

Key Points
Naturally selected CD7 CAR T cells manufactured without additional genetic manipulations contained a high percentage of CAR+ cells.
Naturally selected CD7 CAR T cells were safe and effective among T-ALL/LBL patients in a first-in-human phase 1 trial.

Abstract
Derivation of CD7-targeted chimeric antigen receptor (7CAR) T cells often requires genetic manipulations to ablate the CD7 gene or block CD7 cell surface expression. Our novel approach deriving naturally selected 7CAR (NS7CAR) T cells from bulk T cells was able to overcome major fratricide by minimizing accessible CD7 epitopes. The CD7 molecules of NS7CAR T cells were masked or sequestered by the CD7-targeting CAR. Compared with sorted CD7-negative 7CAR T cells and CD7 knocked-out 7CAR T cells, NS7CAR exhibited similar or superior therapeutic properties, including a greater percentage of CAR+ cells and a higher proportion of CD8+ central memory T cells. In our first-in-human phase 1 trial (NCT04572308), 20 patients with relapsed/refractory T-cell acute lymphoblastic leukemia (T-ALL) (n = 14) and T-cell lymphoblastic lymphoma (T-LBL) (n = 6) were treated with NS7CAR. Nineteen patients achieved minimal residual disease negative complete remission (CR) in the bone marrow (BM) by day 28, and 5 of 9 patients achieved extramedullary CR. With a median follow-up of 142.5 (32-311) days after infusion, 14 patients subsequently received allogeneic hematopoietic stem cell transplant (10 consolidative, 4 salvage) following NS7CAR infusion with no relapses to date. Of the 6 patients who did not receive a transplant, 4 remained in CR at a median time of 54 (32-180) days. Eighteen patients experienced mild cytokine release syndrome (CRS) (grade ≤2), 1 developed grade 3 CRS, and 2 had grade 1 neurotoxicity. These results indicate that NS7CAR-T therapy is a safe and highly effective treatment for T-ALL/LBL. More patients and longer follow-up are needed for validation.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma (T-LBL) are highly aggressive T-lineage malignancies characterized by an infiltration of immature T cells in the bone marrow (BM) and peripheral blood (PB) and/or extramedullary organ involvements including central nervous system (CNS) infiltration.1-9 Effective treatments were elusive and long-term survival of patients with refractory/relapsed (R/R) T-ALL/LBL remains poor despite allogeneic hematopoietic stem cell transplantation (allo-HSCT).2-4,10-12 CAR-T therapies for T-cell malignancies remain underdeveloped due to the shared antigenicity between normal and malignant T cells.10,13-21
CD7 is highly expressed on the surface of T-ALL/LBL T cells and is considered a viable CAR-T therapeutic target.22,23 Although CD7 is expressed on natural killer (NK) cells and normal T cells at various stages of maturation, CD7 knockout mice develop normal lymphoid organs and immune responses, suggesting that CD7 is dispensable for T/NK cell development and functions.24-27
CD7-targeting immunotherapies may be compromised by T-cell fratricide and extermination.24-26 As such, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)-mediated gene ablation and other techniques have been used to produce CD7-targeted CAR T cells that no longer express CD7.23,28,29 The costs of the additional genetic manipulations make such therapies prohibitive. Collecting sufficient autologous (auto) functional T cells to manufacture CD7-targeted CAR T cells remains challenging.23,28-30 While donor-derived or universal CAR T is an option, suitable donors are difficult to find, and CAR T duration and persistence remain to be addressed.18,19,30,31
Here, we describe a novel approach using patient- or donor-derived “naturally selected” CD7-targeted CAR T cells (NS7CAR) without additional CD7 gene ablation or protein expression blockade. We investigated the safety and efficacy of the NS7CAR therapy in a phase 1 clinical trial for T-ALL/LBL patients.
Methods
Construct design and generation of anti-CD7 CAR T products
The 7CAR gene cassette consisted of a CD7-specific mouse monoclonal antibody (TH69)32 cDNA sequence, a 12AA short hinge-only domain,33 fused to the coding sequence of the CD28TM-41BB-CD3 ζ signal domains, and the T2A-linked truncated epidermal growth factor receptor.34 The cassette was placed under the control of the EF1α promoter (Figure 1A) and cloned into a lentiviral vector. PB mononuclear cells (PBMCs) from donors or patients were isolated by density gradient centrifugation. T cells were purified using CD3 magnetic beads or, if needed, CD4 and CD8 magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany) per the manufacturer’s protocol. T cells were cultured in TexMACS good manufacturing practice medium (MACS) at 1 × 106/mL with 200 IU/mL interleukin-2 (IL-2) (SL PHARM, Beijing, China). T cells were stimulated at a 1:1 (bead/cell) ratio with anti-CD3/CD28 Dynabeads (Thermo Fisher Scientific, Vilnius, Lithuania) at a 1:1 (bead/cell) ratio and incubated at 37°C in 5% CO2. 7CAR lentiviral transduction of bulk T cells (NS7CAR) or sorted CD7− T cells (Neg7CAR) were performed 2 days after CD3/CD28 activation. CD7-ablated 7CAR T (KO7CAR) were derived by electroporating bulk T cells with CD7-targeting Cas9-guide RNA ribonucleoprotein 24 hours before 7CAR transduction. CAR T cells were routinely kept in culture for 12 days in TexMACS good manufacturing practice medium (MACS) with 200 IU/mL IL-2 (SL PHARM, Beijing, China). The levels of CD7 messenger RNA (mRNA), protein, and surface expression were determined by qualitative/quantitative reverse transcription-polymerase chain reaction (RT-PCR), Western blotting (WB), and flow cytometry (FCM). In vitro cytotoxicity of CAR T cells against CD7+ tumor cells was determined using an FCM-based assay. To demonstrate the in vivo antileukemia activity of CAR T cells, a CCRF-CEM luciferase reporter cell line (CCRF-CEM-Luc) was IV-injected into B-NDG (NOD-Prkdcscid IL2rgtm1/Bcgen) mice. Five days later, the engrafted mice were given a single dose of CAR T cells and monitored by bioluminescence imaging (supplemental Figure 1E, available on the Blood Web site). Details of the 7CAR manufacturing processes and preclinical studies are described in the supplemental Methods.
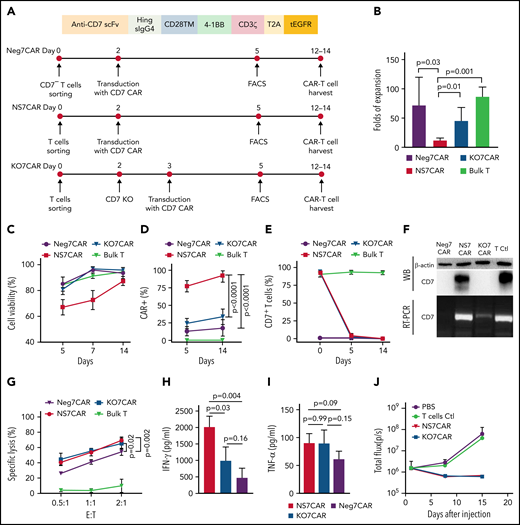
A comparative study of 3 different approaches for generating 7CAR T cells. (A) Schematic diagram of CD7CAR constructs and schemes of the optimized protocol for generating Neg7CAR, NS7CAR, and KO7CAR T cells. (B) A comparison of the fold expansion of bulk T and 3 different CAR T cells, including Neg7CAR, NS7CAR, and KO7CAR T cells. (C) In vitro cell culture viability of Neg7CAR, NS7CAR, KO7CAR T cells, and bulk T cells at days 5, 7, and 14. (D) CAR expression was determined by FCM using tagged truncated epidermal growth factor receptor. (E) The percentages of CD7+ T cells in bulk T, and Neg7CAR, NS7CAR, and KO7CAR T cells are shown. Data are presented as the mean ± SD. A 2-tailed, unpaired 2-sample t test was used for statistical analysis (n = 4 for Neg7CAR and KO7CAR, n = 5 for NS7CAR, and n = 3 for bulk T cells). (F) CD7 protein (upper panel) and mRNA (lower panel) expression in Neg7CAR, NS7CAR, KO7CAR T, and control T cells were determined by RT-PCR and WB. The photographs of the gels were edited to show only the lanes relevant to this experiment. (G-I) Significant levels of CD7 mRNA and protein were detected by RT-PCR and WB from NS7CAR and control T-cell samples but not from Neg7CAR or KO7CAR samples. CAR T cells were cocultured with CFSE-labeled CCRF-CEM at the indicated E:T ratio. Cytolytic activities (G) and the cytokines concentrations (H-I) of Neg7CAR, NS7CAR, and KO7CAR are indicated. Data are presented as the mean ± SD. (J) B-NDG mice (n = 6 per group) were IV-injected with 1 × 106 CCRF-CEM-Luc cells, followed by a single IV injection of 1 × 107 control samples, NS7CAR, or KO7CAR cells 3 days later. Total photon flux after CAR-T injection was plotted. Data are presented as mean ± SEM (n = 6). 7CAR T, anti-CD7 CAR T; Ctl, control; E:T, effector cells: target cells; FACS, fluorescence-activated cell sorting; KO7CAR, CD7 knocked-out CAR T cells; Neg7CAR, CD7-negative T cells derived 7CAR T cells; NS7CAR, naturally selected anti-CD7 CAR T cells; PBS, phosphate-buffered saline.
A comparative study of 3 different approaches for generating 7CAR T cells. (A) Schematic diagram of CD7CAR constructs and schemes of the optimized protocol for generating Neg7CAR, NS7CAR, and KO7CAR T cells. (B) A comparison of the fold expansion of bulk T and 3 different CAR T cells, including Neg7CAR, NS7CAR, and KO7CAR T cells. (C) In vitro cell culture viability of Neg7CAR, NS7CAR, KO7CAR T cells, and bulk T cells at days 5, 7, and 14. (D) CAR expression was determined by FCM using tagged truncated epidermal growth factor receptor. (E) The percentages of CD7+ T cells in bulk T, and Neg7CAR, NS7CAR, and KO7CAR T cells are shown. Data are presented as the mean ± SD. A 2-tailed, unpaired 2-sample t test was used for statistical analysis (n = 4 for Neg7CAR and KO7CAR, n = 5 for NS7CAR, and n = 3 for bulk T cells). (F) CD7 protein (upper panel) and mRNA (lower panel) expression in Neg7CAR, NS7CAR, KO7CAR T, and control T cells were determined by RT-PCR and WB. The photographs of the gels were edited to show only the lanes relevant to this experiment. (G-I) Significant levels of CD7 mRNA and protein were detected by RT-PCR and WB from NS7CAR and control T-cell samples but not from Neg7CAR or KO7CAR samples. CAR T cells were cocultured with CFSE-labeled CCRF-CEM at the indicated E:T ratio. Cytolytic activities (G) and the cytokines concentrations (H-I) of Neg7CAR, NS7CAR, and KO7CAR are indicated. Data are presented as the mean ± SD. (J) B-NDG mice (n = 6 per group) were IV-injected with 1 × 106 CCRF-CEM-Luc cells, followed by a single IV injection of 1 × 107 control samples, NS7CAR, or KO7CAR cells 3 days later. Total photon flux after CAR-T injection was plotted. Data are presented as mean ± SEM (n = 6). 7CAR T, anti-CD7 CAR T; Ctl, control; E:T, effector cells: target cells; FACS, fluorescence-activated cell sorting; KO7CAR, CD7 knocked-out CAR T cells; Neg7CAR, CD7-negative T cells derived 7CAR T cells; NS7CAR, naturally selected anti-CD7 CAR T cells; PBS, phosphate-buffered saline.
Successful NS7CAR-T manufacture for clinical application
For clinical application, we generated the NS7CAR product from patient blood or the blood of their transplant donors. Autologous CAR T cells were used for those patients who never received allo-HSCT. For patients who had relapsed after allo-HSCT at enrollment, CAR T cells were manufactured either from the blood of each patient or that of the transplant donor, based primarily on tumor burden of the patient, pancytopenia status, chimerism status, as well as donor availability. For clinical products, assays performed before infusion included sterility assessments (Gram stain, bacterial culture, mycoplasma PCR), endotoxin levels, residual bead count, viability, and transduction efficiency determined by FCM. The 7CAR T cells were tested for CAR copy numbers per cell by quantitative PCR (qPCR). FCM was used to confirm the exclusion of malignant T cells from the CAR-T preparation. When applicable, levels of minimal residual malignant cells were assessed using qPCR (supplemental Methods).
Study design
A phase 1 study (ClinicalTrials.gov: NCT04572308) was conducted to evaluate the safety and efficacy of NS7CAR for CD7+ R/R T-ALL or T-LBL patients as defined by the NCCN (National Comprehensive Cancer Network) guideline.35 The trial was not designed to find a maximum tolerated dose. Patients between 2 and 65 years of age whose disease failed ≥2 lines of therapies and had ≥80% CD7 expression were eligible. Detailed eligibility criteria are provided in the supplemental Methods. Consolidative allo-HSCT after remission was allowed after day 28 after NS7CAR infusion at the discretion of the physician and patient preference (supplemental Methods).
For NS7CAR manufacturing, PBMCs were collected from patient blood or their transplant donors. When necessary, bridging chemotherapy was given to patients to control their disease progression. A lymphodepletion regimen was administered to all patients using fludarabine (30 mg/m2 per day) and cyclophosphamide (300 mg/m2 per day) for 3 consecutive days (day −5 to day −3) before CAR-T infusion. Patients received 1 single infusion of NS7CAR T cells at 1 of 3 dose levels: a low dose (0.5 × 106/kg), a medium dose (1 to 1.5 × 106/kg), or a high dose (2 × 106/kg). The protocol was designed with 3 dose levels for attending physicians to select based on the patient’s condition (ie, performance status, disease burden, and extramedullary disease [EMD] status after the first low-dose level proved to be safe [supplemental Methods]).
Endpoints and assessments
The primary endpoint of the study was to evaluate the safety of NS7CAR therapy up to at least day 28 after infusion. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome were graded according to the ASTCT (American Society for Transplantation and Cellular Therapy) consensus.36 Other adverse events were graded with CTCAE (Common Terminology Criteria for Adverse Events) version 5.0.37
The secondary endpoint was to evaluate the efficacy and observe the cytokinetics of NS7CAR T cells in patients. Efficacy assessment was conducted according to the NCCN guidelines.35 Minimal residual disease (MRD) was assessed by FCM (sensitivity of threshold of 1 × 10−4). Response of EMD was assessed by positron emission tomography–computed tomography (PET-CT) and/or contrast CT and magnetic resonance imaging (supplemental Methods).35,38,39 The proliferation and persistence of NS7CAR T cells were measured by qPCR and FCM. The variations in cytokines were detected by FCM (supplemental Methods).
Ethics
The study protocol was approved by the Ethics Committee of Hebei Yanda Lu Daopei Hospital and compliant with the Declaration of Helsinki. Written informed consent was obtained from all study participants or their guardians.
Statistical analysis
Clinical safety, efficacy, and after infusion follow-up data are summarized and analyzed using descriptive statistical methods. GraphPad Prism 5.0 was used to conduct the data analysis. Associations between NS7CAR expansion and persistence and different patient subgroups were examined by a 2-tailed t test. P < .05 was considered to be statistically significant.
Results
Comparative preclinical analyses of anti-CD7 CAR T cells revealed potent antileukemia activity of NS7CAR in vitro and in vivo
We used 3 independent approaches to generate CD7-targeted CAR T cells and compared their functionalities in preclinical studies: (1) NS7CAR T cells were derived by transducing bulk T cells with 7CAR first and then subjecting them to “natural selection” in cell culture, (2) Neg7CAR were naturally occurring CD7-negative T cells (3%-10% of CD3+ T cells)40,41 that were purified and transduced with 7CAR, and (3) KO7CAR were generated by transducing 7CAR into bulk T cells whose CD7 genes had been ablated using CRISPR/Cas9 ribonucleoprotein. Over a 2-week culture period, on average, we observed a 12-fold expansion of NS7CAR cells. This was significantly lower than for Neg7CAR and KO7CAR (70- and 45-fold, respectively; NS7CAR vs Neg7CAR: P = .03; NS7CAR vs KO7CAR: P = .01) (Figure 1B). However, 87.3% of NS7CAR T cells in culture were viable, making further studies feasible (Figure 1C). Notably, when harvested, an average of 92.4% of T cells in the NS7CAR group expressed the 7CAR molecule, which was significantly higher than that of Neg7CAR (mean = 17.9%; P < .0001) or KO7CAR (mean = 34.0%; P < .0001) (Figure 1D). Three days after 7CAR lentiviral transduction, T cells in the NS7CAR group had a rapid and dramatic phenotypic transition from CD7+CAR− to CD7−CAR+ per FCM (Figure 1E). Interestingly, although the T cells in the NS7CAR group did not show detectable surface expression of CD7 per FCM after in vitro culture (Figure 1E), significant CD7 mRNA and protein levels were detected by RT-PCR and WB. As expected, no CD7 mRNA or protein expression was observed in Neg7CAR or KO7CAR cells (Figure 1F). These results support the notion that although NS7CAR T cells expressed CD7 mRNA and protein, they were CD7-negative immunologically and escaped fratricidal cytotoxic killing because of antigenic masking/intracellular sequestration by CD7CAR. Indeed, expression of CD7 mRNA in NS7CAR persisted in patients on day 28 after CAR-T infusion (supplemental Figure 2). Furthermore, NS7CAR contained a significantly larger CD8+ subset (CD4/CD8 ratio: NS7CAR vs Neg7CAR: P = .001; NS7CAR vs KO7CAR: P = .02) (supplemental Table 1 and supplemental Figure 1B) and an increased central memory phenotype compared with the other 2 groups (NS7CAR vs Neg7CAR: P = .009; NS7CAR vs KO7CAR: P = .049) (supplemental Table 1 and supplemental Figure 1C). There were no significant differences in programmed cell death protein 1 (PD1), T-cell immunoglobulin and mucin domain containing-3 (TIM3), and lymphocyte-activation gene 3 (LAG3) expression in NS7CAR and KO7CAR cells (P > .05) (supplemental Figure 3).
We next compared the in vitro antileukemia activities of each method. Remarkably, NS7CAR and KO7CAR both showed comparable cytotoxicity against CCRF-CEM cells and released high levels of interferon γ and tumor necrosis factor α, but Neg7CAR cells to a lesser extent (Figure 1G-I). Finally, we used the B-NDG mice engrafted with CCRF-CEM-Luc cells to compare the in vivo antileukemia activities. Consistent with the cytotoxicity analysis, within the first 2 weeks after CAR-T IV injection, both NS7CAR and KO7CAR conferred robust protection against leukemia progression by markedly reducing leukemia burden in the mouse model (Figure 1J and supplemental Figure 1). These data demonstrated the feasibility of generating robust anti-CD7 CAR T directly from bulk 7CAR-transduced T cells without additional genetic manipulation. Prompted by these results, we proceeded to use the NS7CAR T cells in a phase 1 clinical trial.
Patient characteristics and clinical information
Twenty eligible patients were enrolled between November 2020 and September 2021 (Table 1 and Figure 2). NS7CAR T cells were infused into all 20 patients. The median age of the patients was 22 (3-47) years. Fourteen patients were diagnosed with R/R T-ALL and 6 with R/R T-LBL. High-risk subtypes included Ph-positive T-ALL (n = 1) and early T-cell precursor ALL (n = 2). Moreover, 11 patients had high-risk mutations or gene fusion, namely STIL-TAL1, TP53, BCR-ABL, JAK1, JAK3, EZH2, RUNX1, NRAS, CDKN2A, and DNMT3A. Five patients had a history of HSCT before enrollment (3 allo-HSCT and 2 auto-HSCT). Patients were heavily pretreated with a median of 4.5 (2-8) lines of therapies. At enrollment, the median proportion of blasts in 17 patients was 21.49% (0.18%-87.27%) in BM by FCM, while 3 T-LBL patients did not have BM blasts. Five T-LBL patients presented with EMD with diffuse involvement (n = 3) or bulky mediastinal masses >7 cm in diameter (n = 2), while 4 T-ALL patients had EMD including diffuse involvement (n = 1), optic nerve involvement along with CNS leukemia (n = 2), or CNS leukemia alone (n = 1). Three patients with CNS leukemia were graded as CNS-2 (n = 1) or CNS-3 (n = 2). All but 1 received bridging chemotherapy.
Patient characteristics at baseline and response after NS7CAR T-cell infusion
| Patient no. | Age (y) | Sex (F/M) | Diagnosis | History of prior HSCT | PB blasts at enrollment (by morphology), % | BM blasts at enrollment (by FCM), % | EMD at enrollment | Source of CAR T cells | Dose infused (CAR T cells per kg) | BM & PB evaluation at day 28 | Best EMD response | CRS | ICANS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt.01 | 37 | F | T-LBL | Yes | 0 | 0.00 | Yes | Donor | 0.5 × 106 | MRD(−) Cri | CR | 1 | 0 |
| Pt.02 | 3 | M | T-ALL | No | 0 | 1.17 | No | Patient | 1 × 106 | MRD(−) CR | — | 0 | 0 |
| Pt.03 | 14 | M | T-ALL | No | 0 | 0.18 | Yes | Patient | 1 × 106 | MRD(−) CR | CR | 1 | 0 |
| Pt.04 | 28 | M | T-ALL | Yes | 1 | 7.27 | No | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.05 | 30 | M | T-ALL | Yes | 4 | 33.72 | No | Donor | 1 × 106 | MRD(−) CRi | — | 1 | 1 |
| Pt.06 | 47 | F | T-LBL | No | 3 | 87.27 | No* | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.07 | 42 | F | T-ALL | No | 33 | 65.03 | No | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.08 | 15 | F | T-ALL | No | 4 | 15.78 | No | Patient | 1 × 106 | MRD(−) CR | — | 1 | 0 |
| Pt.09 | 25 | M | T-LBL | No | 0 | 0.00 | Yes | Patient | 1 × 106 | MRD(−) CRi | SD | 1 | 0 |
| Pt.10 | 19 | M | T-ALL | No | 62 | 60.23 | Yes | Patient | 1 × 106 | MRD(−) CR | CR | 1 | 0 |
| Pt.11 | 16 | M | T-LBL | No | 0 | 0.00 | Yes | Patient | 2 × 106 | MRD(−) CRi | PR | 1 | 0 |
| Pt.12 | 40 | M | T-LBL | Yes | 0 | 0.78 | Yes | Patient | 1 × 106 | MRD(−) CRi | CR | 3 | 1 |
| Pt.13 | 11 | M | T-ALL | No | 0 | 53.67 | Yes | Patient | 1.5 × 106 | NR | NR | 2 | 0 |
| Pt.14 | 7 | M | T-ALL | No | 0 | 46.37 | No | Patient | 1 × 106 | MRD(−) CRi | — | 2 | 0 |
| Pt.15 | 15 | M | T-ALL | No | 1 | 4.42 | Yes | Patient | 1 × 106 | MRD(−) CRi | CR | 1 | 0 |
| Pt.16 | 13 | M | T-ALL | No | 0 | 4.28 | No | Patient | 1 × 106 | MRD(−) CRi | — | 2 | 0 |
| Pt.17 | 21 | F | T-ALL | No | 0 | 56.01 | No | Patient | 0.5 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.18 | 32 | M | T-ALL | No | 0 | 21.49 | No | Patient | 0.5 × 106 | MRD(−) CR | — | 2 | 0 |
| Pt.19 | 36 | F | T-LBL | Yes | 6 | 10.76 | Yes | Patient | 1 × 106 | MRD(−) CRi | PD | 1 | 0 |
| Pt.20 | 22 | M | T-ALL | No | 0 | 56.63 | No | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
| Patient no. | Age (y) | Sex (F/M) | Diagnosis | History of prior HSCT | PB blasts at enrollment (by morphology), % | BM blasts at enrollment (by FCM), % | EMD at enrollment | Source of CAR T cells | Dose infused (CAR T cells per kg) | BM & PB evaluation at day 28 | Best EMD response | CRS | ICANS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt.01 | 37 | F | T-LBL | Yes | 0 | 0.00 | Yes | Donor | 0.5 × 106 | MRD(−) Cri | CR | 1 | 0 |
| Pt.02 | 3 | M | T-ALL | No | 0 | 1.17 | No | Patient | 1 × 106 | MRD(−) CR | — | 0 | 0 |
| Pt.03 | 14 | M | T-ALL | No | 0 | 0.18 | Yes | Patient | 1 × 106 | MRD(−) CR | CR | 1 | 0 |
| Pt.04 | 28 | M | T-ALL | Yes | 1 | 7.27 | No | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.05 | 30 | M | T-ALL | Yes | 4 | 33.72 | No | Donor | 1 × 106 | MRD(−) CRi | — | 1 | 1 |
| Pt.06 | 47 | F | T-LBL | No | 3 | 87.27 | No* | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.07 | 42 | F | T-ALL | No | 33 | 65.03 | No | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.08 | 15 | F | T-ALL | No | 4 | 15.78 | No | Patient | 1 × 106 | MRD(−) CR | — | 1 | 0 |
| Pt.09 | 25 | M | T-LBL | No | 0 | 0.00 | Yes | Patient | 1 × 106 | MRD(−) CRi | SD | 1 | 0 |
| Pt.10 | 19 | M | T-ALL | No | 62 | 60.23 | Yes | Patient | 1 × 106 | MRD(−) CR | CR | 1 | 0 |
| Pt.11 | 16 | M | T-LBL | No | 0 | 0.00 | Yes | Patient | 2 × 106 | MRD(−) CRi | PR | 1 | 0 |
| Pt.12 | 40 | M | T-LBL | Yes | 0 | 0.78 | Yes | Patient | 1 × 106 | MRD(−) CRi | CR | 3 | 1 |
| Pt.13 | 11 | M | T-ALL | No | 0 | 53.67 | Yes | Patient | 1.5 × 106 | NR | NR | 2 | 0 |
| Pt.14 | 7 | M | T-ALL | No | 0 | 46.37 | No | Patient | 1 × 106 | MRD(−) CRi | — | 2 | 0 |
| Pt.15 | 15 | M | T-ALL | No | 1 | 4.42 | Yes | Patient | 1 × 106 | MRD(−) CRi | CR | 1 | 0 |
| Pt.16 | 13 | M | T-ALL | No | 0 | 4.28 | No | Patient | 1 × 106 | MRD(−) CRi | — | 2 | 0 |
| Pt.17 | 21 | F | T-ALL | No | 0 | 56.01 | No | Patient | 0.5 × 106 | MRD(−) CRi | — | 1 | 0 |
| Pt.18 | 32 | M | T-ALL | No | 0 | 21.49 | No | Patient | 0.5 × 106 | MRD(−) CR | — | 2 | 0 |
| Pt.19 | 36 | F | T-LBL | Yes | 6 | 10.76 | Yes | Patient | 1 × 106 | MRD(−) CRi | PD | 1 | 0 |
| Pt.20 | 22 | M | T-ALL | No | 0 | 56.63 | No | Patient | 1 × 106 | MRD(−) CRi | — | 1 | 0 |
F, female; M, male; NR, no response; Pt., patient.
EMD remitted at enrollment due to prior mediastinal radiotherapy.

Consort diagram of patient flow. Twenty patients were enrolled in the study and underwent leukapheresis. NS7CAR T cells were successfully manufactured and infused into all patients at a low dose (0.5 × 106/kg, n = 3), medium dose (1-1.5 × 106/kg, n = 16), or high dose (2 × 106/kg, n = 1). All were available for day 28 evaluation after NS7CAR T-cell infusion.
Consort diagram of patient flow. Twenty patients were enrolled in the study and underwent leukapheresis. NS7CAR T cells were successfully manufactured and infused into all patients at a low dose (0.5 × 106/kg, n = 3), medium dose (1-1.5 × 106/kg, n = 16), or high dose (2 × 106/kg, n = 1). All were available for day 28 evaluation after NS7CAR T-cell infusion.
Characteristics of infused NS7CAR cell products
Details of the NS7CAR cell products for each patient are provided in supplemental Tables 7 and 8. Using a CD3-positive selection strategy, the malignant cells of 16 of 20 patients were eliminated. Lymphocytes of the 4 remaining patients with CD3 expression on blasts were sorted using the CD4+CD8+ selecting protocol. The target dose of NS7CAR T cells was successfully produced from patients (n = 18) or their allo-HSCT donors (n = 2, due to lymphopenia of the patients) with a median transfection efficiency of 95.10% (59.60%-99.90%). The CAR T cells of each patient were tested for the copy number of the CAR vector (<5 copies per cell) before infusion. NS7CAR-T products were skewed toward a CD8+ subpopulation with a median CD4/CD8 ratio of 0.42 (0.07 to 2.06). By FCM for all patients and confirmed by qPCR for some patients, we saw no evidence of malignant contamination of the NS7CAR end products (supplemental Figures 5 and 6). On day 0, a single dose of 1 of 3 different dose levels of NS7CAR at 0.5 × 106, 1 to 1.5 × 106, or 2 × 106/kg cells per kg were infused to 3, 16, and 1 patient(s), respectively. All CAR T cells were infused fresh, except for 1 patient whose cells were frozen due to holiday interruption.
NS7CAR-T therapy exhibited an excellent safety profile
NS7CAR were well tolerated with 19 of 20 patients having no or mild CRS (no CRS, n = 1; grade 1, n = 14; grade 2, n = 4). One patient experienced grade 3 CRS. The median time to CRS onset was 1 (0-15) day with a median duration of 13 (3-25) days. The most common manifestation of CRS was fever. Most (n = 18) patients did not show symptoms of neurotoxicity, including those 3 who had considerable CNS blasts; grade 1 neurotoxicity occurred in the other 2 patients. Of the 19 patients who had CRS, 3 resolved spontaneously without intervention; 16 patients were given steroids (n = 5), tocilizumab (n = 4), or both (n = 7). Preemptive use of tocilizumab was allowed. Tocilizumab was administered at an early stage after CAR-T infusion in 11 patients, including 7 with grade 1 CRS who had a persistently high fever (≥39°C) for 24 hours.
The other common adverse effect was pancytopenia, which was due to a combination of bridging chemotherapy, lymphodepletion, and CAR-T therapy (Table 2). Between the interval of NS7CAR infusion and transplantation, 2 patients (patients 17 and 19) had sepsis from Gram-positive cocci infections (1 from a catheter-related infection and 1 from a perianal infection). Patient 01, who received a prior allo-HSCT approximately 7 months before CAR-T therapy, had cytomegalovirus (CMV) reactivation on day 30 after CAR-T infusion. No patient had Epstein-Barr virus activation.
CRS, ICANS, and adverse events after NS7CAR T-cell infusion*
| CRS/ICANS | Grade 1 (n) | Grade 2 (n) | Grade 3 (n) | Grade 4 (n) |
|---|---|---|---|---|
| CRS | 14 | 4 | 1 | 0 |
| Fever (≥38°C) | 19 | |||
| Hypotension† | 0 | 4 | 0 | 0 |
| Hypoxia | 0 | 3 | 1 | 0 |
| ICANS | 2 | 0 | 0 | 0 |
| Decreased level of consciousness | 1 | 0 | 0 | 0 |
| Seizure | 0 | 0 | 0 | 0 |
| Impairment of motor functions | 1 | 0 | 0 | 0 |
| Elevated ICP or cerebral edema | 0 | 0 | 0 | 0 |
| CRS/ICANS | Grade 1 (n) | Grade 2 (n) | Grade 3 (n) | Grade 4 (n) |
|---|---|---|---|---|
| CRS | 14 | 4 | 1 | 0 |
| Fever (≥38°C) | 19 | |||
| Hypotension† | 0 | 4 | 0 | 0 |
| Hypoxia | 0 | 3 | 1 | 0 |
| ICANS | 2 | 0 | 0 | 0 |
| Decreased level of consciousness | 1 | 0 | 0 | 0 |
| Seizure | 0 | 0 | 0 | 0 |
| Impairment of motor functions | 1 | 0 | 0 | 0 |
| Elevated ICP or cerebral edema | 0 | 0 | 0 | 0 |
| Adverse events | Grade 1 (n) | Grade 2 (n) | Grade 3 (n) | Grade 4 (n) |
|---|---|---|---|---|
| Hematologic event | ||||
| Anemia | 0 | 2 | 17 | 1 |
| Neutropenia | 0 | 0 | 0 | 20 |
| Lymphopenia | 0 | 0 | 0 | 20 |
| Thrombocytopenia | 0 | 1 | 3 | 15 |
| Gastrointestinal event | ||||
| Loss of appetite | 8 | 8 | 0 | 0 |
| Nausea/vomiting/diarrhea | 6 | 2 | 0 | 0 |
| Abdominal bloating | 1 | 0 | 0 | 0 |
| Cardiovascular event | ||||
| Chest pain | 1 | 0 | 0 | 0 |
| Tachycardia | 10 | 2 | 1 | 0 |
| Heart failure | 1 | 0 | 0 | 0 |
| General conditions | ||||
| Fatigue | 4 | 6 | 1 | 0 |
| Flu-like symptom | 5 | 4 | 5 | 0 |
| Rash | 1 | 1 | 3 | 0 |
| Peripheral edema | 5 | 0 | 0 | 0 |
| Laboratory values | ||||
| AST increase | 2 | 5 | 3 | 2 |
| ALT increase | 3 | 1 | 5 | 2 |
| Bilirubin increase | 2 | 1 | 3 | 0 |
| Creatine increase | 1 | 1 | 0 | 0 |
| LDH increase | 19 | 0 | 0 | 0 |
| Infection | ||||
| Sepsis | 0 | 0 | 2 | 0 |
| Lung infection | 0 | 0 | 0 | 0 |
| Skin infection | 0 | 0 | 1 | 0 |
| Oral ulcer | 1 | 2 | 3 | 0 |
| CMV activation | 1 | 0 | 0 | 0 |
| Coagulopathy | 0 | 0 | 1 | 0 |
| Adverse events | Grade 1 (n) | Grade 2 (n) | Grade 3 (n) | Grade 4 (n) |
|---|---|---|---|---|
| Hematologic event | ||||
| Anemia | 0 | 2 | 17 | 1 |
| Neutropenia | 0 | 0 | 0 | 20 |
| Lymphopenia | 0 | 0 | 0 | 20 |
| Thrombocytopenia | 0 | 1 | 3 | 15 |
| Gastrointestinal event | ||||
| Loss of appetite | 8 | 8 | 0 | 0 |
| Nausea/vomiting/diarrhea | 6 | 2 | 0 | 0 |
| Abdominal bloating | 1 | 0 | 0 | 0 |
| Cardiovascular event | ||||
| Chest pain | 1 | 0 | 0 | 0 |
| Tachycardia | 10 | 2 | 1 | 0 |
| Heart failure | 1 | 0 | 0 | 0 |
| General conditions | ||||
| Fatigue | 4 | 6 | 1 | 0 |
| Flu-like symptom | 5 | 4 | 5 | 0 |
| Rash | 1 | 1 | 3 | 0 |
| Peripheral edema | 5 | 0 | 0 | 0 |
| Laboratory values | ||||
| AST increase | 2 | 5 | 3 | 2 |
| ALT increase | 3 | 1 | 5 | 2 |
| Bilirubin increase | 2 | 1 | 3 | 0 |
| Creatine increase | 1 | 1 | 0 | 0 |
| LDH increase | 19 | 0 | 0 | 0 |
| Infection | ||||
| Sepsis | 0 | 0 | 2 | 0 |
| Lung infection | 0 | 0 | 0 | 0 |
| Skin infection | 0 | 0 | 1 | 0 |
| Oral ulcer | 1 | 2 | 3 | 0 |
| CMV activation | 1 | 0 | 0 | 0 |
| Coagulopathy | 0 | 0 | 1 | 0 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ICP, increased intracranial pressure; LDH, lactate dehydrogenase.
CRS/ICANS and adverse events reported from NS7CAR T-cell infusion to day 30 after infusion.
Four patients had mild hypotension, but vasopressors were not required.
Fourteen of the 20 patients received a subsequent allo-HSCT (12 haploidentical, 2 match unrelated donor), and 3 of 14 patients had a second allo-HSCT. Seven of 14 patients had developed acute or chronic graft-versus-host disease (aGvHD, n = 5; aGvHD/cGvHD, n = 2). Two patients who had prior allo-HSCT before CAR-T therapy received transplant donor-derived CAR T cells. One of them developed transient mild skin aGvHD without any need for intervention.
NS7CAR-T therapy is effective for T-ALL/LBL
On day 28, 16 of 17 (94.12%) patients who had BM involvement achieved MRD-negative complete remission (CR; n = 5) or CR with incomplete hematologic recovery (Cri; n = 11); 3 of 20 patients with T-LBL without BM involvement at enrollment remained CRi (Table 1). Sixteen patients achieved MRD-negative CRi in their BM as early as day 14. Two of 3 patients with the STIL-TAL1 fusion gene achieved molecular-level remission. The 1 T-ALL patient (patient 13) who had no response on day 28 had STIL-TAL1 fusion gene, 53.67% BM blasts, and diffuse bulky EMD before CAR-T therapy and had lost CD7 expression on day 28. Among the 9 patients with EMD (T-ALL, n = 4; T-LBL, n = 5), 5 achieved extramedullary CR at a median time of 29 (15-51) days (Figures 3 and 4; supplemental Table 5), while the 2 patients manifested with bulky mediastinal lesions (>7 cm) achieved extramedullary partial remission (PR) and stable disease (SD), respectively, on day 29, and the PR patient had further improvement of EMD on day 104. Patient 13, who had no response in his BM, also had no response for his EMD. Another T-LBL patient (patient 19) achieved MRD-negative CRi in BM on day 28 but had new-onset disease in the spleen on day 28. The 2 patients (patient 03 and patient 10) who had CNS blasts and/or optic nerve involvement achieved CNS CR on day 15 and day 28, respectively. Except for LAG3 (P = .01) (supplemental Figure 4), the relative expression levels of TIM3 and PD1 in NS7CAR were not significantly different among patients who achieved CR compared with those with less than CR (P > .05). However, the case number included were too small to draw a firm conclusion.

Clinical response to NS7CAR T cells and postinfusion allo-HSCT. The clinical responses of patient BM and EMD after NS7CAR T infusion and transplantation after infusion are shown by swimmer plots. For the 9 patients (Pt.01, Pt.03, Pt.09, Pt.10, Pt. 11, Pt.12, Pt.13, Pt.15, and Pt.19) with EMD, the EMD response statuses are shown in separate bars and different colors from the BM response. Patients who had been followed up to the cutoff date and were no longer followed due to death or withdrawal are indicated. After transplant, Pt.09 did not have an EMD evaluation until 92 days after allo-HSCT, which was proven to be EMD CR. NR, no response. *Red asterisk indicates patients who relapsed from previous transplantation before NS7CAR T-cell infusion.
Clinical response to NS7CAR T cells and postinfusion allo-HSCT. The clinical responses of patient BM and EMD after NS7CAR T infusion and transplantation after infusion are shown by swimmer plots. For the 9 patients (Pt.01, Pt.03, Pt.09, Pt.10, Pt. 11, Pt.12, Pt.13, Pt.15, and Pt.19) with EMD, the EMD response statuses are shown in separate bars and different colors from the BM response. Patients who had been followed up to the cutoff date and were no longer followed due to death or withdrawal are indicated. After transplant, Pt.09 did not have an EMD evaluation until 92 days after allo-HSCT, which was proven to be EMD CR. NR, no response. *Red asterisk indicates patients who relapsed from previous transplantation before NS7CAR T-cell infusion.
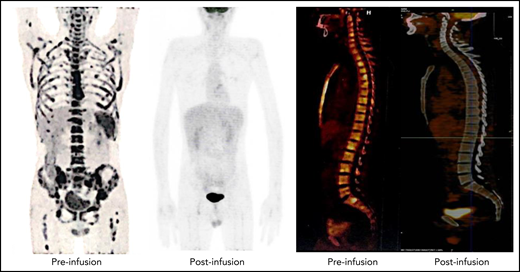
Representative PET-CT images of patient 12 before and after NS7CAR T-cell infusion. (Left) As shown in the PET-CT, the patient had diffuse extramedullary lesions before NS7CAR T-cells infusion. (Right) The patient achieved an extramedullary complete remission on day 51 after infusion.
Representative PET-CT images of patient 12 before and after NS7CAR T-cell infusion. (Left) As shown in the PET-CT, the patient had diffuse extramedullary lesions before NS7CAR T-cells infusion. (Right) The patient achieved an extramedullary complete remission on day 51 after infusion.
Promising duration of remission after NS7CAR-T therapy
At a cutoff date of October 25, 2021, the median follow-up time was 142.5 (32-311) days (Figure 3). Six out of 20 patients did not receive additional antileukemia therapy or transplant following CAR T-cell infusion. Four of them remained progression-free at a median time of 54 (32-180) days. Patient 14 relapsed on day 43, had lost CD7 expression, and died on day 83. After NS7CAR infusion, 10 patients with CR received consolidative allo-HSCT (8 haploidentical donors, 2 match-unrelated donors, including 3 second allo-HSCT) at a median time of 57.5 (42-92) days after infusion (Figure 3). Patient 01, who had a second transplant, developed grade 3 aGvHD and died on day 82. Patient 04 (second transplant) and patient 06 withdrew on days 102 and 147, respectively, due to personal and financial issues. Both patients were at CR at the time of withdrawal. With a median follow-up of 210 (91-311) days after transplantation, the remaining 7 of 10 CR patients maintained MRD-negative status and were alive after transplantation. Patient 15, with STIL-TAL1 fusion gene, relapsed on day 55 due to new onset of cervical lymph nodes and relapsed CNS leukemia without CD7 lost and, who along with the other 3 less than CR patients (1 with relapse, 1 with NR, 1 with SD, and 1 in PR) received salvage transplantation and all achieved CR.
Cytokinetics of NS7CAR and cytokines in vivo
NS7CAR T-cell expansion started at a median of 4.5 (0-15) days after infusion (supplemental Table 9). The median CAR peak copy numbers were 1.37 × 105 copies/μg DNA (0.2 × 105 − 5.36 × 105 copies/μg DNA), and the median time required to reach the peak copy number was 19 (10-42) days. The median time of NS7CAR+ DNA persistence was 90 (28-195) days (supplemental Table 9) at the last follow-up and remained detectable by qPCR in 12 of 14 patients at a median time of 39.5 (5-146) days after allo-HSCT, albeit at a considerably low median level of 3.5 × 101 copies/μg DNA (1.7 × 101 − 6.2 × 102 copies/μg DNA) (supplemental Figure 9). For the 6 patients who did not receive allo-HSCT, CAR T cells remained detectable at their last follow-up at a median of 48.5 (29-120) days, with a median CAR copy number of 3.15 × 103 copies/μg DNA (0.73 × 102 to 3.75 × 104 copies/μg DNA). Patients who had a prior history of HSCT had significantly higher peak NS7CAR values than those who did not (P = .0289) (Figure 5A). There was no significant difference in NS7CAR-T expansion among the subgroups of disease type or the presence of EMD (P > .05) (Figure 5B-C).
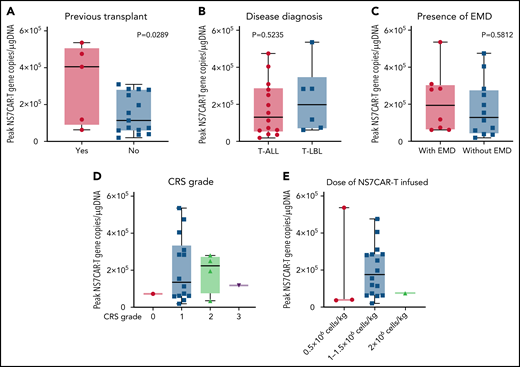
Peak values of NS7CAR T-cell expansion in subgroups of patients as determined by qPCR. The peak value of NS7CAR expansion was significantly associated with previous transplant history (P = .0289) (A) but not with the subgroups of the disease diagnosed (B) or the presence of EMD (C) (P > .05). Statistical significance could not be established due to the small sample size (n < 2) of the CRS grade subgroups (D) or for the doses of NS7CAR T cells infused (E).
Peak values of NS7CAR T-cell expansion in subgroups of patients as determined by qPCR. The peak value of NS7CAR expansion was significantly associated with previous transplant history (P = .0289) (A) but not with the subgroups of the disease diagnosed (B) or the presence of EMD (C) (P > .05). Statistical significance could not be established due to the small sample size (n < 2) of the CRS grade subgroups (D) or for the doses of NS7CAR T cells infused (E).
NS7CAR proliferation reached a level as high as 84.71% of lymphocytes as determined by FCM, with a median peak proportion of 67.76% (36.6%-84.71%) and a median peak time of 17 (7-29) days (supplemental Table 10).
Among the 24 cytokines tested, 13 increased to different degrees in all patients. The median peak time of all 24 cytokines was 13.5 (0-29) days after infusion (supplemental Table 11).
The number of CD7+ cells in the PB dramatically decreased after CAR-T infusion with the subsequent adequate recovery of normal T and NK cells
The most dramatic decrease of malignant and normal CD7+ cells in PB by FCM occurred at a median time of 7 (4-21) days after NS7CAR infusion (supplemental Table 10). The absolute numbers of T/B/NK cells (excluding CAR+ and leukemia T cells) showed a transient decline, to different extents, for each patient during the peak of CAR-T proliferation (Figure 6). CD7+ cells were rapidly eliminated by CAR T, with predominantly CD7-negative cell subsets remaining (Figures 6 and 7). T/B/NK cells then recovered to the baseline levels but decreased shortly after allo-HSCT, as expected. There was an adequate recovery of normal CD7+ T and NK cells after allo-HSCT (Figure 7).
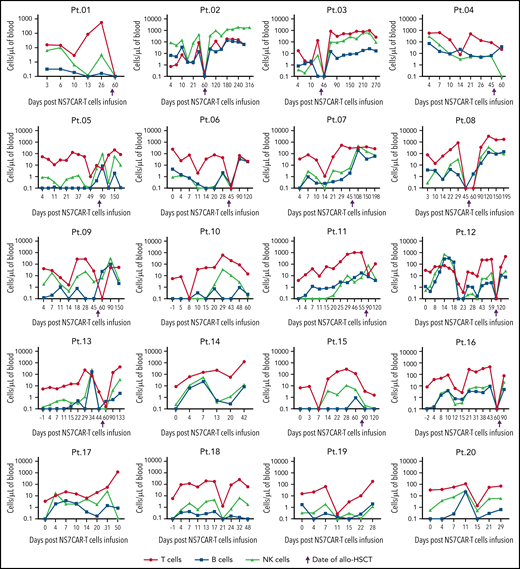
The dynamics of the absolute count of the T, B, and NK cells among individual patients after NS7CAR T-cell infusion and allo-HSCT. The absolute numbers of T, B, and NK cells of each patient were measured after NS7CAR T-cell therapy. CAR T and malignant T cells were excluded from T-cell counting. The absolute numbers of T/B/NK cells showed a temporary decline, to varying levels, for each patient during the peak of CAR-T proliferation. T/B/NK cells then recovered to the baseline levels but decreased shortly again after allo-HSCT. The 4 patients (Pt.05, Pt.07, Pt.11, and Pt.15) missed the time window for blood check within the 1 week after transplant to observe the decline in T and NK cells. There was an adequate recovery of normal T and NK cells after allo-HSCT.
The dynamics of the absolute count of the T, B, and NK cells among individual patients after NS7CAR T-cell infusion and allo-HSCT. The absolute numbers of T, B, and NK cells of each patient were measured after NS7CAR T-cell therapy. CAR T and malignant T cells were excluded from T-cell counting. The absolute numbers of T/B/NK cells showed a temporary decline, to varying levels, for each patient during the peak of CAR-T proliferation. T/B/NK cells then recovered to the baseline levels but decreased shortly again after allo-HSCT. The 4 patients (Pt.05, Pt.07, Pt.11, and Pt.15) missed the time window for blood check within the 1 week after transplant to observe the decline in T and NK cells. There was an adequate recovery of normal T and NK cells after allo-HSCT.

The dynamics of CD7+ T cells, NK cells, and CD7CAR gene copy numbers in each patient. (Left y-axis) The percentages of CD7+ T cells and NK cells as determined by FCM. (Right y-axis) CD7CAR gene copy number by qPCR. CD7+ T and NK cells were eliminated by NS7CAR T cells. An adequate recovery was observed among normal CD7+ T and NK cells after allo-HSCT. The 4 patients (Pt.05, Pt.07, Pt.11, and Pt.15) missed the time window for blood check within the 1 week after transplant to observe the decline in T and NK cells.
The dynamics of CD7+ T cells, NK cells, and CD7CAR gene copy numbers in each patient. (Left y-axis) The percentages of CD7+ T cells and NK cells as determined by FCM. (Right y-axis) CD7CAR gene copy number by qPCR. CD7+ T and NK cells were eliminated by NS7CAR T cells. An adequate recovery was observed among normal CD7+ T and NK cells after allo-HSCT. The 4 patients (Pt.05, Pt.07, Pt.11, and Pt.15) missed the time window for blood check within the 1 week after transplant to observe the decline in T and NK cells.
Discussion
CD7-targeted CAR T cells with disrupted CD7 expression, including by CD7 gene ablation, have been explored in T-ALL preclinical studies.23,28,29 Here, we have described a method to generate “naturally selected” anti-CD7 CAR T cells (NS7CAR) by transducing an anti-CD7 CAR into bulk peripheral T lymphocytes via a lentivirus vector. The NS7CAR T cells were then allowed to emerge naturally in vitro. These NS7CAR T cells were functionally active in vitro and in vivo. While CD7 molecules on the surface of NS7CAR T cells were not detectable by FCM, NS7CAR T cells continue to express CD7 mRNA and protein at levels comparable to, albeit lower than, those in control normal T cells. We think the CD7 molecules of many NS7CAR T cells are most likely antigenically masked or intracellularly sequestered by CD7CAR. Minimizing available surface CD7 antigen from NS7CAR T precluded major fratricide. The possibility that a small fraction of NS7CAR T cells emerged from fratricide during the in vitro “natural selection” process could not be ruled out. NS7CAR-T contrasts with bona fide CD7-null Neg7CAR T or KO7CAR T cells. Thus, many NS7CAR T cells are not CD7-negative per se but are functionally CD7-negative. As CAR signaling likely requires protein complex formation, we posit that CAR T cells are activated via the interaction between the free CD7CAR on the surface of NS7CAR T cells and the CD7 expressed on target T cells, and intracellular CD7− CD7CAR complexes are unable to activate T cells. Whether intracellular CD7CAR-CD7 interaction resulted in attenuated CAR signaling and contributed to the lower extent of NS7CAR cell expansion in vitro vis-à-vis Neg7CAR and KO7CAR cells remains to be determined. Finally, whether fratricide played a role in the reduced expansion of NS7CAR also awaits further investigation.
In our preclinical study, we compared the anti-CD7 CAR T cells derived from 3 different approaches. NS7CAR and KO7CAR showed comparable cytotoxicity and cytokine release in the in vitro functional assay and conferred robust antitumor protection in a mouse xenograft model. A significantly higher number of CD8+ central memory CAR T cells were present in the NS7CAR population. The CD8 bias of NS7CAR may afford more sustainable disease control compared with the other engineered T cells.42-45 Importantly, because no gene editing or additional gene transfer is involved in deriving NS7CAR, the risk of off-target genetic alteration is obviated. Thus, the biological properties of NS7CAR and the ease of its production facilitate its clinical application.
To date, there have been only limited clinical trials and case reports on anti-CD7 CAR-T therapy that targets T-cell malignancies.18-20,46,47 NS7CAR can be generated from the donor or patient PBMCs with a median transfection efficiency of >95%, and approximately 90% of the NS7CAR T cells were viable. One of the safety concerns of infusing autologous CAR T cells prepared from patients with T-cell malignancies is the contamination of malignant cells in the final cell products.13,48 Based on the phenotype of circulating tumor cells in each patient, the use of either CD3 selection or CD4/CD8 selection coupled with in vitro “natural selection” and potent antileukemia activity from NS7CAR can successfully exclude malignant cells from the NS7CAR preparation.
In our clinical trial, all but 1 patient achieved MRD-negative CR/CRi in their BM by day 28. Initial high extramedullary CR could also be achieved in the majority of patients, including patients with CNS leukemia or diffuse extramedullary involvements. Two patients with bulky mediastinal masses achieved PR and SD, respectively. For patients with bulky masses to achieve CR, a longer time may be necessary as the patient in PR was found to have further clinical improvement on day 104 based on PET-CT evaluation. Combined treatment approaches likely will be needed as well.
Patients enrolled in our trial were heavily pretreated. In view of the high relapse rate among B-ALL patients treated with anti-CD19 CAR-T monotherapy and given T-ALL/LBL are more aggressive than B-ALL in general, our study allowed postremission consolidative allo-HSCT.4,8,46,49 A total of 10 patients with both CR in BM and EMD pursued consolidative allo-HSCT, which limited our ability to assess the durability of NS7CAR-T monotherapy independently. This notwithstanding, with a median follow-up time of >4.5 months (up to 10 months), no relapse was observed in the CAR-T followed by the allo-HSCT group. Of 6 CR patients who did not receive a transplant, 4 remained CR at last follow-up with a median time of 1.5 months and up to 6 months. CD7 marker loss, however, was observed in some patients with less than a CR or relapsed disease.
STIL-TAL1 gene fusion occurs in up to about 25% of T-ALL patients and is associated with a higher frequency of relapse and less favorable outcomes.50-53 Indeed, all 3 patients with a STIL-TAL1 fusion gene in our trial had a poor prognosis with 1 no response and 2 early relapses, suggesting that CAR-T therapy alone may not reverse the poor clinical outcome. Once such patients achieve a CR from CAR T, transplantation should be considered early in the treatment course.
The expansion of NS7CAR T cells reached a median peak on day 19, with persistence detectable up to 195 days as of the last follow-up. NS7CAR T cells remained detectable at low levels by qPCR in 12 of 14 patients at a median time of 1.2 months (up to 4.9 months) at the last follow-up after allo-HSCT. Reexpansion of CAR T cells or depletion of CD7+ cells after allo-HSCT was not observed, suggesting that little CAR-T activity remained following transplantation.
NS7CAR exhibited a superior safety profile with only mild CRS in most patients and no significant neurotoxicity. Tocilizumab was used early in our protocol for grade 1 CRS when high fever persisted. Such early intervention and management likely play a role in reducing CRS, as previously reported.54,55
Two patients who had a prior history of allo-HSCT at enrollment received CAR T cells generated from transplant donors. One developed mild skin aGvHD. Thus, special attention should be paid to such patients. Seven of 14 patients received a subsequent allo-HSCT, including 2 with a second allo-HSCT who developed GvHD. While CAR T cells remained detectable in the 7 patients who developed GvHD after infusion, the levels of residual CAR T cells were low and unlikely to be responsible for the GvHD. The incidence of GvHD reported here is expected and consistent with our previous clinical experiences and studies.56
Immunodeficiency associated with the depletion/impairment of normal T/NK cells is a concern following anti-CD7 CAR-T therapy. In our trial, no apparent increase in infection rate was observed between the interval of NS7CAR infusion and transplantation. After NS7CAR therapy, the absolute number of T (excluding CAR T and tumor cells) and NK cells showed only a patient-dependent transient decline of variable degrees during the peak of CAR T proliferation. One case of CMV reactivation was observed in a patient with prior allo-HSCT. No fungal infections or flare-ups were observed.
In conclusion, NS7CAR demonstrated robust antitumor effects both in vitro and in vivo. Our first-in-human clinical trial showed that NS7CAR T cells could be successfully generated without additional genetic manipulations to prevent CD7 expression. Overall, NS7CAR is safe and highly effective for treating R/R T-ALL/LBL. The inclusion of more patients and extended observation time is needed for further evaluation of NS7CAR T-cell therapy.
Acknowledgments
The authors thank all the medical staff in Hebei Yanda Lu Daopei Hospital for their clinical and technical assistance, especially their care for patients, and Hebei Senlang Biotechnology Co, Ltd, for their contributions to the preclinical investigations.
This study was supported (in part) by grant funding from the S&T Program of Hebei (Grant number: 21372410D).
Authorship
Contribution: P.L. and X.H. designed and supervised the clinical trial; Y.L. and J.L. designed and conducted the preclinical study and analyzed preclinical data; Y.L., L.W., and Q.W. oversaw the CAR T-cell production and quality control; J.Y., X.Z., and P.L. conducted the clinical study and provided patient care; J.Y., X.Z., X.Y., and H.W. collected and analyzed clinical data and performed statistical analyses; P.L., Y.L., X.Y., X.H., and J.L. wrote the manuscript; and P.L., X.H., J.L., and D.J. revised the manuscript.
Conflict-of-interest disclosure: Y.L., L.W., Q.W., and J.L. are employees of Hebei Senlang Biotechnology Co, Ltd. The remaining authors declare no competing financial interests.
Correspondence: Peihua Lu, Hebei Yanda Lu Daopei Hospital, Sipulan Road, Yanjiao Economic and Technological Development Zone, Langfang City, Hebei 065201, China; e-mail: peihua_lu@126.com; Jianqiang Li, Hebei Senlang Biotechnology Co, Ltd, Room 512 and 513, Building 1, 136 Yellow River Ave, Shijiazhuang High-tech Development Zone, Shijiazhuang, Hebei 050000, China; e-mail: lijianqiang@senlangbio.com; and Xiaojun Huang, Peking University People's Hospital, Peking University Institute of Hematology, No.11 Xizhimen South St, Xicheng District, Beijing 100044, China; e-mail: xjhrm@medmail.com.cn.
Detailed data are provided in the supplemental Appendix. Other data are available from the corresponding author upon reasonable request and via the data transfer agreement.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal