

Key Points
PKCβ phosphorylation of occludin S490 is a downstream mediator of tPA-induced cerebrovascular permeability in ischemic stroke.
Inhibition of PKCβ prevents hemorrhagic transformation in ischemic stroke associated with delayed thrombolysis in mice.

Abstract
The current standard of care for moderate to severe ischemic stroke is thrombolytic therapy with tissue plasminogen activator (tPA). Treatment with tPA can significantly improve neurologic outcomes; however, thrombolytic therapy is associated with an increased risk of intracerebral hemorrhage (ICH). The risk of hemorrhage significantly limits the use of thrombolytic therapy, and identifying pathways induced by tPA that increase this risk could provide new therapeutic options to extend thrombolytic therapy to a wider patient population. Here, we investigate the role of protein kinase Cβ (PKCβ) phosphorylation of the tight junction protein occludin during ischemic stroke and its role in cerebrovascular permeability. We show that activation of this pathway by tPA is associated with an increased risk of ICH. Middle cerebral artery occlusion (MCAO) increased phosphorylation of occludin serine 490 (S490) in the ischemic penumbra in a tPA-dependent manner, as tPA−/− mice were significantly protected from MCAO-induced occludin phosphorylation. Intraventricular injection of tPA in the absence of ischemia was sufficient to induce occludin phosphorylation and vascular permeability in a PKCβ-dependent manner. Blocking occludin phosphorylation, either by targeted expression of a non-phosphorylatable form of occludin (S490A) or by pharmacologic inhibition of PKCβ, reduced MCAO-induced permeability and improved functional outcome. Furthermore, inhibiting PKCβ after MCAO prevented ICH associated with delayed thrombolysis. These results show that PKCβ phosphorylation of occludin is a downstream mediator of tPA-induced cerebrovascular permeability and suggest that PKCβ inhibitors could improve stroke outcome and prevent ICH associated with delayed thrombolysis, potentially extending the window for thrombolytic therapy in stroke.
Introduction
Stroke was the fifth leading cause of death in the United States in 20191 and is a major cause of adult disability.2 Approximately 87% of strokes are ischemic, and 13% are hemorrhagic.3 Compared with ischemic stroke, hemorrhagic strokes have a worse prognosis and higher mortality,4 and the hemorrhagic conversion of an ischemic stroke can significantly increase disability and mortality.5-7 The only pharmacologic therapy approved by the US Food and Drug Administration for acute ischemic stroke remains tissue plasminogen activator (tPA). However, its use is limited because current guidelines indicate it must be administered within 4.5 hours of stroke onset. This restriction is due in part to the risk of intracerebral hemorrhage (ICH) associated with thrombolytic tPA therapy, and it is estimated that only 5% to 7% of patients with ischemic stroke receive intravenous tPA, with another 1% to 2% receiving intra-arterial therapy.8,9 Importantly, the risk of ICH increases with time after stroke onset.10
Previous studies have indicated that tPA induces vessel permeability through activation of platelet-derived growth factor-C (PDGF-CC) and signaling through the PDGF receptor-α (PDGFRα) on glial fibrillary acidic protein (GFAP)-positive cells associated with the parenchymal side of the neurovascular unit (NVU).11,12 Furthermore, a number of studies have implicated this pathway in multiple central nervous system (CNS) disorders in which loss of blood–brain barrier (BBB) integrity is known to occur11-15 (as reviewed elsewhere16,17). Cleavage of the latent PDGF-CC dimer by tPA converts latent PDGF-CC (PDGF-CCL) to active PDGF-CC (PDGF-CCa) capable of binding to the PDGFRα and triggering signaling.18,19 In patients with ischemic stroke treated with thrombolytic tPA, there is a highly significant positive correlation between hemorrhagic transformation and the plasma concentrations of PDGF-CC.20 In addition, a phase 2 clinical trial in patients treated with intravenous tPA for ischemic stroke established that inhibition of PDGFRα signaling with the antagonist imatinib is safe and tolerable and may reduce neurologic disability in patients treated with thrombolysis after ischemic stroke.21 A randomized phase 3 clinical trial is underway (registered at ClinicalTrials.gov as #NCT03639922). However, how PDGFRα signaling in the NVU leads to the loss of endothelial barrier integrity and increases the risk of ICH is not known. Thus, understanding the downstream effects of tPA-mediated PDGFRα signaling has important clinical implications and may accelerate the development of adjuvant treatments that could reduce the risk of hemorrhagic complications associated with thrombolytic therapy.
An important component of the BBB is the well-developed tight junction complex present in endothelial cells of CNS vessels.22 Disruption of the tight junctions contributes to the underlying pathology of a number of diseases of the brain, including ischemic stroke, yet the pathologic mechanisms that lead to the loss of the tight junctions remain incompletely understood.23 Hypoxia-induced expression of vascular endothelial growth factor A (VEGF) contributes significantly to increased BBB permeability in stroke and to an increased risk of ICH.24,25 Blocking this pathway reduces ischemic stroke–associated hemorrhage,25 and importantly, inhibition of VEGF signaling attenuates the risk of hemorrhage associated with tPA-mediated thrombolysis.26
Studies of the blood–retinal barrier have shown that phosphorylation of the tight junction protein occludin regulates VEGF-induced vascular permeability.27,28 VEGF receptor-2 (VEGFR2) activation acts through protein kinase Cβ (PKCβ) to stimulate phosphorylation of occludin at serine 490 (S490). Occludin phosphorylation leads to its ubiquitination by the ligase Itch and trafficking of occludin to the cytoplasm where it is degraded.28,29 Other junctional proteins involved in barrier properties such as claudin-5 are internalized with occludin, resulting in increased paracellular gaps and permeability in retinal vessels. Expression of an S490A phospho-inhibitory mutant of occludin acts in a dominant manner to block VEGF-induced endothelial permeability,27,28 showing that stabilizing occludin at the junctional complex prevents or reduces VEGF-induced barrier breakdown and paracellular permeability. Recently, the role of occludin phosphorylation in regulation of the blood–retinal barrier was shown in vivo through the use of a transgenic mouse model expressing the S490AOCC stable mutant of occludin in vascular endothelial cells.30 VEGF-induced retinal permeability was reduced by one-half in mice expressing S490A occludin in vascular endothelial cells. Furthermore, expression of the point mutant completely blocked diabetes-induced increases in retinal vascular permeability and, importantly, prevented diabetes-induced visual loss. In the current report, we test the hypothesis that PKCβ-mediated phosphorylation of occludin in endothelial cells is a downstream pathway regulated by tPA-induced PDGF-CCa signaling in the NVU during ischemic stroke.
Materials and methods
Animals
All animal procedures were performed in accordance with local legislation and approved by the Institutional Animal Care and Use Committees at the University of Michigan or the Swedish National Board for Laboratory Animals and European Union Directive (2010/63/EU). Wild-type (WT) C57BL/6 male and female mice were from The Jackson Laboratory (Bar Harbor, ME) or Charles River Laboratories (Erkrath, Germany). Mice with human occludin S490 mutated to Ala (S490AOCC) under a floxed stop targeted to the ROSA26 locus were recently described.30 For dominant conditional vascular expression of the transgene, mice were crossed with PDGFb-(i)CreER mice.31 tPA-deficient (tPA−/−) (Plattm1Mlg) mice were backcrossed into C57BL/6 background.32
Middle cerebral artery occlusion stroke model
Middle cerebral artery occlusion (MCAO) was performed in age- and sex-matched C57BL/6 WT mice or transgenic mice as described previously.15,33 Late thrombolysis was as described,15 with modifications.12,33 Infarct and hemorrhage volume were determined as explained elsewhere.33,34 Vascular permeability was assessed by Evans blue dye as described,12,33 or by image analysis of 70-kDa dextran-Texas Red. Full details of the MCAO model are presented in the supplemental Materials and Methods (available on the Blood Web site).
Intracerebroventricular injection for immunoblot analysis
Intracerebroventricular (ICV) injection of phosphate-buffered saline (PBS), tPA, ANGPTL4, imatinib, PKCβ inhibitor LY-333531, or VEGFR2 kinase inhibitor I SU-5408 was performed in WT C57BL/6 mice, and permeability was determined 6 hours later by using Evans blue in one hemisphere. Immunoblotting was performed on the other hemisphere by using an anti–occludin-phospho-S490 specific antibody.35 Full details are presented in the supplemental Materials and methods.
Immunohistochemistry
Immunostaining of 10 µm coronal sections were quantified by using Imaris software (Bitplane, Belfast, United Kingdom). For S490 occludin phosphorylation, image intensity colocalizing with ZO-1 was quantified. Full details are presented in the supplemental Materials and methods.
Vascular fragment isolation and microarray analysis
PDGF-CCa protein or PBS was injected into the left lateral ventricle of C57BL/6 mice. Four hours later, the left-brain hemispheres were used for vascular fragment isolation and preparation of RNA. Total RNA from each sample was used to generate amplified and biotinylated sense-strand complementary DNA for Affymetrix analysis (Thermo Fisher Scientific, Waltham, MA). Full details are presented in the supplemental Materials and methods.
Corridor task
Lateralized sensory-motor integration was measured by using an adapted corridor task36,37 and is described in full in the supplemental Materials and Methods.
Results
Occludin S490 phosphorylation is induced by tPA after ischemic stroke
To determine whether stroke induces S490 phosphorylation of occludin, and if phosphorylation is regulated by tPA-induced PDGF-CCa signaling, we used a murine photo-thrombotic model of ischemic stroke.15 Brain regions ipsilateral and contralateral to MCAO were examined 3 and 24 hours after occlusion for occludin S490 phosphorylation by immunofluorescence microscopy with a previously characterized phospho-specific antibody.38 Total occludin staining of the cortical vasculature was evident in both the ischemic and contralateral hemispheres (Figure 1A). In contrast, staining of phosphoserine 490 (pS490) occludin at the junctional complex was largely absent in the nonischemic hemisphere but was increased adjacent to the ischemic core of the ipsilateral hemisphere at both 3 and 24 hours, indicating that MCAO induces occludin S490 phosphorylation.
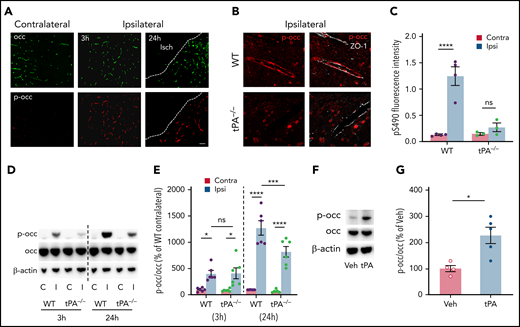
MCAO induces occludin S490 phosphorylation in a tPA-dependent manner. Mice were subjected to MCAO and killed at 3 and 24 hours after MCAO. (A) Stitched images of the region adjacent to the ischemic core (Isch) showing vessel pS490 occludin (red) and total occludin (green) staining in WT mice compared with contralateral hemisphere. Scale bar, 60 µm. (B) Confocal images zoomed in on individual vessels showing pS490 occludin (red) and ZO-1 (white) staining in tPA−/− mice after MCAO compared with WT mice 24 hours’ post-MCAO. Scale bar, 17 µm. (C) Quantification of pS490 fluorescence intensity at the junction 24 hours after MCAO in WT and tPA−/− mice in the contralateral (Contra) and ipsilateral (Ipsi) regions. (D) The amount of pS490 and total occludin in extracts from the ipsilateral (I) and contralateral (C) hemispheres was determined by western blot in WT and tPA−/− mice at 3 and 24 hours’ post-MCAO. (E) Quantification is expressed as relative to WT contralateral from each time point. (F) ICV injection with either PBS or tPA was performed in WT mice; after 6 hours, protein levels of pS490 and total occludin were determined by western blot of the whole brain extract. (G) Quantification is shown. One-way analysis of variance followed by Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. *P < .05, ***P < .001, ****P < .0001. ns, nonsignificant; Veh, vehicle.
MCAO induces occludin S490 phosphorylation in a tPA-dependent manner. Mice were subjected to MCAO and killed at 3 and 24 hours after MCAO. (A) Stitched images of the region adjacent to the ischemic core (Isch) showing vessel pS490 occludin (red) and total occludin (green) staining in WT mice compared with contralateral hemisphere. Scale bar, 60 µm. (B) Confocal images zoomed in on individual vessels showing pS490 occludin (red) and ZO-1 (white) staining in tPA−/− mice after MCAO compared with WT mice 24 hours’ post-MCAO. Scale bar, 17 µm. (C) Quantification of pS490 fluorescence intensity at the junction 24 hours after MCAO in WT and tPA−/− mice in the contralateral (Contra) and ipsilateral (Ipsi) regions. (D) The amount of pS490 and total occludin in extracts from the ipsilateral (I) and contralateral (C) hemispheres was determined by western blot in WT and tPA−/− mice at 3 and 24 hours’ post-MCAO. (E) Quantification is expressed as relative to WT contralateral from each time point. (F) ICV injection with either PBS or tPA was performed in WT mice; after 6 hours, protein levels of pS490 and total occludin were determined by western blot of the whole brain extract. (G) Quantification is shown. One-way analysis of variance followed by Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. *P < .05, ***P < .001, ****P < .0001. ns, nonsignificant; Veh, vehicle.
The presence of endogenous tPA activity has been shown to increase rapidly within the ischemic hemisphere after MCAO,39,40 and the early increase in tPA activity contributes to an increase in vascular permeability.15,40 To examine if endogenous tPA plays a role in occludin phosphorylation after MCAO, tPA−/− mice were also subjected to MCAO. Figure 1B presents confocal images of cortical vessels in WT and tPA−/− mice 24 hours after MCAO. A dramatic increase in occludin pS490 in the vessel wall was observed in the ischemic hemisphere of WT mice, which colocalizes with the tight junction–organizing protein ZO-1. However, in the peri-infarct area of tPA−/− mice, pS490 staining at the junctional complex was largely absent. Figure 1C shows the quantification of pS490 staining colocalizing with ZO1 in the peri-infarct area and a similar region from the contralateral hemisphere of both WT and tPA−/− mice. In WT mice, there was a significant increase in S490 phosphorylation in vessels adjacent to the ischemic core after MCAO that was not apparent in tPA−/− mice.
Western blotting of the cortex from each hemisphere for pS490 occludin in WT and tPA−/− mice at 3 and 24 hours after MCAO confirmed the immunofluorescence microscopy results indicating a critical role for tPA in stimulating occludin phosphorylation. S490 phospho-occludin was essentially undetectable in the nonischemic contralateral hemisphere but was clearly observed in the ipsilateral hemisphere at 3 hours after MCAO in WT mice and displayed an even greater increase at 24 hours. In tPA−/− mice, phosphorylation of S490 was significantly attenuated at the 24-hour time point but not the 3-hour time point compared with WT mice (Figure 1D-E). The apparent difference from immunostaining in tPA−/− mice with induced stroke likely results from the western blot detecting phospho-occludin in the entire peri-infarct area of the hemisphere and throughout the cell. Nevertheless, tPA−/− mice clearly had reduced occludin phosphorylation consistent with multiple factors inducing phosphorylation, with an increasing role for tPA over time. To determine if tPA was sufficient to induce occludin phosphorylation, ICV injection of tPA was performed in WT mice. These results showed that in the absence of an ischemic insult, tPA could directly induce a significant increase in occludin phosphorylation by 6 hours compared with vehicle (Figure 1F-G). Together, these results reveal that tPA is both necessary to induce robust occludin S490 phosphorylation following an ischemic insult and sufficient to induce phosphorylation in the absence of cerebral ischemia.
tPA/PDGF-CCa signaling increases occludin phosphorylation and permeability
Previously, it was shown that during ischemic stroke, endogenous tPA increases BBB permeability through activation of latent PDGF-CCL and subsequent signaling by the PDGFRα15 in GFAP-positive perivascular astrocytes on the parenchymal side of the NVU.11,12 To explore how PDGFRα signaling in perivascular astrocytes leads to occludin phosphorylation in endothelial cells and increased BBB permeability, we performed expression profiling of isolated vascular fragments containing endothelial cells, pericytes/vascular smooth muscle cells, and perivascular astrocytes41 4 hours after ICV injection of PDGF-CCa in naive WT mice. RNA expression was analyzed by using an Affymetrix mouse GeneChip 2.0 ST array (Thermo Fisher Scientific). Using a P value <.05 and log 2-fold change >0.5 or <0.5, a total of 207 messenger RNAs (mRNAs) were identified that were significantly differentially expressed in the mice treated with PDGF-CCa compared with control, with the expression of 69 genes increasing and 138 decreasing (supplemental Table 1). Functional clustering and pathway analysis did not identify any highly significant pathways or reactome interactions, with most showing only 3 or 4 linked genes.
However, cytokines known to alter vascular permeability were identified in the array analysis. Both Tnf, which was upregulated by 2.3-fold, and Angptl4, which was downregulated by 3.4-fold, were 2 of the top 10 most-altered mRNAs (Figure 2A). Interestingly, Angptl4 was the second most affected mRNA, with only an uncharacterized open reading frame showing a greater fold change in expression. Furthermore, Angptl4 was previously reported to inhibit VEGF receptor signaling.42,43 The change in Angptl4 mRNA expression was confirmed by quantitative reverse transcription polymerase chain reaction from a different set of vascular fractions collected 4 hours after PDGF-CCa or tPA ICV injection and compared with animals injected with vehicle revealing a 3.2-fold decrease with PDGF-CCa or a 3.0-fold decrease with tPA (Figure 2B). Furthermore, vascular fragments were isolated from ipsilateral hemispheres collected 24 hours after MCAO and compared with vascular fragments from sham-operated mice for Angptl4 mRNA. These data show that by 24 hours after MCAO, the expression of Angptl4 is significantly downregulated by 2.7-fold (Figure 2C). Collectively, these data suggest that tPA-induced PDGFRα signaling during ischemic stroke can inhibit the expression of Angptl4 and that this action could potentially enhance VEGF activity in the NVU.

tPA/PDGF-CCa signaling increases occludin phosphorylation and permeability. (A) Gene expression heatmap of the top differentially expressed genes in isolated vascular fragments from WT mice 4 hours after ICV injection of either PBS or PDGF-CCa. (B) The vascular fragments from WT mice were isolated 4 hours after ICV injection of PBS, PDGF-CCa, or tPA and were analyzed by using quantitative polymerase chain reaction for differential expression of Angptl4. (C) The isolated vascular fraction from WT mice, either control without MCAO or isolated 24 hours after MCAO, was analyzed by using quantitative polymerase chain reaction for the expression of Angptl4. (D) WT mice were given an ICV injection with PBS, tPA, or tPA in combination with imatinib, or ANGPTL4, or VEGFR2 inhibitor (VEGFR2i), or PKCβ inhibitor (PKCβi). At 5 hours, Evans blue was injected intravenously; at 6 hours, animals were perfused with PBS. One brain hemisphere was blotted to determine pS490 and total occludin protein levels. (E) Quantification of blot S490 phosphorylation. (F) The other brain hemisphere was processed for BBB permeability by measuring Evans blue dye extravasation. (G) BBB permeability was assessed after 6 hours of either saline or tPA ICV injection in PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice by measuring Evans blue dye extravasation. One-way analysis of variance followed by a Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, nonsignificant; Veh, vehicle.
tPA/PDGF-CCa signaling increases occludin phosphorylation and permeability. (A) Gene expression heatmap of the top differentially expressed genes in isolated vascular fragments from WT mice 4 hours after ICV injection of either PBS or PDGF-CCa. (B) The vascular fragments from WT mice were isolated 4 hours after ICV injection of PBS, PDGF-CCa, or tPA and were analyzed by using quantitative polymerase chain reaction for differential expression of Angptl4. (C) The isolated vascular fraction from WT mice, either control without MCAO or isolated 24 hours after MCAO, was analyzed by using quantitative polymerase chain reaction for the expression of Angptl4. (D) WT mice were given an ICV injection with PBS, tPA, or tPA in combination with imatinib, or ANGPTL4, or VEGFR2 inhibitor (VEGFR2i), or PKCβ inhibitor (PKCβi). At 5 hours, Evans blue was injected intravenously; at 6 hours, animals were perfused with PBS. One brain hemisphere was blotted to determine pS490 and total occludin protein levels. (E) Quantification of blot S490 phosphorylation. (F) The other brain hemisphere was processed for BBB permeability by measuring Evans blue dye extravasation. (G) BBB permeability was assessed after 6 hours of either saline or tPA ICV injection in PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice by measuring Evans blue dye extravasation. One-way analysis of variance followed by a Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, nonsignificant; Veh, vehicle.
ICV injections were used to examine the contribution of tPA to occludin phosphorylation and vascular permeability through its action on PDGFRα signaling and the downstream effects on ANGPTL4, VEGFR2, and the signal transduction kinase PKCβ, which phosphorylates occludin S490. For this analysis, tPA was injected via ICV in WT mice together with specific inhibitors of PDGFRα, VEGFR2, or PKCβ, or with recombinant ANGPTL4. These data show that the ICV injection of tPA in naive WT mice induces a significant increase in occludin phosphorylation compared with saline-injected mice (Figure 2D-E), coincident with an expected12,15 increase in cerebral permeability to Evans blue dye 6 hours after tPA injection (Figure 2F). However, coinjection of tPA with the PDGFRα inhibitor imatinib, the VEGFR2 inhibitor SU-5408, the PKCβ inhibitor LY-333531, or ANGPTL4 protein blocked both occludin phosphorylation and cerebral permeability (Figure 2D-F).
To directly test the hypothesis that occludin phosphorylation in endothelial cells regulates BBB permeability in response to tPA, a transgenic mouse model with vascular endothelial cell conditional expression of an S490A mutant of occludin was used. The occludin S490A mutant is incorporated at the Rosa26 site under a floxed stop.30 By crossing these mice with the tamoxifen-inducible endothelial cell–specific PDGFb-(i)CreER mice, the transgene was expressed in a vascular endothelial cell–restricted manner by tamoxifen injection at postnatal day 3 (supplemental Figure 1). ICV injections of tPA in PDGFbiCre+; S490AOCC+/+ mice vs littermate control PDGFbiCre+ mice showed that mice expressing the dominant occludin S490A mutant in endothelial cells were completely protected from tPA-induced Evans blue dye extravasation (Figure 2G). These data support a pathway in which tPA-mediated cleavage and activation of PDGF-CCL induces PDGFRα signaling, with subsequent reduction of ANGPTL4 expression promoting VEGFR2 signaling in endothelial cells and PKCβ phosphorylation of S490 on occludin27,44 leading to the loss of tight junction integrity.
Inhibition of occludin S490 phosphorylation prevents MCAO-induced permeability
We next examined if PKCβ-mediated occludin phosphorylation plays a role in vascular permeability after ischemic stroke. For these studies, MCAO was induced in control PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice, and the extravasation of fluorescently labeled (Texas red) 70 kDa dextran was determined after PBS perfusion 24 hours later. As expected, MCAO induced a highly significant increase in permeability to 70-kDa dextran after MCAO in the PDGFiCre+ control mice compared with the nonischemic contralateral hemisphere as determined by quantitative fluorescent microscopy. In contrast, expression of S490A occludin completely prevented the increase in BBB permeability (Figure 3A).

Inhibition of occludin S490 phosphorylation prevents MCAO-induced permeability. (A) PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice were subjected to MCAO, and BBB permeability was determined 24 hours later by quantifying a 70-kDa fluorescent dextran leak in cross-sections from both the ipsilateral (Ipsi) and the contralateral (Contra) region. (B) WT mice were given vehicle (Veh) or the PKCβ inhibitor (PKCβi; 10 mg/kg) once a day for 3 days and then subjected to MCAO followed by one additional dose of the inhibitor 1 hour later. pS490 occludin (red) and total occludin (green) were detected by immunostaining in brain sections 24 hours after MCAO. Scale bar, 25 µm. Quantification of pS490 at the junction (C) and total occludin from confocal images (D) 24 hours after MCAO. (E) Image of BBB permeability to 70-kDa dextran in entire coronal sections in both contralateral and ipsilateral hemispheres 24 hours after MCAO. Scale bar, 600 µm. (F) Quantification of dextran leak in cross-sections 24 hours after MCAO. (G) BBB permeability to 70-kDa dextran was also assessed by quantifying the amount of extravasated dye in brain homogenates 24 hours after MCAO. One-way analysis of variance followed by a Holm-Šídák post hoc test. *P < .05, ****P < .0001. ns, nonsignificant.
Inhibition of occludin S490 phosphorylation prevents MCAO-induced permeability. (A) PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice were subjected to MCAO, and BBB permeability was determined 24 hours later by quantifying a 70-kDa fluorescent dextran leak in cross-sections from both the ipsilateral (Ipsi) and the contralateral (Contra) region. (B) WT mice were given vehicle (Veh) or the PKCβ inhibitor (PKCβi; 10 mg/kg) once a day for 3 days and then subjected to MCAO followed by one additional dose of the inhibitor 1 hour later. pS490 occludin (red) and total occludin (green) were detected by immunostaining in brain sections 24 hours after MCAO. Scale bar, 25 µm. Quantification of pS490 at the junction (C) and total occludin from confocal images (D) 24 hours after MCAO. (E) Image of BBB permeability to 70-kDa dextran in entire coronal sections in both contralateral and ipsilateral hemispheres 24 hours after MCAO. Scale bar, 600 µm. (F) Quantification of dextran leak in cross-sections 24 hours after MCAO. (G) BBB permeability to 70-kDa dextran was also assessed by quantifying the amount of extravasated dye in brain homogenates 24 hours after MCAO. One-way analysis of variance followed by a Holm-Šídák post hoc test. *P < .05, ****P < .0001. ns, nonsignificant.
Because PKCβ inhibition could block tPA-induced permeability after ICV injection by preventing occludin phosphorylation, we reasoned that PKCβ inhibition would also reduce occludin phosphorylation and BBB permeability after MCAO. For these studies, the PKCβ-specific inhibitor was injected daily at 10 mg/kg intraperitoneally for 4 days, beginning 3 days before MCAO, and permeability was measured 24 hours after MCAO. The results showed that the PKCβ inhibitor significantly blocked MCAO-induced S490 occludin phosphorylation in the peri-infarct regions but did not alter total occludin (Figure 3B-D). BBB permeability to Texas red–conjugated 70-kDa dextran was clearly observed in whole brain cross-sections after MCAO and was nearly completely blocked by PKCβ inhibitor pretreatment (Figure 3E-F). These results were verified by repeating the experiment and extracting the dextran for quantification, and again the PKCβ inhibitor–treated group had significantly reduced permeability to dextran after MCAO compared to control animals (Figure 3G). Collectively, these studies show that PKCβ phosphorylation of occludin S490 contributes to MCAO-induced permeability.
Inflammatory cells, including neutrophils, are recruited to the brain parenchyma in response to ischemia, contributing to the pathogenesis of stroke.45 We next tested if PKCβ inhibition also reduced the inflammation induced by MCAO. Brain sections from animals treated with either vehicle or PKCβ inhibitor, as noted earlier, were stained for CD45 (leukocyte marker) and IB4 (vessel marker, which is known to also bind to some leukocytes46) or Ly6G (neutrophil marker). By 24 hours after MCAO, there was a significant increase in CD45+cells in the infarct area as previously shown,12 with both increased vessel leukostasis and infiltration. Treatment with the PKCβ inhibitor significantly reduced the overall number of both CD45+ (Figure 4A-B) and Ly6G+(Figure 4C-D) cells. However, it was not clear if this treatment reduced the extent of leukostasis, as leukocytes were present in the larger vessels both with and without treatment.
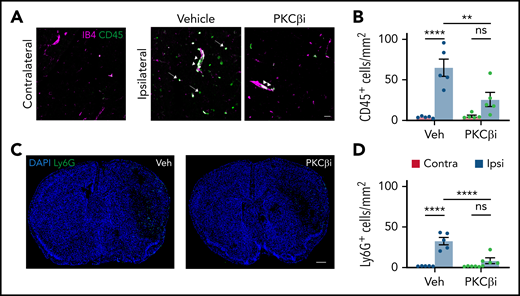
PKCβ inhibition reduces MCAO-induced inflammation. Mice were treated with either vehicle (Veh) or the PKCβ inhibitor (PKCβi; 10 mg/kg) once a day for 3 days and then subjected to MCAO followed by 1 additional dose of the inhibitor 1 hour later. (A) Twenty-four hours after MCAO, representative confocal images were obtained from the contralateral and ipsilateral (ischemic core) regions stained with vessel marker IB4 (purple) and leukocyte marker CD45 (green) showing leukostasis (arrowheads) and infiltration of CD45+ cells to the parenchyma (arrows). Scale bar, 30 µm. (C) Stitched confocal images of the whole brain section showing the contralateral region on the left and ipsilateral on the right, stained with neutrophil marker Ly6G (green) and nuclei marker 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bar, 500 µm. The number of CD45+ (B) and Ly6G+ (D) cells was counted in both contralateral (red bars) and ipsilateral (blue bars) hemispheres. Data are represented as mean ± standard error of the mean. One-way analysis of variance followed by a Holm-Šídák post hoc test. **P < .01, ****P < .0001. ns, nonsignificant.
PKCβ inhibition reduces MCAO-induced inflammation. Mice were treated with either vehicle (Veh) or the PKCβ inhibitor (PKCβi; 10 mg/kg) once a day for 3 days and then subjected to MCAO followed by 1 additional dose of the inhibitor 1 hour later. (A) Twenty-four hours after MCAO, representative confocal images were obtained from the contralateral and ipsilateral (ischemic core) regions stained with vessel marker IB4 (purple) and leukocyte marker CD45 (green) showing leukostasis (arrowheads) and infiltration of CD45+ cells to the parenchyma (arrows). Scale bar, 30 µm. (C) Stitched confocal images of the whole brain section showing the contralateral region on the left and ipsilateral on the right, stained with neutrophil marker Ly6G (green) and nuclei marker 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bar, 500 µm. The number of CD45+ (B) and Ly6G+ (D) cells was counted in both contralateral (red bars) and ipsilateral (blue bars) hemispheres. Data are represented as mean ± standard error of the mean. One-way analysis of variance followed by a Holm-Šídák post hoc test. **P < .01, ****P < .0001. ns, nonsignificant.
PKCβ inhibition decreases infarct volume and improves functional outcome
To determine if PKCβ inhibition can improve stroke outcome, the cerebral infarct area was determined by 2,3,5-triphenyltetrazolium chloride staining at 3 and 7 days after MCAO in the vehicle or PKCβ inhibitor–treated mice. For this study, the PKCβ-specific inhibitor was injected daily at 10 mg/kg intraperitoneally for 6 days, beginning 3 days before MCAO and continuing for 3 days after MCAO. These data indicate that compared with vehicle-treated mice, the infarct volume was significantly reduced 3 days after MCAO in the PKCβ inhibitor–treated mice group (Figure 5A), and this benefit was sustained 7 days after MCAO (Figure 5B-C).

PKCβ inhibition decreases infarct volume and improves functional outcome. WT mice were given vehicle or the PKCβ inhibitor (PKCβi; 10 mg/kg) once a day for 3 days and then subjected to MCAO followed by 3 additional daily doses of the inhibitor. Infarct volume was assessed by using 2,3,5-triphenyltetrazolium chloride (TTC) staining at 72 hours (A) and 7 days (B) after MCAO. (C) Representative images of TTC staining 7 days after MCAO. (D) Functional outcome was measured by assessing the lateralized bias in the corridor test 7 days after MCAO. (E) Pearson’s correlation between the lateralized bias and infarct volume in vehicle- and PKCβi-treated mice, 7 days after MCAO. One-way analysis of variance followed by a Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. **P < .01, ****P < .0001. Contra, contralateral; Ipsi, ipsilateral; ns, nonsignificant; Veh, vehicle.
PKCβ inhibition decreases infarct volume and improves functional outcome. WT mice were given vehicle or the PKCβ inhibitor (PKCβi; 10 mg/kg) once a day for 3 days and then subjected to MCAO followed by 3 additional daily doses of the inhibitor. Infarct volume was assessed by using 2,3,5-triphenyltetrazolium chloride (TTC) staining at 72 hours (A) and 7 days (B) after MCAO. (C) Representative images of TTC staining 7 days after MCAO. (D) Functional outcome was measured by assessing the lateralized bias in the corridor test 7 days after MCAO. (E) Pearson’s correlation between the lateralized bias and infarct volume in vehicle- and PKCβi-treated mice, 7 days after MCAO. One-way analysis of variance followed by a Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. **P < .01, ****P < .0001. Contra, contralateral; Ipsi, ipsilateral; ns, nonsignificant; Veh, vehicle.
To assess whether inhibition of PKCβ rescues behavioral deficits after ischemic stroke, we performed a lateralized, sensory motor integration test. This behavioral test measures the lateralized bias in exploring sugar pellets in a corridor task.36,37,47 MCAO was induced on the left side and caused damage to the left cerebral hemisphere, which consequently affected sensory-motor functions on the right side; therefore, a bias in ipsilateral explorations (left-side explorations) is expected after MCAO. We observed a significant increase in ipsilateral bias 7 days after MCAO in WT mice compared with sham-operated mice. In contrast, PKCβ inhibitor treatment in WT mice showed a significant recovery in contralateral explorations compared with vehicle-treated WT mice. No significant difference was observed in exploration bias between sham-operated mice without MCAO and PKCβ inhibitor–treated WT mice with MCAO, indicating complete rescue of the lateralized bias in WT mice by PKCβ treatment (Figure 5D). Importantly, there was a significant positive correlation between brain infarct volume and ipsilateral bias, validating the corridor test as a measure of MCAO functional outcome (Figure 5E).
Inhibition of PKCβ prevents hemorrhagic transformation after delayed tPA treatment
We have previously shown that delayed thrombolysis, 5 hours after photo-thrombotic MCAO, is associated with an increase in ICH.12,15 To determine if occludin phosphorylation is necessary for the observed increase in ICH associated with delayed tPA thrombolysis, PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice expressing the S490A mutant of occludin were subjected to MCAO and then treated with thrombolytic tPA 5 hours after stroke. The mice were then analyzed for hemorrhage volume in serial brain section images 72 hours after MCAO. Figure 6A shows a clear increase in hemorrhagic transformation associated with delayed tPA treatment in the PDGFiCre+ animals compared with animals with induced stroke without tPA. However, expression of S490A occludin profoundly inhibited ICH associated with delayed tPA compared with control PDGFiCre+ mice treated identically (Figure 6A-B).

Inhibition of PKCβ prevents hemorrhagic transformation after delayed tPA treatment. (A) PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice were subjected to MCAO and then treated intravenously with either PBS or tPA (10 mg/kg) 5 hours after MCAO. (B) The volume of hemorrhage was quantified from serial brain sections 72 hours after MCAO. (C) Mice were subjected to MCAO; 1 hour (light blue bars) or 5 hours (green bars) later, animals were treated with vehicle (Veh) or PKCβ inhibitor (PKCβi; 10 mg/kg) daily for 3 days. Delayed tPA thrombolysis was performed 5 hours after MCAO, and brains were analyzed at 72 hours after MCAO. (D) Hemorrhage volume was measured 72 hours after MCAO. One-way analysis of variance followed by a Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. ***P < .001, ****P < .0001. ns, nonsignificant.
Inhibition of PKCβ prevents hemorrhagic transformation after delayed tPA treatment. (A) PDGFiCre+ mice and PDGFiCre+; S490AOCC+/+ mice were subjected to MCAO and then treated intravenously with either PBS or tPA (10 mg/kg) 5 hours after MCAO. (B) The volume of hemorrhage was quantified from serial brain sections 72 hours after MCAO. (C) Mice were subjected to MCAO; 1 hour (light blue bars) or 5 hours (green bars) later, animals were treated with vehicle (Veh) or PKCβ inhibitor (PKCβi; 10 mg/kg) daily for 3 days. Delayed tPA thrombolysis was performed 5 hours after MCAO, and brains were analyzed at 72 hours after MCAO. (D) Hemorrhage volume was measured 72 hours after MCAO. One-way analysis of variance followed by a Holm-Šídák post hoc test was used for comparison of ≥3 groups; a t test was used for comparison between 2 groups. ***P < .001, ****P < .0001. ns, nonsignificant.
Finally, the efficacy of PKCβ inhibition for preventing hemorrhagic transformation in WT mice with delayed thrombolysis was measured after MCAO. For these studies, a therapeutic treatment regimen was used instead of prophylactic treatment. MCAO was induced by photo-thrombosis, and PKCβ inhibitor was given daily (10 mg/kg) starting either 1 hour after MCAO or 5 hours after MCAO (Figure 6D) and continued for 3 days. Thrombolysis was initiated with tPA at 5 hours after MCAO, and hemorrhage was assessed at 72 hours as described earlier. Again, delayed thrombolysis with tPA induced significant ICH compared with no tPA (vehicle control), whereas treatment with the PKCβ inhibitor (either 1 hour post-MCAO or 5 hours’ post-MCAO) prevented ICH that was not significantly different from no tPA (Figure 6C-D). These results suggest that therapeutic PKCβ inhibition given after the ischemic event can prevent stroke-induced hemorrhagic transformation associated with delayed tPA treatment by blocking occludin S490 phosphorylation.
Discussion
Recent studies have shown that preserving the BBB significantly improves functional outcome after stroke in mice.15,34,48 The current work points to PKCβ phosphorylation of occludin as a major regulator of barrier permeability in the CNS in response to ischemia. MCAO induces occludin phosphorylation on S490 downstream of PKCβ, and this phosphorylation was required for cerebral vascular permeability in a thrombotic stroke model. Preventing occludin S490 phosphorylation either genetically by expressing the S490A point mutant or targeting the relevant kinase, PKCβ, with a specific inhibitor blocks MCAO-induced permeability. The signaling pathway induced by MCAO involves endogenous tPA activation of PDGF-CCL, Angptl4 downregulation, and VEGFR2 activation of PKCβ and occludin phosphorylation (Figure 7). Furthermore, the high systemic concentrations of recombinant tPA achieved during therapeutic thrombolysis can further increase permeability in an occludin phosphorylation–dependent manner. The extent to which thrombolytic tPA exacerbates barrier permeability and increases the risk of ICH is likely related to its penetration across the BBB, as the effects of thrombolytic tPA on the BBB seem to increase with time after stroke as spontaneous barrier dysfunction is intensifying.49 Importantly, our data show that PKCβ inhibitor treatment given after stroke was effective in preventing delayed tPA-induced ICH, suggesting the possibility of repurposing previously developed PKCβ inhibitors for stroke therapy.
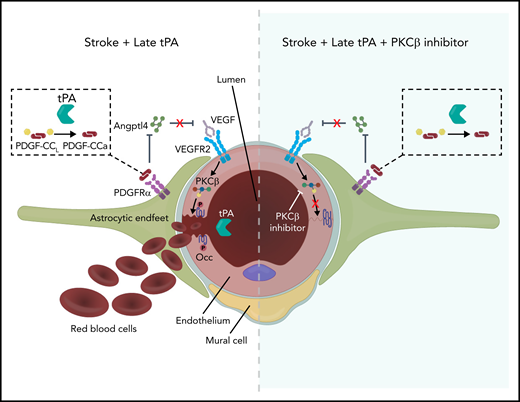
Model of tPA signaling to occludin phosphorylation and BBB permeability. Ischemic stroke induces release of endogenous tPA that cleaves latent PDGF-CCL to active PDGF-CCa, which induces PDGFRα signaling in perivascular astrocytes. This results in decreased expression of ANGPTL4, relieving the ANGPTL4 inhibition of VEGF signaling, which further promotes ischemia-induced VEGF signaling through VEGFR2, which leads to activation of PKCβ and occludin phosphorylation on S490. This phosphorylation site regulates endocytosis of occludin with other junctional proteins, resulting in increased paracellular permeability. Recombinant thrombolytic tPA may leak into the brain parenchyma and further activate this system, leading to ICH when given beyond 4.5 hours after stroke. However, inhibiting PKCβ may provide a therapeutic option to maintain the BBB, extending the tPA therapeutic window. Note: Mural cell was retracted to emphasize VEGFR2 on the endothelial cell abluminal membrane.
Model of tPA signaling to occludin phosphorylation and BBB permeability. Ischemic stroke induces release of endogenous tPA that cleaves latent PDGF-CCL to active PDGF-CCa, which induces PDGFRα signaling in perivascular astrocytes. This results in decreased expression of ANGPTL4, relieving the ANGPTL4 inhibition of VEGF signaling, which further promotes ischemia-induced VEGF signaling through VEGFR2, which leads to activation of PKCβ and occludin phosphorylation on S490. This phosphorylation site regulates endocytosis of occludin with other junctional proteins, resulting in increased paracellular permeability. Recombinant thrombolytic tPA may leak into the brain parenchyma and further activate this system, leading to ICH when given beyond 4.5 hours after stroke. However, inhibiting PKCβ may provide a therapeutic option to maintain the BBB, extending the tPA therapeutic window. Note: Mural cell was retracted to emphasize VEGFR2 on the endothelial cell abluminal membrane.
Earlier studies have shown that the release of endogenous tPA in the ischemic brain occurs as early as 1 hour after MCAO and that this action is associated with an increase in BBB permeability that is prevented in tPA−/− mice.40 The mechanism of action of tPA on the BBB is not fully understood. However, it is known to require tPA proteolytic cleavage of latent PDGF-CCL to its active form, PDGF-CCa (Figure 7), which can bind to the PDGFRα and induce receptor phosphorylation, as inhibition of PDGF-CCL activation or PDGFRα phosphorylation both prevent stroke-induced permeability in mice.12,15,40 We have also shown that tPA-mediated activation of PDGFRα occurs in GFAP+ perivascular astrocytes on the parenchymal side of the NVU.11,12 The current studies have focused on identifying factors downstream of the tPA-induced PDGFRα pathway that affect ICH, with a focus on the tight junction complex and changes to vascular integrity.
Tight junction alterations have been shown to be a major cause of BBB permeability after ischemic stroke,34,48,50 particularly at later time points.51 The current study shows that occludin phosphorylation at S490 plays a critical regulatory role in barrier properties downstream of tPA. The increase in occludin phosphorylation is mediated, at least in part, by tPA, as direct ICV injection of tPA induced an increase in occludin phosphorylation that was commensurate with increased permeability, and this was blocked in animals expressing occludin S490A. MCAO induced an increase in occludin S490 phosphorylation at 3 hours that dramatically increased by 24 hours. Furthermore, tPA deficiency or inhibition of PKCβ reduced occludin S490 phosphorylation at 24 hours after MCAO. These data are consistent with an increasing contribution of tPA to vascular permeability over time. Collectively, these data indicate that tPA alone is sufficient to induce a signaling pathway leading to occludin phosphorylation on S490 through PKCβ, promoting disruption of the BBB.
We also found that inhibition of PKCβ reduced leukocyte infiltration into the ipsilateral hemisphere. This observation is consistent with a recent report showing that thrombolytic tPA can increase the transmigration of neutrophils and other inflammatory cells into the CNS after stroke.52 Although future studies are needed to determine the mechanism of PKCβ inhibition on reducing inflammation, the studies are consistent with tPA signaling to occludin regulating BBB permeability.
Our data suggest that the pathway from parenchymal tPA signaling to endothelial occludin phosphorylation involves activation of PDGFRα signaling in the NVU11,12 that results in a decrease in Angtpl4 expression and increased VEGFR2 activation (Figure 7). Co-ICV injection of tPA with ANGPTL4, or a PDGFRα inhibitor, or a VEGFR2 inhibitor, or inhibition of PKCβ, completely blocked tPA-induced occludin phosphorylation and BBB permeability. Previous studies have shown that blocking PDGFRα significantly reduces thrombolysis-associated ICH and improves outcome in rodent models of stroke.12,15,53 Here, we show that PDGF-CCa downregulation of Angptl4 is correlated with increased BBB permeability. Although VEGF signaling is well established as contributing to ischemia-induced BBB permeability,54 the use of a selective PDGFRα inhibitor, imatinib, does not inhibit VEGFR2,55 suggesting that PDGFRα activation is parallel or upstream of VEGF signaling. This is further supported by PDGF-CCa–induced downregulation of Angptl4, a modulator of vascular permeability, which has been shown to inhibit both VEGFR2 signaling42 and permeability.56 Gene deletion of Angptl4 leads to a 3- to 4-fold increase in paracellular permeability in the retina at postnatal day 7.57 Furthermore, the C-terminal natural cleavage products of ANGPTL4 inhibits both basic fibroblast growth factor and VEGF-induced signaling in endothelial cells,58 and ANGPTL4 has been shown to provide protective effects at the BBB, preventing tPA-induced permeability after ischemic stroke using transient MCAO43 and reducing brain edema and neurologic deficits after bacterial collagenase–induced ICH in mice.59
The contribution of ANGPTL4 to vascular permeability is complex, however. Several publications provide evidence for a role of ANGPTL4 in angiogenesis and vascular permeability downstream of hypoxia-inducible factor.60 This may be either through reported direct interaction with neuropilin61 or through interaction with integrin α5β1 and Rac1/PAK activation or by direct interaction with tight junction proteins claudin-5 and ZO-1.62 It is also important to note that Angptl4 is not the only factor regulated by PDGF-CCa, and other pathways that could regulate BBB permeability may also be activated. For example, proinflammatory factors such as IL6 and Cxcl3 are both downregulated by PDGF-CCa, whereas Tnf and Il1a expression is upregulated by PDGF-CCa (Figure 2A; supplemental Table 1). The increased expression of Tnf and Il1a by PDGF-CCa has previously been shown in a model of experimental autoimmune encephalomyelitis,41 suggesting that PDGF-CCa regulation of local inflammation may also play a role in BBB permeability. Several genes associated with cell metabolism were also differentially regulated by PDGF-CCa, including Cox8b, Ddit4, Cdkn1a, and Rbp7. However, whether changes in the expression of these genes affect BBB permeability has not been determined. Another limitation of our study is that we have not shown directly in ischemic stroke that PDGF-CCa–mediated downregulation of Angptl4 is responsible for the increase in occludin phosphorylation and the increase in BBB permeability.
In the current context, we found that ANGPTL4 effectively blocked tPA-induced BBB permeability and occludin phosphorylation after ICV injection. These data suggest that in stroke, tPA activation of PDGF-CCa/PDGFRα and subsequent downregulation of Angptl4 could significantly enhance hypoxia-induced VEGF signaling in endothelial cells leading to increased PKCβ activation, occludin phosphorylation, and loss of tight junction integrity. This loss of tight junctions leads to increased BBB permeability and contributes significantly to thrombolytic tPA–associated ICH (Figure 7).
Clinically, PKCβ inhibitors have been developed and tested for toxicity and effectiveness in large-scale clinical trials to control VEGF-induced retinal vascular permeability.63,64 In our study, inhibition of PKCβ proved to have beneficial effects after ischemic stroke, preventing tPA-induced permeability and ICH. Others have reported that PKCβ inhibition was also effective in preventing BBB permeability and edema formation during hyperglycemic stroke.65 We also observed that PKCβ inhibition decreased infarct volume. Studies have highlighted that increased BBB permeability contributes to the expanding infarct during an ischemic stroke50 and that preserving the BBB significantly improves outcome after stroke in mice.15,34,48 Importantly, the current studies indicate that the PKCβ inhibitor given up to 5 hours after MCAO could prevent delayed tPA-induced ICH, as did expression of occludin S490A, showing that PKCβ phosphorylation of occludin is an important downstream target in tPA-induced ICH.
In summary, the findings of this study show the importance of PKCβ-induced occludin phosphorylation in CNS vascular permeability. In addition, these results hold translational potential for repurposing of PKCβ inhibitors as adjuvant therapy to extend tPA treatment in ischemic stroke, allowing tPA-induced thrombolysis without adverse effects in the vasculature.
Acknowledgments
The authors thank Alyssa Dreffs for experimental support and Tiago Figueiredo for the visual abstract scientific illustration.
This research was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL055374, D.A.L. and D.A.A.), National Institute on Aging (AG074552, D.A.L.), and National Eye Institute (EY012021, D.A.A.); the American Heart Association (19TPA34880040, D.A.L.); the Vision Research Core Grant (P30EY007003); the Michigan Diabetes Research and Training Center Grant (P30DK020572); Research to Prevent Blindness (D.A.A.); the Swedish Research Council (2016-02593, 2017-01794, U.E.); the Swedish Brain Foundation (F2020-0034 and F2021-0039, U.E., I.N., and L.F.); and the Hållsten Research Foundation (U.E., I.N., and L.F.).
Authorship
Contribution: D.A.L. and D.A.A. conceived the study; A.G., E.J.S., A.M., M.Z., D.T., I.N., J.P., and L.F. designed and performed experiments, and analyzed data; A.G., E.J.S., D.A.A., and D.A.L. interpreted results and wrote the manuscript; M.Z., I.N., L.F., and U.E. contributed to discussions and edited the manuscript; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: U.E., L.F., E.J.S., and D.A.L. hold a patent on modulating the blood–neural barrier using a PDGFRα antagonist. D.A.L. and D.A.A. have a patent pending for the use of protein kinase C inhibition with tPA treatment for ischemic stroke. The remaining authors declare no competing financial interests.
Correspondence: Daniel A. Lawrence, Department of Internal Medicine 7301 MSRB III, University of Michigan, Ann Arbor, MI 48109-5644; e-mail: dlawrenc@umich.edu; or David A. Antonetti, University of Michigan Kellogg Eye Center, 1000 Wall St, Rm 7317, Ann Arbor, MI 48105; e-mail: dantonet@med.umich.edu.
The microarray data sets presented in this publication have been deposited in the Gene Expression Omnibus database (accession number GSE176245).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal