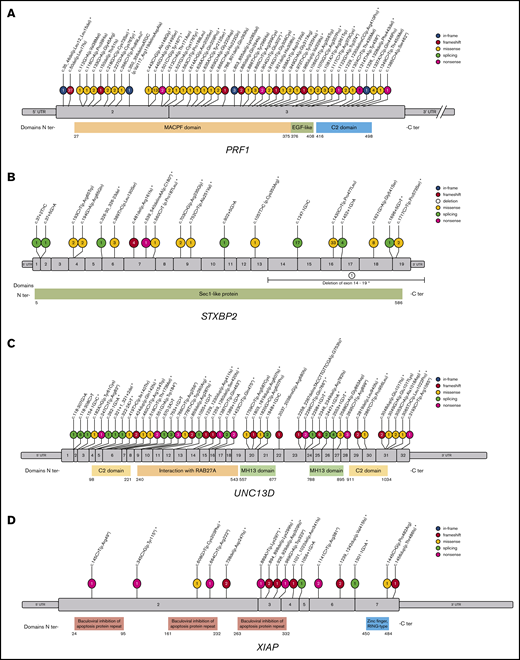
Distributions and frequencies of pathogenic or likely pathogenic variants in the most frequently affected genes associated with HLH in our cohort. (A) Distributions and frequencies of pathogenic or likely pathogenic variants in PRF1. (B) Distributions and frequencies of pathogenic or likely pathogenic variants in STXBP2. (C) Distributions and frequencies of pathogenic or likely pathogenic variants in UNC13D. Note: the 253-kb inversion is not shown in the graph. (D) Distributions and frequencies of pathogenic or likely pathogenic variants in XIAP. (A-D) The total number of alleles affected with each variant is indicated in the circles. *Novel variants.
Distributions and frequencies of pathogenic or likely pathogenic variants in the most frequently affected genes associated with HLH in our cohort. (A) Distributions and frequencies of pathogenic or likely pathogenic variants in PRF1. (B) Distributions and frequencies of pathogenic or likely pathogenic variants in STXBP2. (C) Distributions and frequencies of pathogenic or likely pathogenic variants in UNC13D. Note: the 253-kb inversion is not shown in the graph. (D) Distributions and frequencies of pathogenic or likely pathogenic variants in XIAP. (A-D) The total number of alleles affected with each variant is indicated in the circles. *Novel variants.
The second most frequently mutated gene in our cohort was the STXBP2 gene but with fewer unique variants (87 alleles, 20 unique variants). One-half (10/20) of unique STXBP2 variants were missense changes. As shown in Figure 4B, the missense variant c.1430C>T (p.Pro477Leu) and the splicing variant c.1247-1G>C were the 2 most frequent variants identified in STXBP2, accounting for 33 alleles in 18 patients and 17 alleles in 12 patients, respectively. Of the 18 patients carrying the c.1430C>T (p.Pro477Leu) variant, 13 were Middle Eastern and 5 were unknown. This result implies that this particular mutation in the STXBP2 gene identified in these patients might be identical by descent (ie, they might be inherited from a common ancestry). On the other hand, among the 12 patients who carry at least 1 splicing variant c.1247-1G>C, 8 were European-American, 1 was Latino-Spanish, and 3 were unknown (Tables 1 and 2; Figures 3 and 4B).
UNC13D was identified with a similar number of causal alleles to STXBP2 but with the doubled number of unique variants (82 alleles, 45 unique variants). Fourteen of 45 unique UNC13D variants were splicing mutations. The intronic variant c.118-308C>T and the 253-kb inversion were each identified in 6 patients in the compound heterozygous state along with a diverse second mutation, much higher than a recently reported Chinese cohort16 (Table 1; Figures 3 and 4C). As shown in Table 1, among the 39 patients with the UNC13D pathogenic or likely pathogenic variants, besides common HLH symptoms, cases 95 and 130 were also reported to have dysmorphic facies, case 98 seizures, and case 119 hypertelorism. Eleven mutated alleles and 7 unique alterations were identified in the STX11 gene in our cohort. Although most unique variants (5/7) are truncating variants, the missense variant c.173T>C (p.Leu58Pro) was the most frequently identified in STX11, appearing as 2 homozygotes in 2 patients with unknown relationship. Twenty-nine and 20 pathogenic or likely causal variants representing 11 and 13 unique genetic alterations were also identified in RAB27A and LYST, respectively. Most patients (7/9; 77.8%) with a molecular variation finding in the LYST gene were reported to have an albinism phenotype including silver hair, abnormal pigmentation, and oculocutaneous albinism (Tables 1 and 2; Figure 3; supplemental Figure 2).
Thirty unique pathogenic and likely pathogenic variants were identified in the 3 genes associated with an X-linked condition: 16, 9, and 5 unique variants in XIAP, SH2D1A, and MAGT1 were identified in 20, 10, and 5 male patients, respectively. Of these unique variants, 83% (25/30) were truncating variants which would presumably result in loss of function of the protein products (Table 1; Figures 2A, 3, and 4D; supplemental Figure 2).
Discussion
In this study, we analyzed 1892 samples tested for a panel of 15 HLH-associated genes, which were received between September 2013 and June 2018 at CCHMC. Both known and novel pathogenic or likely pathogenic variants have been identified in this population, and their frequency and distributions were analyzed. We found that 12% of the patients had at least 1 pathogenic or likely pathogenic variant in 1 of the 15 genes, and 10.4% of the patients had a definite molecular diagnosis in an HLH-associated gene. To our knowledge, this is the largest cohort of genetically diagnosed HLH, with the highest number of targeted HLH disease-causing genes tested simultaneously.
Patients with a definite genetic diagnosis were significantly younger than patients with only 1 heterozygote finding, with a median age of 14 months (197 cases) compared with 10 years (30 cases) (P = .007). When including all patients analyzed, we found that samples from younger patients were more likely to result in a genetic diagnosis (see "Results"). This observation in our cohort is consistent with previous reports in other similar studies.16-18
Together, the genes responsible for degranulation defects (UNC13D, STXBP2, STX11, LYST, RAB27A) accounted for the majority of cases (107/197; 54.3%). As seen in southern Europe,17 FHL2 and FHL3 account for a significant proportion of our cases (91/197; 46.2%). However, whereas FHL5 accounted for a minor proportion of HLH in Europeans, it is much more prevalent in our cohort (42/197; 21.3%). PRF1 was the most frequent causal gene (52/197; 26.4%). Although this finding is not surprising, other groups have described PRF1 defects in 40% to 60% of their cases.19,20 Nevertheless, all cohorts reported to date used different methods and criteria to define their cases; some included the variants of unknown significance, and others reported only the known significant biallelic or hemizygous variants. These inconsistent criteria make it difficult to compare the results among different groups. The lower percentage of PRF1 gene mutations described in our cohort may possibly reflect the use of immunological screening (perforin detection by flow cytometry) before sending for genetic diagnosis.
Compared with other large cohorts previously reported,16,17,21 our results show an extremely wide range of different mutations, reflecting the vast multiethnic population living in North America. Among the 227 cases, mutations were found in 10 different genes (PRF1, UNC13D, STX11, STXBP2, RAB27A, LYST, XIAP, SH2D1A, MAGT1, and SLC7A7), with a total of 175 different mutations. Of these unique mutations, 105 were found in only 1 patient. Only 4 variants were observed in more than 7 patients (PRF1: c.50del, c.445G>A; STXBP2: c.1247-1G>C, c.1430C>T). The well-described frameshift variant commonly found in patients with African American background, c.50del in PRF1,19,22 was the most common variant found in our cohort. The second most common mutation in PRF1 was c.445G>A, which has been previously described by multiple groups in different ethnic backgrounds, including Caucasian, Hispanic, Portuguese, German, and Chinese,16,19,20,23,24 reflecting the multiethnic population in North America. Interestingly, the commonly reported mutation in the Turkish population,20,25,26 c.1122G>A, was found in only 1 patient in our cohort. Similarly, we have only 1 case with c.1349C>T, the most common PRF1 mutation among the Chinese population,16 and no cases of c.1090_1091del or c.207del, variants commonly found in the Japanese population.27,28 One of the most prevalent mutations in our cohort, c.1247-1G>C in STXBP2, was previously described by other groups and found in multiple ethnic background including Caucasian, Turkish, Northern European, and Pakistani.5,29,30
Splicing mutations accounted for an important proportion of our UNC13D variants, with 14 of 45 unique mutations,31,32 which represents a different mutation spectrum of UNC13D variation from a Chinese cohort.16 We observed 3 cases of c.753+1G>T, a predominant mutation found in the German population20 as well as other parts of Europe (Italy33 and Croatia15 ). The deep intronic variant c.118-308C>T was observed in 6 unrelated patients, being one of the most frequent UNC13D mutations in our cohort. This is one of the most commonly reported mutations among the Korean population, but interestingly we had no cases of c.754-1G>C, the other very common mutation in Korea.32,34
The NGS-based approach is highly accurate in capturing point mutations and small deletions and insertions (such as <10 base pair deletions, duplication or insertion), which are the vast majority of sequence changes causing HLH in this 15-gene panel. However, our NGS-based diagnostic pipeline could not reliably identify large structural variations such as large deletions, duplications, or insertions occurring in the genes in this panel because of the limitation of sensitivity and specificity. Better algorithms that could use enriched NGS data to reliably identify these types of variation are warranted. It may be helpful for making a definite genetic diagnosis in the 30 patients in whom only 1 suspicious heterozygous variant in the genes associated with autosomal recessive conditions was identified.
Overall, a genetic diagnosis could be made in 10.4% of the patients using this HLH panel approach. The relatively low diagnostic rate can be explained by multiple factors. Complete clinical information and fulfillment of HLH clinical criteria is crucial to understanding our genetic results. As a heterogeneous disease with different disease mechanisms, many HLH-associated genes have yet to be discovered. In addition, some newly discovered HLH-associated genes such as NLRC435,36 and CDC4237 were not included in our panel and therefore could cause a missing genetic diagnosis for some of those negative cases. Some additional primary immune deficiencies have been reported to present with HLH, for example severe combined immunodeficiency, DiGeorge syndrome, Wiskott-Aldrich syndrome, chronic granulomatous disease, and STAT1 gain of function, among others.18,38-40 Several metabolic diseases also predispose to the development of HLH.41-44 With sequencing costs and analysis times going down, whole exome sequencing and even whole genome sequencing will provide more comprehensive solutions for detecting underlying genetic causes of HLH. Whole exome and whole genome sequencing would also identify genetic disorders that are associated with clinical phenotypes that mimic HLH. Indeed, a broader use of whole exome sequencing has already been recommended for patients with HLH.18 On the other hand, a significant proportion of the 1859 patients in this cohort likely had a secondary form of acquired HLH that developed in the context of malignancy, autoimmune disease, or infection, and whole exome and whole genome sequencing may not be a cost-effective approach for all patients with HLH at present. As the field of genetics continues to make clinical advances, clinicians should continue to weigh the ease, cost, completeness, and timeliness of genetic panel testing options for patients with HLH.
Data sharing requests can be e-mailed to the corresponding authors, Miao Sun (miao.sun@cchmc.org) or Rebecca A. Marsh (rebecca.marsh@cchmc.org).
Acknowledgments
The authors thank the physicians and genetic counselors that referred patients to our laboratory for HLH testing and the patients and their families that participated in this testing.
Authorship
Contribution: K.A.R., R.A.M., and M.S. designed the research; V.G.-L., L.D., R.S., J.C., K.Z., K.A.R., R.A.M., and M.S. analyzed data; V.G.-L. and M.S. interpreted results; V.G.-L., J.C., R.A.M., and M.S. wrote the manuscript; and all authors edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Miao Sun, Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: miao.sun@cchmc.org; and Rebecca A. Marsh, Division of Bone Marrow Transplant and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: rebecca.marsh@cchmc.org.

