

Key Points
rVWF prophylaxis reduced spontaneous bleeding rates in patients previously on VWF on-demand therapy.
Patients switching from prophylaxis with plasma-derived VWF to rVWF experienced a similar level of control over their spontaneous bleeding.

Abstract
International guidelines conditionally recommend long-term prophylaxis in patients with von Willebrand disease (VWD) and severe and frequent bleeding. As recombinant von Willebrand factor (rVWF; vonicog alfa) may reduce the frequency of treated spontaneous bleeding events (BEs), we investigated the efficacy and safety of rVWF prophylaxis in adults with severe VWD. Patients with BEs requiring VWF therapy in the past year (on-demand VWF therapy [prior on-demand group] or plasma-derived VWF prophylaxis [pdVWF; switch group]) were enrolled in a prospective, open-label, nonrandomized, phase 3 study. The planned duration of rVWF prophylaxis was 12 months; starting rVWF dose was 50 ± 10 VWF: ristocetin cofactor (VWF:RCo) IU/kg twice weekly (prior on-demand group) or based on prior pdVWF weekly dose/dosing frequency (switch group). The primary endpoint was annualized bleeding rate (ABR) of treated spontaneous BEs (sABR) during rVWF prophylaxis. Over the 12-month study period, treated sABR decreased by 91.5% on-study vs historical sABR in 13 patients in the prior on-demand group, and by 45.0% in 10 patients in the switch group (model-based analysis ratio, 0.085; 95% confidence interval [CI], 0.021-0.346 and 0.550; 95% CI, 0.086-3.523, respectively). No treated spontaneous BEs were recorded in 84.6% (11/13) and 70.0% (7/10) of patients, respectively. The safety profile of rVWF was consistent with the previously established profile, with no new adverse drug reactions identified. Findings suggest that rVWF prophylaxis can reduce treated spontaneous BEs in patients previously receiving on-demand VWF therapy and maintains at least the same level of hemostatic control in patients who switch from prophylaxis with pdVWF to rVWF, with a favorable safety profile. This trial was registered at www.clinicaltrials.gov (#NCT02973087) and www.clinicaltrialsregister.eu (#EudraCT 2016-001478-14).
Introduction
von Willebrand disease (VWD) is an inherited autosomal bleeding disorder characterized by deficiencies in von Willebrand factor (VWF), a plasma glycoprotein that mediates platelet adhesion/aggregation and stabilizes coagulation factor VIII (FVIII) in the circulation.1-3 VWD is the most common inherited bleeding disorder (prevalence based on VWF activity: 0.6% to 1.3%).4,5 Symptomatic VWD requiring treatment is less common, with a prevalence of 23 to 113 per million population, or as high as 1 in 1000 in some clinical settings.2,6
VWD is associated with a heterogenous bleeding phenotype, including easy bruising, prolonged bleeding from wounds or surgeries, mucocutaneous bleeds, heavy menstrual bleeding, and joint bleeds.1,7 Patients with VWD, especially those with the severe bleeding phenotype (mostly type 2 or 3, but also some type 1), have a poorer health-related quality of life (HRQoL) than the general population and are at increased risk of bleeding, which can be life-threatening (eg, gastrointestinal events) or lead to long-term complications (eg, arthropathy).1,8-10 These bleeding events (BEs) require immediate on-demand and/or long-term VWF prophylaxis to induce and/or maintain hemostatic levels of VWF and FVIII.1,7 Long-term VWF prophylaxis could also help reduce/prevent recurrent bleeds,11-14 thereby improving HRQoL and reducing the risk of long-term complications.15 International management guidelines for VWD published in 2021 conditionally recommend long-term prophylaxis in patients with VWD and a history of severe and frequent bleeds. However, these guidelines acknowledge the low certainty in the evidence of effects and the need for further studies to develop evidence-based guidelines for prophylaxis in VWD.7
Recombinant VWF (rVWF; vonicog alfa, Vonvendi [United States]/Veyvondi [Europe], Takeda Pharmaceuticals USA, Lexington, MA)16,17 has previously demonstrated efficacy with a consistent safety profile when used for on-demand treatment of hemorrhage and perioperative management of bleeding in patients with VWD.18,19 Because patients with severe VWD may also benefit from rVWF prophylaxis to reduce the frequency of spontaneous BEs requiring VWF treatment,11,20 this trial investigated the efficacy, safety, pharmacokinetics (PKs), and pharmacodynamics (PDs) of rVWF prophylaxis in this population.
Methods
Trial summary
This phase 3, prospective, open-label, nonrandomized, multicenter study (ClinicalTrials.gov NCT02973087; EudraCT 2016-001478-14) evaluated the annualized bleeding rate (ABR) for treated spontaneous BEs (referred to as sABR hereafter) during on-study rVWF prophylaxis in adults with severe VWD relative to the patients’ historical sABRs. Two patient cohorts were recruited: patients in the prior on-demand group had previously received on-demand VWF therapy, whereas patients in the switch group had received plasma-derived VWF (pdVWF) prophylaxis. The protocol was approved by the respective Institutional Review Boards or Ethics Committees and applicable regulatory authorities before patient enrollment. The study was conducted in compliance with the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference on Harmonization at 32 sites, 18 of which enrolled patients (supplemental Table 1, available on the Blood Web site) in 9 countries from November 2017 to July 2020.
Key patient eligibility criteria
The study enrolled patients aged ≥18 years with a body mass index of 15 to 40 kg/m2 and a diagnosis of severe VWD (baseline VWF:ristocetin cofactor activity [VWF:RCo] <20 IU/dL).11,21 Genetic testing and multimer analysis at screening were used to confirm the VWD type of the patient, with the additional criterion of VWF antigen ≤3 IU/dL required for patients with type 3 VWD. Patients were required to have reliable medical records for BEs for ≥12 months preceding enrollment that documented the requirement for VWF treatment to manage BEs. Patients in the prior on-demand group were receiving on-demand VWF treatment at screening and had ≥3 documented spontaneous BEs (not including menorrhagia) requiring VWF treatment during the past 12 months. Patients in the switch group had received pdVWF prophylaxis for ≥12 months at screening. All patients provided written informed consent. Exclusion criteria are provided in the supplemental Methods.
Treatment
Patients received IV infusions of rVWF. The planned duration for rVWF prophylaxis was 12 months; the actual treatment period was ≤18 months to allow for uninterrupted rVWF prophylaxis in patients enrolling in the phase 3b extension/continuation study (ClinicalTrials.gov NCT03879135; EudraCT 2018-003453-16). In the prior on-demand group, the recommended starting dose regimen was 50 ± 10 VWF:RCo IU/kg twice weekly. In the switch group, the starting dose/dosing frequency was based on the prior pdVWF weekly VWF dose equivalent (within ±10%) divided into 1 to 3 weekly infusions (maximum: 80 VWF:RCo IU/kg per infusion). rVWF dosage could be individualized (maximum: 80 VWF:RCo IU/kg) based on available individual historical PK data, type/severity of historical BEs, and/or monitoring of appropriate clinical and laboratory measures. Study guidelines for the treatment of breakthrough bleeding and planned surgery/dental procedures are discussed in the supplemental Methods.
Outcome measures
The primary outcome measure was the ABR for treated spontaneous/non-traumatic BEs (sABRs; as assessed by the investigator) during the first 12 months of rVWF prophylaxis. On-study treated sABRs were compared within each patient with historical sABRs, as derived from patient medical records and reported at the screening by the investigator, and summarized separately by cohort (ie, prior on-demand or switch cohorts).
Secondary efficacy outcome measures included sABR intrapatient comparison (proportion of patients with reduction or preservation success), categorized sABR (values of 0, >0 to 2, >2 to 5, or >5), rVWF consumption, and ABR by bleed location. For the prior on-demand group, sABR reduction success was defined as a ≥25% reduction in sABR from historical on-demand treatment to on-study rVWF prophylaxis. For the switch group, sABR preservation success was defined as an sABR during on-study rVWF prophylaxis equal to or less than the historical sABR during pdVWF prophylaxis.
Management of spontaneous and traumatic breakthrough bleeds was evaluated as an exploratory efficacy outcome measure and included rVWF consumption (in addition to rVWF prophylaxis) with or without recombinant FVIII (rFVIII) and overall hemostatic efficacy at resolution. If surgery was required, the efficacy of perioperative bleed management was assessed, including intraoperative actual vs predicted blood loss, intraoperative and overall hemostatic efficacy, and daily intraoperative and postoperative weight-adjusted rVWF consumption (in addition to prophylactic rVWF consumption), with or without rFVIII.
Safety (from the first rVWF exposure until study completion) was evaluated by reporting adverse events (AEs), including those of special interest such as hypersensitivity, thrombogenicity, and immunogenicity, and clinically significant changes in vital signs and clinical laboratory parameters relative to baseline. AEs were categorized according to the Medical Dictionary for Regulatory Activities (MedDRA, version 23.0). Assays performed to detect the presence of binding and neutralizing antibodies to VWF and FVIII are described in the supplemental Methods.
PK/PD parameters after a single rVWF prophylactic dose in the prior on-demand group (initial PK/PD assessment) and after multiple rVWF prophylactic doses in both groups (steady-state PK/PD assessments) were derived using noncompartmental methods. Details of the PK/PD assessments are described in the supplemental Methods.
Patients were provided with an electronic diary at the screening to record infusions/rVWF consumption, BEs, patient self-assessments of bleed severity and hemostatic efficacy, untoward events/experiences, concomitant medications, and patient-reported outcomes.
Statistical analysis
Because no formal statistical tests were planned for this study, the sample size was not based on a power calculation for a significance test but was driven mainly by the European Medicines Agency Guideline on the Clinical Investigation of Human pdVWF Products.21 Approximately 22 adults were to be included in the study: ≥8 patients in each cohort and a total of ≥5 patients with type 3 VWD.
The safety analysis set comprised all patients who signed informed consent and received any amount of rVWF. The full analysis set (the primary analysis population for the efficacy outcome measures) comprised all patients who received rVWF prophylaxis. The PK/PD full analysis set comprised all patients who received ≥1 rVWF infusion and provided ≥1 quantifiable PK or PD measurement after administration for the PK or PD analysis.
The primary endpoint analysis provided an estimation of sABR through month 12. The mean and 95% Wald confidence interval (CI) for the historical and on-study sABRs and the ratio of the 2 sABRs (on-study/historical) were estimated within each group using a generalized linear mixed-effects model fitting a negative binomial distribution and a logarithmic link function (the default). The number of unique BEs for each patient by observation period (historical or on-study) was a dependent variable, and the observation period (historical or on-study) an independent term. The model options included the logarithm of the observation period duration (in years) as an offset. Historical sABR was based on reliable medical records for BEs occurring within 12 months before the first on-study rVWF prophylactic infusion. If a bleed occurred >24 hours following resolution of a previous bleed or at a different anatomical location, it was generally counted as a unique BE by the investigator. BEs occurring at the same anatomical location (eg, right knee) with the same etiology (ie, spontaneous vs injury) within 24 hours after the onset of the first bleed or bleeds occurring at multiple locations related to the same injury (eg, knee and ankle bleeds following a fall) were to be reported as a single BE by the investigator. BEs were analyzed as reported by the investigator.
Descriptive statistics were also performed for the primary endpoint and all other outcome measures. Clopper-Pearson CIs at the 95% level were provided for percentages when appropriate.
Results
Study population
In total, 29 patients were screened; 6 patients failed the screening, resulting in 23 patients enrolled in the study who received rVWF prophylaxis (Figure 1). Seventeen patients completed the study (ie, the 12-month treatment period); reasons for discontinuations are provided in Figure 1. Overall, the mean (standard deviation [SD]) age of patients was 40.6 (19.3) years, approximately half were male, and most had type 3 VWD (78.3%) (Table 1). Patient baseline characteristics were similar between both groups, with some exceptions: patients in the switch group had lower historical bleeding rates, fewer concomitant medical conditions, and less frequent use of prior medications and nondrug therapies than those in the prior on-demand group.

Patient disposition. *Primary reason: platelet count <100 000/mL at screening (n = 1), scheduled for surgical intervention (n = 1), history or presence of a VWF inhibitor at screening (n = 1), patient not willing or able to comply with protocol requirements (n = 2), VWD inclusion criteria not met (n = 1). †Patients who were treated on-demand with any VWF during the 12-month period before enrolling in this study. ‡Patients who were treated prophylactically with a pdVWF for ≥12 months before enrolling in this study. §Nonserious headache (moderate intensity), which was considered by the investigator to be possibly related to rVWF and began during an rVWF infusion. ‖One patient in each group withdrew consent for reasons unrelated to efficacy/bleeding, and 1 patient in the prior on-demand group was lost to follow-up. ¶Scheduled for extended treatment with hydrocortisone >10 mg per day (not permitted during the study). **Required treatment with high corticosteroid doses for rheumatoid arthritis (not permitted during the study). ††All patients who were enrolled and received any amount of rVWF. ‡‡All patients who received rVWF prophylaxis. §§All patients who received ≥1 rVWF infusion and provided ≥1 quantifiable PK/PD postdose measurement. FAS, full analysis set; PKFAS, pharmacokinetic full analysis set; SAS, safety analysis set.
Patient disposition. *Primary reason: platelet count <100 000/mL at screening (n = 1), scheduled for surgical intervention (n = 1), history or presence of a VWF inhibitor at screening (n = 1), patient not willing or able to comply with protocol requirements (n = 2), VWD inclusion criteria not met (n = 1). †Patients who were treated on-demand with any VWF during the 12-month period before enrolling in this study. ‡Patients who were treated prophylactically with a pdVWF for ≥12 months before enrolling in this study. §Nonserious headache (moderate intensity), which was considered by the investigator to be possibly related to rVWF and began during an rVWF infusion. ‖One patient in each group withdrew consent for reasons unrelated to efficacy/bleeding, and 1 patient in the prior on-demand group was lost to follow-up. ¶Scheduled for extended treatment with hydrocortisone >10 mg per day (not permitted during the study). **Required treatment with high corticosteroid doses for rheumatoid arthritis (not permitted during the study). ††All patients who were enrolled and received any amount of rVWF. ‡‡All patients who received rVWF prophylaxis. §§All patients who received ≥1 rVWF infusion and provided ≥1 quantifiable PK/PD postdose measurement. FAS, full analysis set; PKFAS, pharmacokinetic full analysis set; SAS, safety analysis set.
Patient demographics and baseline characteristics
| Prior on-demand group* (n = 13) | Switch group† (n = 10) | |
|---|---|---|
| Age, y | ||
| Mean (SD) | 38.0 (17.6) | 43.9 (21.8) |
| Median (range) | 30.0 (20-67) | 34.0 (18-77) |
| Sex, n (%) | ||
| Male | 5 (38.5) | 7 (70.0) |
| Female | 8 (61.5) | 3 (30.0) |
| Body mass index, kg/m2 | ||
| Mean (SD) | 23.3 (3.1) | 23.3 (3.5) |
| Median (range) | 23.6 (17.8-29.3) | 23.7 (17.7-28.6) |
| VWD type, n (%) | ||
| Type 1 | 2 (15.4) | 1 (10.0) |
| Type 2A | 0 | 1 (10.0) |
| Type 2B | 1 (7.7) | 0 |
| Type 3 | 10 (76.9) | 8 (80.0) |
| VWF:RCo, IU/dL | ||
| Mean (SD) | 5.6 (10.7) | 0.8 (2.6) |
| Median (range) | 0 (0-27.8) | 0 (0-8.3) |
| FVIII:C, IU/dL | ||
| Mean (SD) | 25.9 (40.6) | 10.3 (12.5) |
| Median (range) | 3.0 (2-111) | 3.5 (1-40) |
| Prior on-demand group* (n = 13) | Switch group† (n = 10) | |
|---|---|---|
| Age, y | ||
| Mean (SD) | 38.0 (17.6) | 43.9 (21.8) |
| Median (range) | 30.0 (20-67) | 34.0 (18-77) |
| Sex, n (%) | ||
| Male | 5 (38.5) | 7 (70.0) |
| Female | 8 (61.5) | 3 (30.0) |
| Body mass index, kg/m2 | ||
| Mean (SD) | 23.3 (3.1) | 23.3 (3.5) |
| Median (range) | 23.6 (17.8-29.3) | 23.7 (17.7-28.6) |
| VWD type, n (%) | ||
| Type 1 | 2 (15.4) | 1 (10.0) |
| Type 2A | 0 | 1 (10.0) |
| Type 2B | 1 (7.7) | 0 |
| Type 3 | 10 (76.9) | 8 (80.0) |
| VWF:RCo, IU/dL | ||
| Mean (SD) | 5.6 (10.7) | 0.8 (2.6) |
| Median (range) | 0 (0-27.8) | 0 (0-8.3) |
| FVIII:C, IU/dL | ||
| Mean (SD) | 25.9 (40.6) | 10.3 (12.5) |
| Median (range) | 3.0 (2-111) | 3.5 (1-40) |
FVIII:C, factor VIII clotting (activity).
Patients who were treated on-demand with any VWF during the 12-month period before enrolling in this study.
Patients who were treated prophylactically with a pdVWF for ≥12 months before enrolling in this study.
Efficacy
In patients previously receiving on-demand VWF treatment, the model-based mean sABR for treated bleeds was reduced by 91.5% (ratio, 0.085; 95% CI, 0.021-0.346) during the 12-month study period, relative to the mean historical sABR (Table 2). Mean sABR for treated bleeds decreased from 6.54 (95% CI, 2.52-17.00) to 0.56 (95% CI, 0.15-2.05). In patients who switched from prior pdVWF prophylaxis to rVWF prophylaxis in this study, the model-based mean sABR for treated bleeds was reduced by 45% (ratio, 0.550; 95% CI, 0.086-3.523) from 0.51 (95% CI, 0.04-6.31) to 0.28 (95% CI, 0.02-3.85).
Primary efficacy analysis: comparison of on-study sABRs with historical estimates
| Prior on-demand group (n = 13) | Switch group (n = 10) | |
|---|---|---|
| Historical | ||
| No. of treated spontaneous BEs | 201 | 50 |
| sABR mean (95% CI)* | 6.54 (2.52 to 17.00) | 0.51 (0.04 to 6.31) |
| rVWF prophylaxis (on-study treatment) | ||
| No. of treated spontaneous BEs | 9 | 18 |
| sABR mean (95% CI)* | 0.56 (0.15 to 2.05) | 0.28 (0.02 to 3.85) |
| Comparison (rVWF prophylaxis vs historical sABR) | ||
| sABR rVWF prophylaxis:historical ratio (95% CI) | 0.085 (0.021 to 0.346) | 0.550 (0.086 to 3.523) |
| sABR percentage change from historical (95% CI)† | −91.5% (−97.9% to −65.4%) | −45.0% (−91.4% to 252.3%) |
| Prior on-demand group (n = 13) | Switch group (n = 10) | |
|---|---|---|
| Historical | ||
| No. of treated spontaneous BEs | 201 | 50 |
| sABR mean (95% CI)* | 6.54 (2.52 to 17.00) | 0.51 (0.04 to 6.31) |
| rVWF prophylaxis (on-study treatment) | ||
| No. of treated spontaneous BEs | 9 | 18 |
| sABR mean (95% CI)* | 0.56 (0.15 to 2.05) | 0.28 (0.02 to 3.85) |
| Comparison (rVWF prophylaxis vs historical sABR) | ||
| sABR rVWF prophylaxis:historical ratio (95% CI) | 0.085 (0.021 to 0.346) | 0.550 (0.086 to 3.523) |
| sABR percentage change from historical (95% CI)† | −91.5% (−97.9% to −65.4%) | −45.0% (−91.4% to 252.3%) |
Estimated using a generalized linear mixed-effects model for the full analysis set through month 12. Only BEs treated with VWF infusions are included. Six BEs of unknown cause (4 historical [all in the prior on-demand group] and 2 on-study [switch group]) were counted as spontaneous BEs for this analysis.
Percentage change in sABR was calculated directly from the sABR ratio (RR): 100 × (RR - 1).
The observed reductions in sABR based on descriptive statistics were consistent with the model-based analysis of sABR. Median (range) change from historical to on-study (while on rVWF prophylaxis) sABR for treated bleeds was −3.0 (−155.0 to 2.8) in the prior on-demand group and 0 (−33.9 to 3.9) in the switch group (Figure 2). The proportions of patients with a treated sABR of 0 during rVWF prophylaxis (on-study through month 12) was 84.6% (11/13) in the prior on-demand group and 70.0% (7/10) in the switch group (Figure 2). In the prior on-demand group, 92.3% (12/13) of patients (95% CI, 64.0% to 99.8%) had a reduction of ≥25% in sABR during on-study rVWF prophylaxis (through month 12) relative to their historical sABR. In the switch group, 90% (9/10) of patients (95% CI, 55.5% to 99.7%) had an sABR during on-study rVWF prophylaxis that was not higher than their historical sABR.
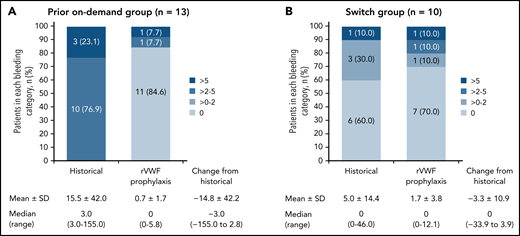
Treated spontaneous ABRs. sABRs for (A) the prior on-demand group and (B) the switch group (FAS). Figures show the proportion of patients in each bleeding category, historically and on-study, as well as mean (SD) and median (range) change from historical to on-study sABR (on-study through month 12). Historically (ie, within the past 12 months), none of the patients in the prior on-demand group had a 0 or >0 to 2 sABR.
Treated spontaneous ABRs. sABRs for (A) the prior on-demand group and (B) the switch group (FAS). Figures show the proportion of patients in each bleeding category, historically and on-study, as well as mean (SD) and median (range) change from historical to on-study sABR (on-study through month 12). Historically (ie, within the past 12 months), none of the patients in the prior on-demand group had a 0 or >0 to 2 sABR.
Most patients (18/23) had 0 treated spontaneous BEs while receiving rVWF prophylaxis through month 12. Of the 5 patients who had treated spontaneous BEs, 3 had reductions from their historical sABR, and 2 had an increase from their historical sABR (supplemental Table 2). Historical and on-study spontaneous BEs are shown by patient in supplemental Figure 1. Most of the on-study–treated spontaneous BEs in the 5 patients were mucosal bleeds and/or menorrhagia (prior on-demand group: 5 oral/other mucosa, 3 menorrhagia, and 1 other location; Switch group: 14 oral/other mucosa, 1 hemarthrosis, and 3 unknown location). There were no muscle/soft tissue, skin, gastrointestinal, central nervous system, or body cavity bleeds, and no hematuria.
rVWF prophylactic consumption
Most patients (prior on-demand: 100%; switch: 80%) started on a twice-weekly rVWF prophylaxis regimen (Table 3). Three patients had ≥1 increase in their dosing regimen (1 in prior on-demand changed from twice weekly to 3 times weekly and 2 in the switch group changed from twice weekly to 3 times weekly or every 3 days). The mean (SD) weight-adjusted weekly rVWF dose per patient was 98.6 (40.6) IU/kg in the prior on-demand group and 94.0 (30.4) IU/kg in the switch group.
Prophylactic consumption of rVWF through month 12
| Prior on-demand group (n = 13) | Switch group (n = 10) | |
|---|---|---|
| Total no. of prophylactic infusions administered | 846 | 878 |
| Initial dosing frequency, n (%) | ||
| Once weekly | 0 (0) | 1 (10.0) |
| Twice weekly | 13 (100) | 8 (80.0) |
| 3 times weekly | 0 (0) | 1 (10.0) |
| Patients with ≥1 increase in dosing regimen, n (%) | 1 (7.7) | 2 (20.0) |
| Infusions per patient | ||
| Mean (SD) | 65.1 (38.4) | 87.8 (30.2) |
| Median (range) | 67.0 (2-116) | 104.5 (31-116) |
| No. of weekly infusions per patient | ||
| Mean (SD) | 1.9 (0.7) | 1.9 (0.4) |
| Median (range) | 1.9 (0.7-3.5) | 2.0 (0.8-2.1) |
| Average weight-adjusted rVWF dose per infusion, IU/kg | ||
| Mean (SD) | 52.2 (3.8) | 52.2 (16.2) |
| Median (range) | 52.9 (44.8-58.8) | 55.9 (24.4-79.4) |
| Weight-adjusted weekly rVWF dose per patient, IU/kg | ||
| Mean (SD) | 98.6 (40.6) | 94.0 (30.4) |
| Median (range) | 95.6 (34.9-187.8) | 106.8 (48.0-128.0) |
| Prior on-demand group (n = 13) | Switch group (n = 10) | |
|---|---|---|
| Total no. of prophylactic infusions administered | 846 | 878 |
| Initial dosing frequency, n (%) | ||
| Once weekly | 0 (0) | 1 (10.0) |
| Twice weekly | 13 (100) | 8 (80.0) |
| 3 times weekly | 0 (0) | 1 (10.0) |
| Patients with ≥1 increase in dosing regimen, n (%) | 1 (7.7) | 2 (20.0) |
| Infusions per patient | ||
| Mean (SD) | 65.1 (38.4) | 87.8 (30.2) |
| Median (range) | 67.0 (2-116) | 104.5 (31-116) |
| No. of weekly infusions per patient | ||
| Mean (SD) | 1.9 (0.7) | 1.9 (0.4) |
| Median (range) | 1.9 (0.7-3.5) | 2.0 (0.8-2.1) |
| Average weight-adjusted rVWF dose per infusion, IU/kg | ||
| Mean (SD) | 52.2 (3.8) | 52.2 (16.2) |
| Median (range) | 52.9 (44.8-58.8) | 55.9 (24.4-79.4) |
| Weight-adjusted weekly rVWF dose per patient, IU/kg | ||
| Mean (SD) | 98.6 (40.6) | 94.0 (30.4) |
| Median (range) | 95.6 (34.9-187.8) | 106.8 (48.0-128.0) |
Over 80% of patients received ≥70% of the planned infusions. Prophylactic compliance (100 × number of actual infusions/number of planned infusions) per patient ranged from 36% to 100% in the prior on-demand group and from 67% to 100% in the switch group (mean [SD], 81.6 [22.0] and 87.2 [13.4], respectively).
Management of breakthrough bleeds
There were 31 treated, all-cause BEs during the 12-month study period: 12 BEs in 4 patients in the prior on-demand group and 19 BEs in 3 patients in the switch group. Most were mild or moderate (75% in the prior on-demand group and 89% in the switch group), spontaneous (75% in the prior on-demand group and 95% in the switch group), and required only 1 infusion of rVWF (66.7% and 88.9%, respectively) with or without rFVIII. The median average rVWF dose for on-demand infusions (with or without rFVIII) was 52.2 and 53.4 IU/kg, respectively. Three patients (prior on-demand: 1; switch: 2) received a single rFVIII infusion in addition to rVWF for the treatment of a total of 7 breakthrough bleeds. The median average rFVIII dose for on-demand infusions was 27.8 and 23.2 IU/kg, respectively. Use of antifibrinolytics was permitted for treatment of minor or moderate breakthrough BEs when additional infusion of rVWF to treat the breakthrough BE was considered not necessary. Two patients (1 each in the prior on-demand and switch groups) had concomitant antifibrinolytics reported with no confirmed association to any BEs. Both patients had zero treated (per investigator’s assessment) on-study BEs of any cause.
Most breakthrough bleeds occurred in mucosal locations, and all occurred in patients with type 3 VWD. The hemostatic efficacy rating was “excellent” or “good” for all 12 BEs in the prior on-demand group and for 7/9 BEs in the switch group that were assessed (10 BEs were not assessed); the rating for the remaining 2 BEs was “fair.”
Perioperative management
Perioperative management of bleeding with rVWF treatment (without rFVIII) was successful for 3 patients who underwent surgical procedures during the study. These included 2 minor, minimally invasive procedures (hyaluronic acid injection on ankle and removal of formations from skin on chest, hand, and face) and a major dental procedure (tooth extraction). In all 3 patients, rVWF infusions were started the day before surgery and repeated on the day of surgery (dental surgery: 2 infusions totaling 98 IU/kg) or for up to 7 days (removal of skin formations: 5 infusions totaling 130 IU/kg) or 18 days (hyaluronic acid injection: 6 infusions totaling 234 IU/kg) after surgery. No patient had surgery-related blood loss greater than predicted. Intraoperative and all postoperative hemostatic efficacy ratings were “excellent” for all 3 patients.
Safety and immunogenicity
Overall, 17 (73.9%) patients experienced a total of 41 AEs (Tables 4 and 5). Three serious AEs were reported in 3 patients; all were considered unrelated to rVWF treatment. In the prior on-demand group, 1 patient experienced multiple injuries from a fall, which resulted in hospitalization. In the switch group, 1 patient had a mild urinary tract infection, which was diagnosed after a brief hospitalization due to abdominal pain and suspicion of appendicitis, and another patient had rheumatoid arthritis resulting in hospitalization. This patient was discharged from the hospital with some improvement, but the condition continued; the 2 other serious AEs resolved.
AEs in patients who received rVWF prophylaxis (safety analysis set)*
| Prior on-demand group (n = 13) n (%)/events | Switch group (n = 10) n (%)/events | Total (n = 23) n (%)/events | |
|---|---|---|---|
| AE† | 10 (76.9)/26 | 7 (70.0)/15 | 17 (73.9)/41 |
| Mild | 7 (53.8)/18 | 4 (40.0)/12 | 11 (47.8)/30 |
| Moderate | 1 (7.7)/5 | 2 (20.0)/2 | 3 (13.0)/7‡ |
| Severe | 2 (15.4)/3 | 1 (10.0)/1 | 3 (13.0)/4§ |
| Serious AE | 1 (7.7)/1 | 2 (20.0)/2 | 3 (13.0)/3 |
| AE considered related to rVWF | 1 (7.7)/1 | 0 | 1 (4.3)/1 |
| Serious AE considered related to rVWF | 0 | 0 | 0 |
| AE considered related to study procedures | 0 | 0 | 0 |
| Serious AE considered related to study procedures | 0 | 0 | 0 |
| AE leading to discontinuation of rVWF | 1 (7.7)/1 | 0 | 1 (4.3)/1 |
| Fatal AE | 0 | 0 | 0 |
| Life-threatening AE | 0 | 0 | 0 |
| AE of special interest‖ | 1 (7.7)/1¶ | 1 (10.0)/1** | 2 (8.7)/2 |
| Prior on-demand group (n = 13) n (%)/events | Switch group (n = 10) n (%)/events | Total (n = 23) n (%)/events | |
|---|---|---|---|
| AE† | 10 (76.9)/26 | 7 (70.0)/15 | 17 (73.9)/41 |
| Mild | 7 (53.8)/18 | 4 (40.0)/12 | 11 (47.8)/30 |
| Moderate | 1 (7.7)/5 | 2 (20.0)/2 | 3 (13.0)/7‡ |
| Severe | 2 (15.4)/3 | 1 (10.0)/1 | 3 (13.0)/4§ |
| Serious AE | 1 (7.7)/1 | 2 (20.0)/2 | 3 (13.0)/3 |
| AE considered related to rVWF | 1 (7.7)/1 | 0 | 1 (4.3)/1 |
| Serious AE considered related to rVWF | 0 | 0 | 0 |
| AE considered related to study procedures | 0 | 0 | 0 |
| Serious AE considered related to study procedures | 0 | 0 | 0 |
| AE leading to discontinuation of rVWF | 1 (7.7)/1 | 0 | 1 (4.3)/1 |
| Fatal AE | 0 | 0 | 0 |
| Life-threatening AE | 0 | 0 | 0 |
| AE of special interest‖ | 1 (7.7)/1¶ | 1 (10.0)/1** | 2 (8.7)/2 |
Table displays the number and percentage of patients who had ≥1 AE and the number of AEs for a given parameter.
AEs starting or worsening after the first dose of rVWF.
Patients were counted once for the highest severity.
Joint (shoulder) injury, supraventricular tachycardia, and ventricular extrasystoles (all in the same patient); joint (knee) injury; headache; arthralgia; gastroenteritis; all events resolved; includes events in 2 patients who also had severe events and so are listed in the severe category.
Fall and multiple injuries from fall (2 events in the same patient and requiring hospitalization); toothache; rheumatoid arthritis. All events resolved except rheumatoid arthritis.
AEs of special interest defined as thromboembolic events, hypersensitivity reactions (including allergic or anaphylactic reactions), the development of neutralizing or binding antibodies to VWF and FVIII, and binding antibodies to trace proteins in rVWF (Chinese hamster ovary immunoglobulin G [IgG], murine IgG, and human Furin IgG).
Purpura, which developed due to trauma, was classified as a thromboembolic event (per broad SMQ search); considered nonserious and nonsevere by investigator, and resolved with no action taken.
Rash pruritic was classified as a hypersensitivity reaction (per broad SMQ search); considered nonserious and nonsevere by investigator, and resolved with no action taken.
AEs occurring in >1 patient (safety analysis set)
| MedDRA preferred term | Prior on-demand group n (%)/events | Switch group n (%)/events |
|---|---|---|
| Headache | 4 (30.8)/4 | 0 |
| Arthralgia | 1 (7.7)/1 | 2 (20.0)/2 |
| ALT increased | 1 (7.7)/1 | 1 (10.0)/1 |
| Ear infection | 2 (15.4)/2 | 0 |
| Gastroenteritis | 0 | 2 (20.0)/2 |
| Joint injury | 2 (15.4)/2 | 0 |
| Urinary tract infection | 1 (7.7)/1 | 1 (10.0)/1 |
| MedDRA preferred term | Prior on-demand group n (%)/events | Switch group n (%)/events |
|---|---|---|
| Headache | 4 (30.8)/4 | 0 |
| Arthralgia | 1 (7.7)/1 | 2 (20.0)/2 |
| ALT increased | 1 (7.7)/1 | 1 (10.0)/1 |
| Ear infection | 2 (15.4)/2 | 0 |
| Gastroenteritis | 0 | 2 (20.0)/2 |
| Joint injury | 2 (15.4)/2 | 0 |
| Urinary tract infection | 1 (7.7)/1 | 1 (10.0)/1 |
ALT, alanine aminotransferase.
Table displays the number and percentage of patients who had AEs that occurred in >1 patient across the 2 groups (prior on-demand and switch) and the number of events with the same preferred term. Only 1 AE (nonserious headache) out of 41 treatment-emergent AEs was considered related to study treatment.
Only 1 patient had an AE (nonserious headache of moderate intensity) considered possibly related to rVWF by the investigator. This event began during an rVWF infusion, improved but reoccurred later, and led to study treatment discontinuation and withdrawal from the study. The patient had a medical history of alcohol abuse, hypertension, and depression, which may be considered alternative etiologies for the headache. The event resolved 3 days after the patient withdrew. No AE was considered related to rFVIII or study procedures. Two patients, 1 per group, experienced AEs of special interest: rash pruritic was classified as a hypersensitivity reaction (per broad standardized MedDRA query [SMQ]), and purpura, which developed due to trauma, was classified as a thromboembolic event (per broad SMQ). Neither event was considered serious or severe by the investigators. No patient developed binding or neutralizing antibodies to rVWF or FVIII. No fatal or life-threatening AEs were reported.
PKs and PDs
In the prior on-demand group, most PK parameters for VWF:RCo were similar between the initial and final assessment (supplemental Table 3). Trough FVIII:C levels increased significantly by almost fivefold (least-squares mean point estimates) from 3.83 IU/dL (initial assessment before first prophylaxis dose) to 18.7 IU/dL (final assessment following 12 months of rVWF prophylaxis; P < .0001). In patients in the switch group, VWF:RCo steady-state PK parameters were generally stable between the 2 assessments (supplemental Table 3). For the 2 patients in the prior on-demand and 3 patients in the switch group who experienced spontaneous BEs, predose FVIII activity ranged from 5 to 141 IU/dL and 6 to 109 IU/dL over the 12-month study period, respectively.
Discussion
This study is the first to assess the efficacy and safety of rVWF prophylaxis and to compare on-study rVWF prophylaxis with intrapatient historical pdVWF prophylaxis based on medical records. In patients previously treated on-demand with VWF products, rVWF prophylaxis reduced the frequency of spontaneous BEs requiring VWF treatment. Patients switching from pdVWF to rVWF prophylaxis experienced levels of control over treated spontaneous BEs similar to their historical levels. Based on the data available in the medical records, 94% of historical bleeds were spontaneous, and >95% were mild or moderate bleeds. Most of these BEs were mucosal. During the study, a high proportion of patients in each group achieved 0 treated spontaneous BEs (prior on-demand: 11/13; switch: 7/10). All treated spontaneous BEs occurred in patients with type 3 VWD, and most were mucosal bleeds and/or menorrhagia. There were no fatal or life-threatening BEs and no spontaneous gastrointestinal BEs. One joint bleed occurred in a patient with historical joint bleeding. One AE (nonserious headache) was considered to be related to rVWF by the investigator, although there were confounding factors (medical history of alcohol abuse, hypertension, and depression).
The current sources of evidence supporting the use of VWF prophylaxis have been reviewed by a multidisciplinary panel as part of the development of the 2021 VWD management guidelines.7 These included 5 pre–post observational studies with an explicit comparison between prophylaxis and no prophylaxis time periods,11-14,20,22-25 8 pre–post studies with an implicit comparison between prophylaxis and no prophylaxis time periods,20,23,24,26-33 and 1 randomized controlled trial (PRO.WILL; EudraCT 2006-001383-23) comparing prophylaxis with on-demand treatment with VWF/FVIII concentrates.34 In this study, patients receiving prophylaxis (n = 10) had significantly (P < .0001) fewer spontaneous BEs than those receiving on-demand treatment (n = 9) with VWF/FVIII concentrates (Fanhdi/Alphanate; Grifols, Research Triangle Park, NC), with an incidence rate of 0.34 per patient-month vs 1.41 per patient-month, respectively.34 The risk of spontaneous BEs was also significantly lower with prophylaxis than with on-demand therapy. Unlike the study reported herein, 13 gastrointestinal BEs were reported, although 9 occurred in 1 patient. In line with the PRO.WILL study outcomes, an analysis of pooled data from observational studies with explicit comparative data demonstrated a reduced risk of BEs with prophylaxis relative to no prophylaxis (relative risk, 0.34; 95% CI, 0.25-0.46).7
Based on this evidence, the multidisciplinary guideline panel conditionally recommended the use of long-term VWF prophylaxis in patients with VWD and a history of severe and frequent bleeds. The panel concluded it is likely that long-term VWF prophylaxis reduces the risk of recurrent BEs and, possibly, complications of recurrent bleeds, such as hemarthrosis.7 The panel also suggested that patients are likely to highly value the reduction in risk of BEs, with the impact of BEs on HRQoL likely to inform their decision-making. However, the recommendation for long-term VWF prophylaxis was conditional based on the low certainty of the evidence of effects. In addition, the guideline panel identified several research needs related to prophylaxis for patients with VWD, including studies on the use of rVWF vs pdVWF concentrate.7 The study reported herein demonstrates a reduction in the frequency of spontaneous BEs with rVWF prophylaxis vs historical on-demand VWF therapy in this patient population and thus adds to the evidence reviewed by the guideline panel.
VWF:RCo mean half-life (t1/2) for rVWF (17.2 hours) at the initial assessment for the prior on-demand group was comparable with previously reported data (between 17.8 and 22.6 hours),18,19 and is longer than the t1/2 values reported for the pdVWF products Humate P (antihemophilic factor/VWF complex [human]; CSL Behring GmbH, Marburg, Germany: mean, 12.8 hours35) and Voncento (human coagulation FVIII/human VWF complex; CSL Behring GmbH, Marburg, Germany: mean, 13.7 hours36). Given that VWF stabilizes FVIII, it was not unanticipated to observe FVIII trough activity increased fivefold from the initial to the final assessment in patients previously receiving on-demand treatment with VWF. The consistency of the steady-state results confirms that rVWF exposure can be sustained over the treatment period and that the PK properties of rVWF are not dependent on the duration of prophylactic treatment.
Most patients received twice-weekly rVWF 50 IU/kg per infusion, which is consistent with recently published data on the prophylactic use of pdVWF.37 As the efficacy and PK/PD data were predominantly obtained at starting regimens of twice-weekly infusions of 50 ± 10 IU/kg in patients previously receiving on-demand therapy, results from this study support a starting rVWF prophylactic dose of 40 to 60 IU/kg twice weekly for most patients. Treating physicians should consider an increase in dose and/or dose frequency based on the occurrence of clinically significant breakthrough bleeds.
Of the 31 treated, all-cause BEs occurring during the 12-month study period, 7 (in 3 patients) were treated with rVWF plus a single infusion of rFVIII. Concomitant use of rVWF plus rFVIII was permitted for breakthrough bleeds if investigators felt rVWF alone was insufficient to establish hemostasis based on the patient’s clinical response. This approach was designed to reflect decision-making in real-world settings. Three surgical procedures during the study were also successfully managed with rVWF treatment (without rFVIII) to prevent and treat perioperative bleeding.
This study had several limitations. It was a nonrandomized, noncomparative trial involving a limited number of patients, although a justified and acceptable number by regulatory guidelines for this rare disease.21 Furthermore, differences between the data collection methods for historical BEs (ie, retrospective extraction from medical records) and on-study BEs (ie, prospective monitoring with an electronic patient diary) may have introduced bias and resulted in some data gaps. Historical BEs were included in the primary and secondary efficacy analyses if there was adequate evidence of a BE occurring and being treated with VWF infusions. However, the investigator assessment of hemostatic efficacy was not available for all historical BEs.
In summary, this prospective, open-label, multicenter, phase 3 study demonstrated the efficacy and safety of rVWF prophylaxis in patients with severe VWD. In patients previously treated on-demand with VWF products, rVWF prophylaxis reduced the frequency of spontaneous BEs requiring VWF treatment. Furthermore, patients switching from prophylaxis with pdVWF to rVWF prophylaxis experienced a similar level of hemostatic control over spontaneous BEs, and a high proportion of patients in both groups had no treated spontaneous BEs during rVWF prophylaxis. The safety profile of rVWF observed in this study was consistent with the previously established safety profile, and no new adverse drug reactions were identified.16,17
Acknowledgments
The authors thank all patients and their caregivers who took part in the phase 3 rVWF prophylaxis study, as well as the study investigators and sites (NCT02973087, https://clinicaltrials.gov/ct2/show/NCT02973087). Under the direction of the authors, medical writing support was provided by Joanne Vaughan, an employee of Excel Medical Affairs (Fairfield, CT), and was funded by Takeda Development Center Americas, Inc (Lexington, MA).
This study was funded by Baxalta US Inc, a Takeda company (Lexington, MA), and Baxalta Innovations GmbH, a Takeda company (Vienna, Austria).
Authorship
Contribution: F.W.G.L. contributed significantly to the analysis and interpretation of data and study conduct; F.P. contributed to the amendment in the design of the study and the analysis and interpretation of the data; M.E., A.T., G.C., M.W., and T.W. contributed to the analysis and interpretation of the data and study conduct; G.Ö. and J.Z. contributed significantly to the conception and design of the study and study conduct as well as to the analysis and interpretation of data; B.M. contributed significantly to the conception and design of the study and analysis and interpretation of data; J.B. and Y.W. contributed significantly to the analysis and interpretation of data; and all authors had access to data outputs, had key roles in the writing/editing of the manuscript, and have primary responsibility for the final approved manuscript.
Conflict-of-interest disclosure: F.W.G.L. has received grants/research funding from CSL Behring, Sobi, Takeda, and uniQure; consultancy fees from BioMarin, CSL Behring, Takeda, and uniQure (all fees to university); and was a Data Safety Monitoring Board Member for Roche. F.P. has participated in advisory boards of BioMarin, Grifols, Roche, Sanofi, Sobi, Spark, and Takeda. M.E. has received consulting fees from BioMarin, CSL Behring, Genentech/Roche, Kedrion, NHF, Novo Nordisk, Pfizer, Sanofi, and Takeda; and received research funding from CSL Behring, Genentech, Novo Nordisk, Sanofi, Takeda, and uniQure. A.T. has received honoraria from Bayer, Biotest, Chugai, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Roche, Sobi, and Takeda. G.C. has served on advisory boards or been a consultant for Bayer, BioMarin, CSL Behring, Grifols, Kedrion, LFB, Novo Nordisk, Roche, Sanofi, Sobi, Takeda, and uniQure. M.W. has served on advisory boards or been a consultant for Bayer, BioMarin, Bioverativ, Catalyst Biosciences, CSL Behring, Genentech, Hema Biologics, Novo Nordisk, Takeda, and uniQure. T.W. has received research support from Sanofi, Takeda, and Genentech. J.B., Y.W., J.Z., B.M., and G.Ö. are employees of Takeda Development Center Americas Inc. and Takeda stockholders.
Correspondence: Frank W. G. Leebeek, Department of Hematology, University Medical Center, Erasmus MC, Office Na-822, Wytemaweg 80, 3015 CN Rotterdam, The Netherlands; e-mail: f.leebeek@erasmusmc.nl.
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results reported in this article, will be made available within 3 months from the initial request to researchers who provide a methodologically sound proposal. The data will be provided after their deidentification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. The study sponsor analyzed the data and conducted the statistical analyses. Data outputs were shared and discussed with all authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal